
Experimental and Theoretical Studies of Dissociative Electron Attachment to Metabolites Oxaloacetic and Citric Acids

 , , and
, , and
Abstract
:1. Introduction
2. Methodology
2.1. Experimental Procedure
2.2. Theoretical Method
3. Results and Discussion
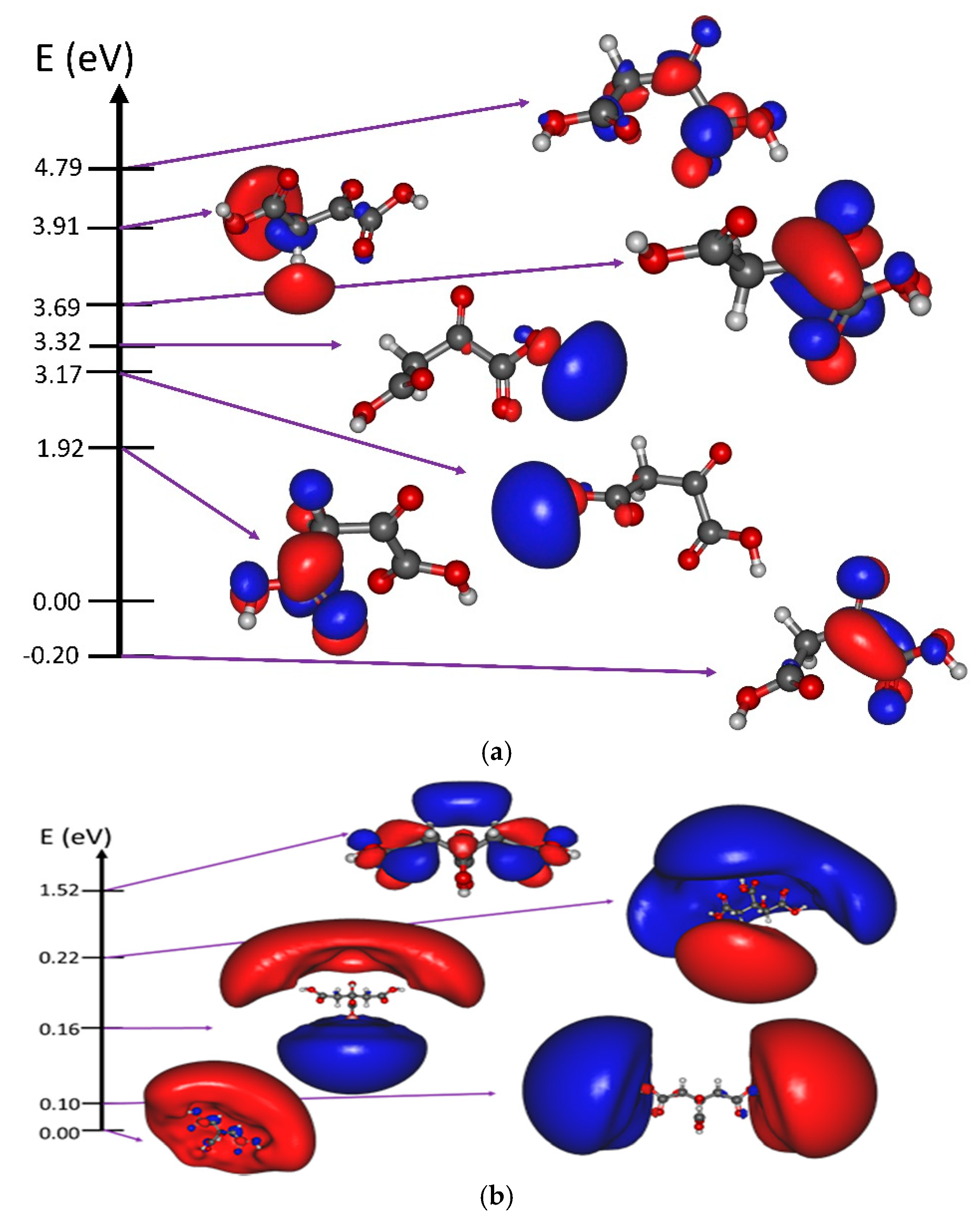
3.1. Resonant Electron Attachment and Formation of the Transitory Negative Anion
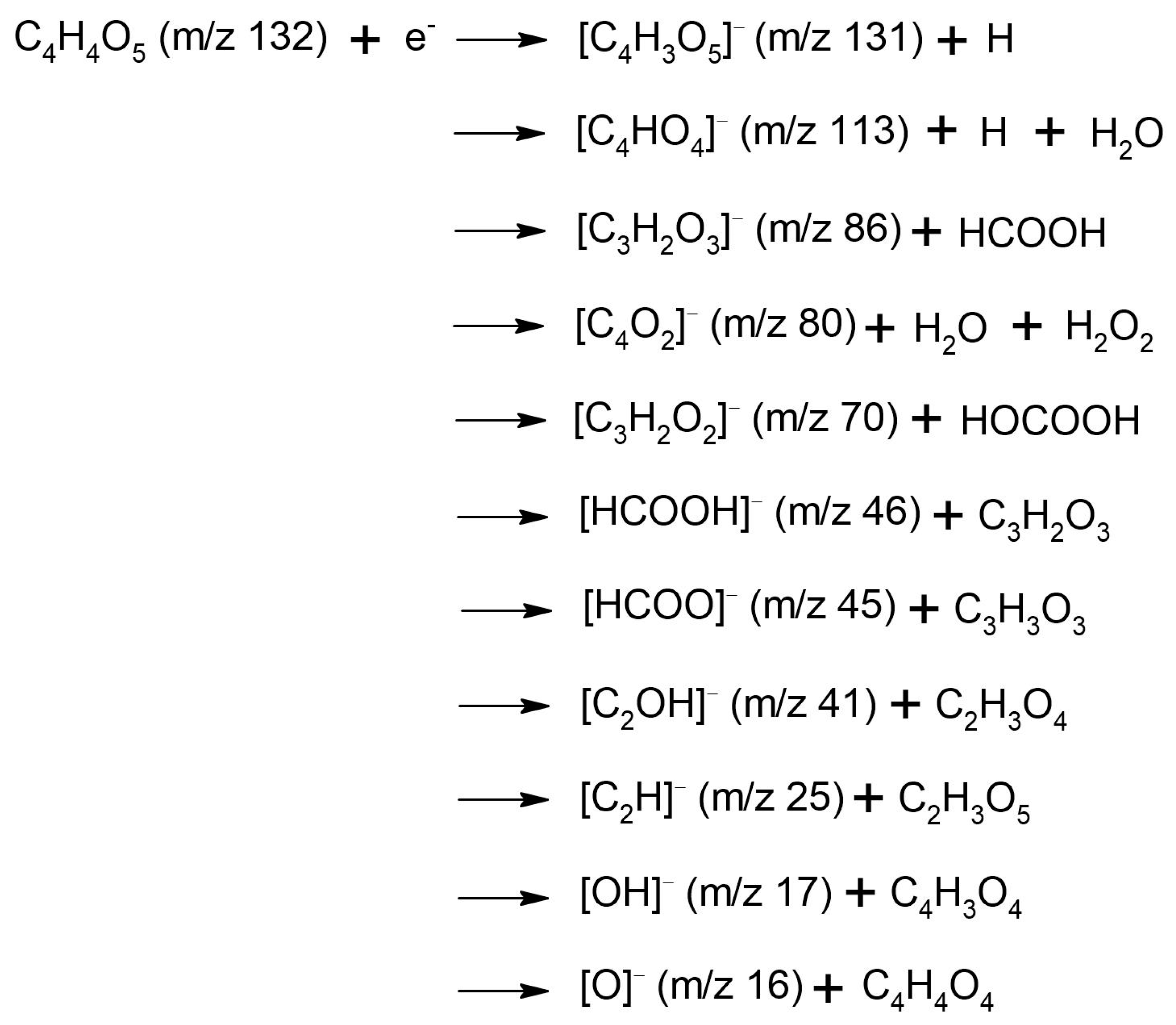
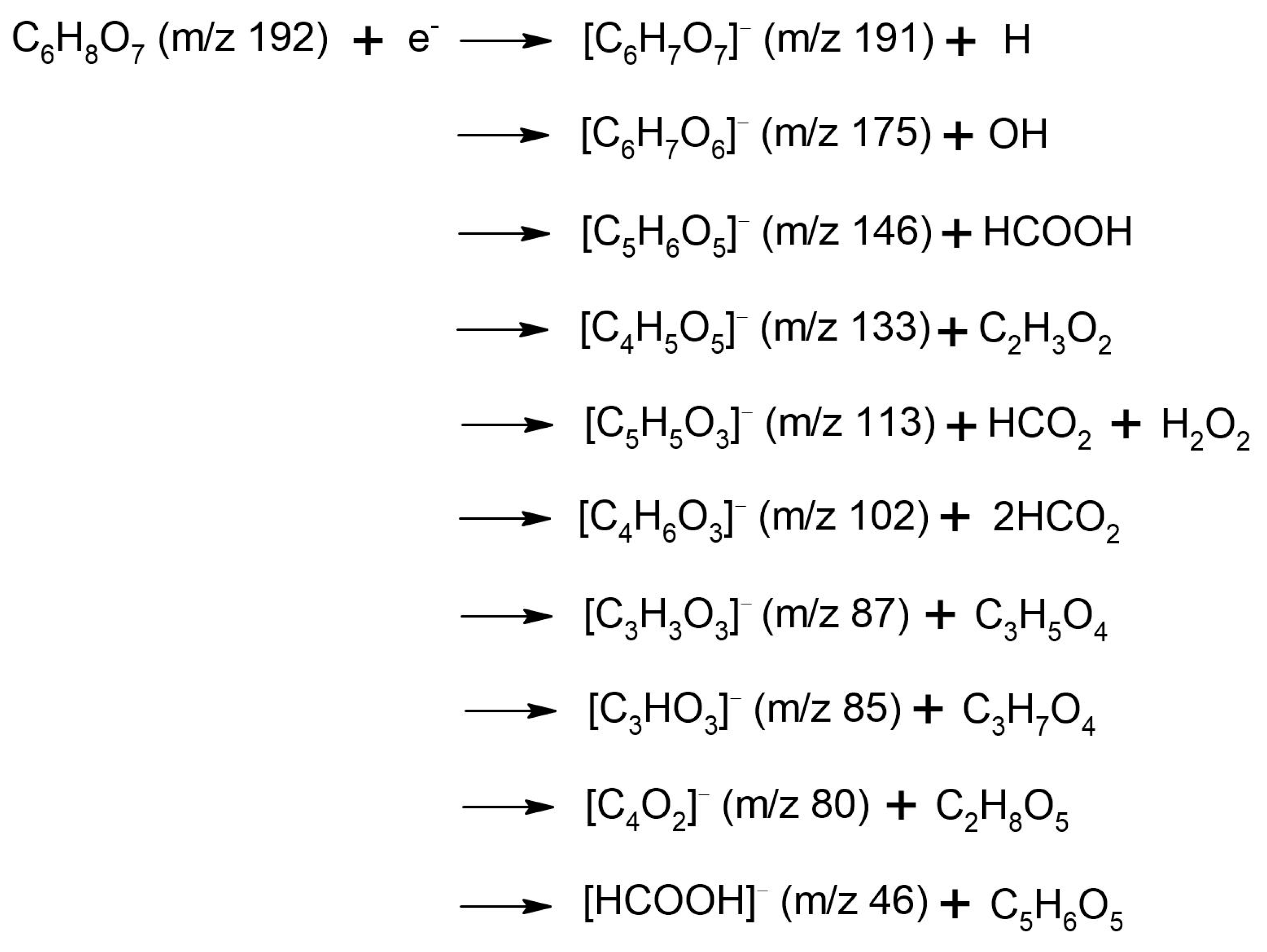
3.2. Fragmentation of the Transitory Negative Anion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pshenichnyuk, A.; Modelli, A.; Komolov, S. Interconnections between dissociative electron attachment and electron-driven biological processes. Int. Rev. Phys. Chem. 2018, 37, 125–170. [Google Scholar] [CrossRef]
- Alizadeh, E.; Orlando, T.M.; Sanche, L. Biomolecular Damage Induced by Ionizing Radiation: The Direct and Indirect Effects of Low-Energy Electrons on DNA. Annu. Rev. Phys. Chem. 2015, 66, 379–398. [Google Scholar] [CrossRef]
- Baccarelli, I.; Bald, I.; Gianturco, F.A.; Illenberger, E.; Kopyra, J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep. 2011, 508, 1–44. [Google Scholar] [CrossRef]
- Sanche, L. Interaction of low energy electrons with DNA: Applications to cancer radiation therapy, Radiat. Phys. Chem. 2016, 128, 36–43. [Google Scholar] [CrossRef]
- Schürmann, R.; Vogel, S.; Ebel, K.; Bald, I. The Physico-Chemical Basis of DNA Radiosensitization: Implications for Cancer Radiation Therapy. Chem. A Eur. J. 2018, 24, 10271–10279. [Google Scholar] [CrossRef] [PubMed]
- Bölher, E.; Wamecke, J.; Swiderek, P. Control of chemical reactions and synthesis by low energy electron. Chem. Soc. Rev. 2013, 42, 9219–9231. [Google Scholar]
- Abdoul-Carime, H.; Bald, I.; Illenberger, E.; Kopyra, J. Selective Synthesis of Ethylene and Acetylene from Dimethyl Sulfide Cold Films Controlled by Slow Electrons. J. Phys. Chem. C 2018, 122, 24137–24142. [Google Scholar] [CrossRef]
- Kopyra, J.; Rabilloud, F.; Wierzbicka, P.; Abdoul-Carime, H. Energy-Selective Decomposition of Organometallic Compounds by Slow Electrons: The Case of Chloro(dimethyl sulfide)gold(I). J. Phys. Chem. A 2021, 125, 966–972. [Google Scholar] [CrossRef]
- Mucke, M.; Braune, M.; Barth, S.; Förstel, M.; Lischke, T.; Ulrich, V.; Arion, T.; Becker, U.; Bradshaw, A.; Hergenhahn, U. A hitherto unrecognized source of low-energy electrons in water. Nat. Phys. 2010, 6, 143–146. [Google Scholar] [CrossRef]
- Boudaiffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 eV) Electrons. Science 2000, 287, 1658–1660. [Google Scholar]
- Meißner, R.; Kočišek, J.; Feketeová, L.; Fedor, J.; Fárník, M.; Limão-Vieira, P.; Illenberger, E.; Denifl, S. Low-energy electrons transform the nimorazole molecule into a radiosensitizer. Nat. Commun. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Rak, J.; Chomicz, L.; Wiczk, J.; Westphal, K.; Zdrowowicz, M.; Wityk, P.; Żyndul, M.; Makurat, S.; Golon, Ł. Mechanisms of Damage to DNA Labeled with Electrophilic Nucleobases Induced by Ionizing or UV Radiation. J. Phys. Chem. B 2015, 119, 8227–8238. [Google Scholar] [CrossRef]
- Zhan, C.; Chen, X.-J.; Yi, J.; Li, J.-F.; Wu, D.-Y.; Tian, Z.-Q. From plasmon-enhanced molecular spectroscopy to plasmon-mediated chemical reactions. Nat. Rev. Chem. 2018, 2, 216–230. [Google Scholar] [CrossRef]
- Schürmann, R.; Bald, I. Real-time monitoring of plasmon induced dissociative electron transfer to the potential DNA radiosensitizer 8-bromoadenine. Nanoscale 2017, 9, 1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlather, A.E.; Manjavacas, A.; Lauchner, A.; Marangoni, V.S.; DeSantis, C.J.; Nordlander, P.; Halas, N.J. Hot Hole Photoelectrochemistry on Au@SiO2@Au Nanoparticles. J. Phys. Chem. Lett. 2017, 8, 2060−2067. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Campbell, J.M.; Deng, W.; Guller, A.; Nisar, S.; Liu, G.; Wilson, B.C.; Goldys, E.M. Mechanisms for Tuning Engineered Nanomaterials to Enhance Radiation Therapy of Cancer. Adv. Sci. 2020, 7, 2003584. [Google Scholar] [CrossRef] [PubMed]
- Kopyra, J. Low energy electron attachment to the nucleotide deoxycytidine monophosphate: Direct evidence for the molecular mechanisms of electron-induced DNA strand breaks. Phys. Chem. Chem. Phys. 2012, 14, 8287–8289. [Google Scholar] [CrossRef]
- Wyrzykowski, D.; Hebanowska, E.; Nowak-Wiczk, G.; Makowski, M.; Chmurzyński, L. Thermal behaviour of citric acid and isomeric aconitic acids. J. Therm. Anal. Calorim. 2011, 104, 731–735. [Google Scholar] [CrossRef] [Green Version]
- Sigma Aldrich Information Product Number 04126.
- Gohlke, S.; Abdoul-Carime, H.; Illenberger, E. Dehydrogenation of adenine induced by slow (<3 eV) electrons. Chem. Phys. Lett. 2003, 380, 595–599. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Sommerfeld, T.; Weber, R.J. Empirical Correlation Methods for Temporary Anions. J. Phys. Chem. A 2011, 115, 6675–6682. [Google Scholar] [CrossRef]
- Illenberger, E.; Momigny, J. Gaseous Molecular Ions. An Introduction to Elementary Processes Induced by Ionization; Baümgartel, H., Franck, E.U., Grünbein, W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Sommerfeld, T. Dipole bound states as doorways in (dissociative) electron attachment. J. Phys. Conf. Ser. 2005, 4, 245–250. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Desfrançois, C. Electron weakly bound to molecules by dipolar, quadrupolar and polarization forces. Eur. Phys. J. D 1998, 2, 149–156. [Google Scholar] [CrossRef]
- Li, Z.; Ryszka, M.; Dawley, M.M.; Carmichael, I.; Bravaya, K.B.; Ptasińska, S. Dipole-Supported Electronic Resonances Mediate Electron-Induced Amide Bond Cleavage. Phys. Rev. Lett. 2019, 122, 73002. [Google Scholar] [CrossRef] [Green Version]
- Kopyra, J.; Rabilloud, F.; Abdoul-Carime, H. Core-excited resonances initiated by unusually low energy electrons observed in dissociative electron attachment to Ni(II)(bis)acetylacetonate. J. Chem. Phys. 2020, 153, 124302. [Google Scholar] [CrossRef]
- Pshenichnyuk, S.A.; Asfandiarov, N.L. Structural rearrangements as relaxation pathway for molecular negative ions formed via vibrational Feshbach resonance. Phys. Chem. Chem. Phys. 2020, 22, 16150–16156. [Google Scholar] [CrossRef]
- Pelc, A.; Sailer, W.; Scheier, P.; Mason, N.; Märk, T. Low energy electron attachment to formic acid. Eur. Phys. J. D 2002, 20, 441–444. [Google Scholar] [CrossRef]
- Sailer, W.; Pelc, A.; Probst, M.; Limtrakul, J.; Scheier, P.; Illenberger, E.; Märk, T.D. Dissociative electron attachment to acetic acid (CH3COOH). Chem. Phys. Lett. 2003, 378, 250–256. [Google Scholar] [CrossRef]
- Zawadzki, M.; Ranković, M.; Kočišek, J.; Fedor, J. Dissociative electron attachment and anion-induced dimerization in pyruvic acid. Phys. Chem. Chem. Phys. 2018, 20, 6838–6844. [Google Scholar] [CrossRef]
- Zawadzki, M.; Wierzbicka, P.; Kopyra, J. Dissociative electron attachment to benzoic acid (C7H6O2). J. Chem. Phys. 2020, 152, 174304. [Google Scholar] [CrossRef]
- Da Silva, F.F.; Varella, M.T.D.N.; Jones, N.C.; Hoffmann, S.V.; Denifl, S.; Bald, I.; Kopyra, J. Electron-Induced Reactions in 3-Bromopyruvic Acid. Chem. A Eur. J. 2019, 25, 5498–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, S.; Hazra, M.K. H2CO3→ CO2+ H2O decomposition in the presence of H2O, HCOOH, CH3COOH, H2SO4 and HO2 radical: Instability of the gas-phase H2CO3 molecule in the troposphere and lower stratosphere. RSC Adv. 2015, 5, 17623–17635. [Google Scholar] [CrossRef]
- Da Silva, F.F.; Matias, C.; Almeida, D.; Garcia, G.; Ingólfsson, O.; Flosadóttir, H.D.; Ómarsson, B.; Ptasinska, S.; Puschnigg, B.; Scheier, P.; et al. NCO−, a Key Fragment Upon Dissociative Electron Attachment and Electron Transfer to Pyrimidine Bases: Site Selectivity for a Slow Decay Process. J. Am. Soc. Mass Spectrom. 2013, 24, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Bald, I.; Langer, J.; Tegeder, P.; Ingólfsson, O. From isolated molecules through clusters and condensates to the building blocks of life. Int. J. Mass Spectrom. 2008, 277, 4–25. [Google Scholar] [CrossRef]
- Schürmann, R.; Tsering, T.; Tanzer, K.; Denifl, S.; Kumar, S.V.K.; Bald, I. Resonant Formation of Strand Breaks in Sensitized Oligonucleotides Induced by Low-Energy Electrons (0.5–9 eV). Angew. Chem. Int. Ed. 2017, 56, 10952–10955. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Bouteiller, Y.; Desfrançois, C.; Philippe, L.; Schermann, J.P.; Niinistö, L.; Styring, S.; Tommos, C.; Warncke, K.; Wood, B.R. Excess Electrons in Polar Cluster Anions. Acta Chem. Scand. 1997, 51, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Blakely, W.F.; Fuciarelli, A.F.; Wegher, B.J.; Dizdaroglu, M. Hydrogen peroxide-induced base damage in deoxyribonucleic acid. Radiat. Res. 1990, 121, 338–343. [Google Scholar] [CrossRef]
- Dizaroglu, M.; Gajewski, E. Selected ion mass spectrometry: Assays of oxidative DNA damage. Methods Enzymol. 1990, 186, 530–544. [Google Scholar]
- Yermilov, V.; Yoshie, Y.; Rubio, J.; Ohshima, H. Effects of carbon dioxide/bicarbonate on induction of DNA single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine and base-propenal mediated by peroxynitrite. FEBS Lett. 1996, 399, 67–70. [Google Scholar] [CrossRef] [Green Version]








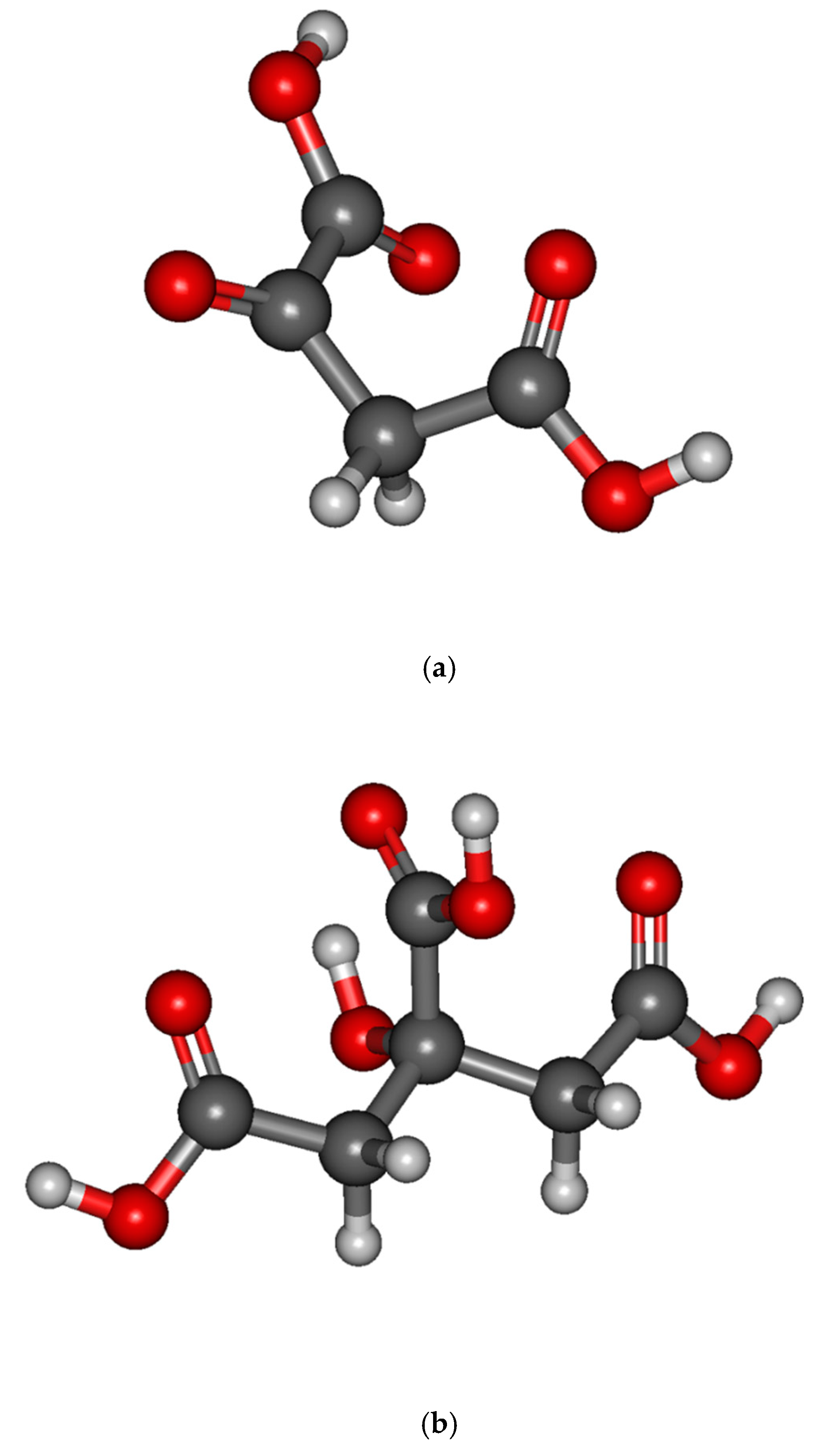{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
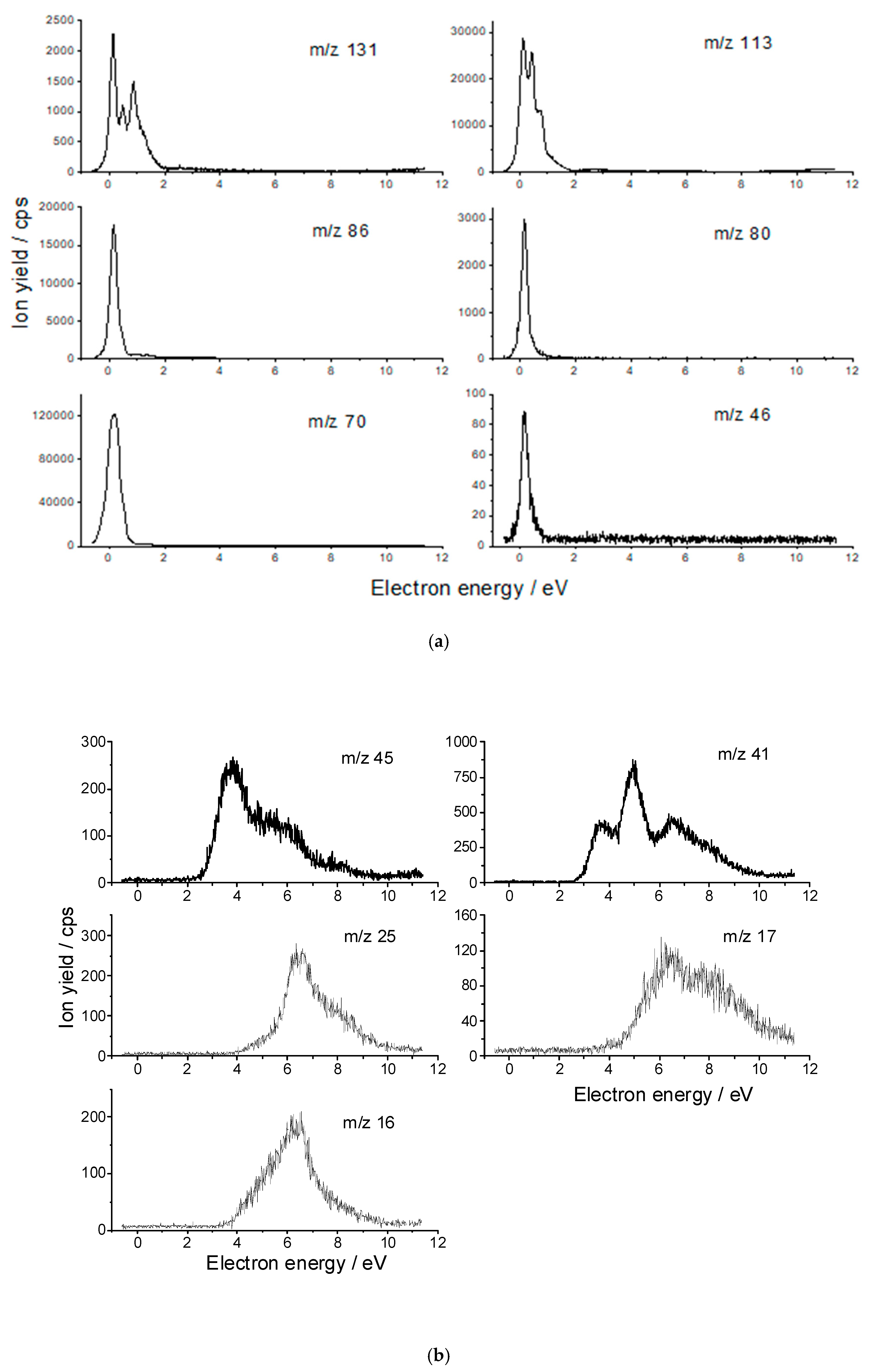
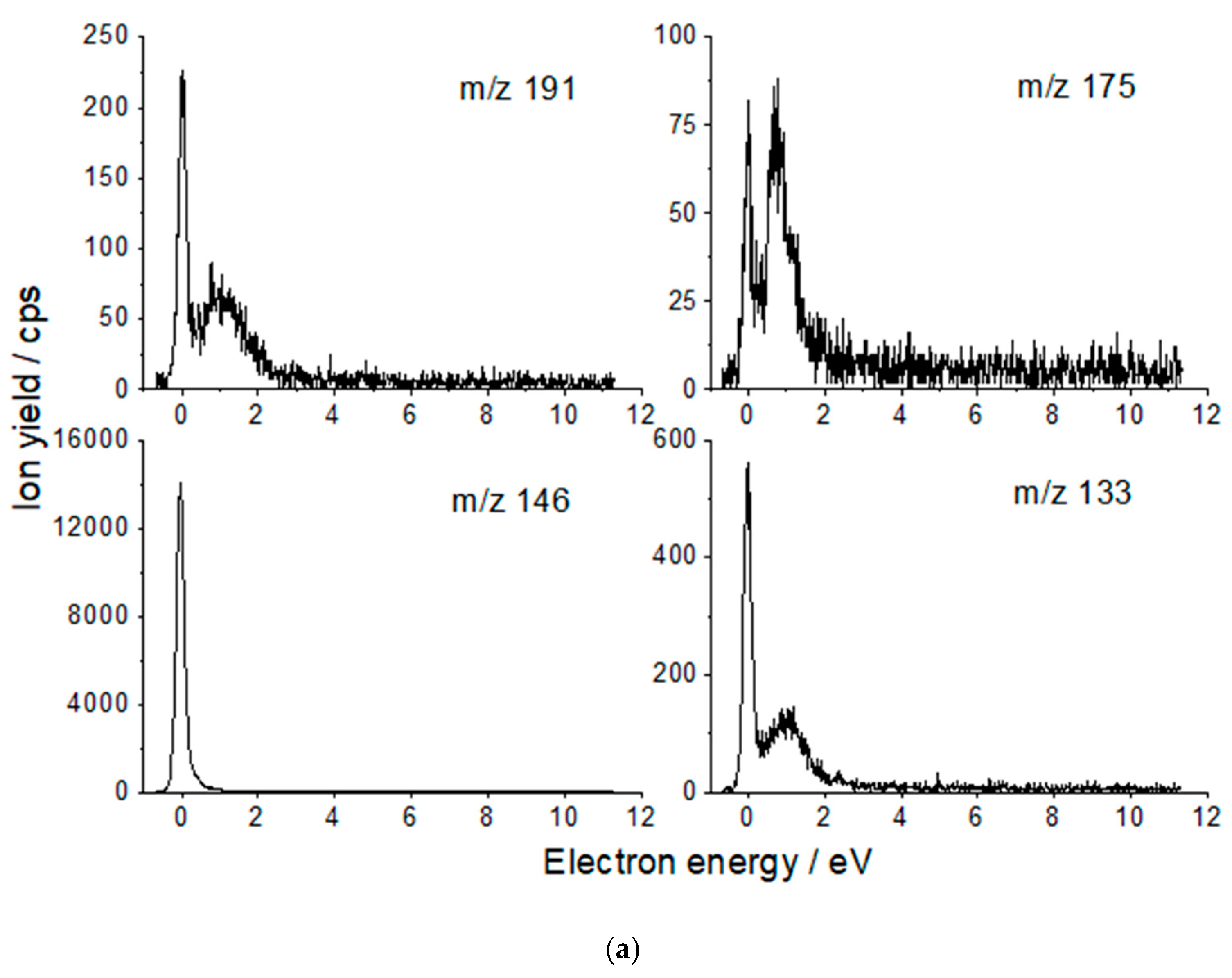
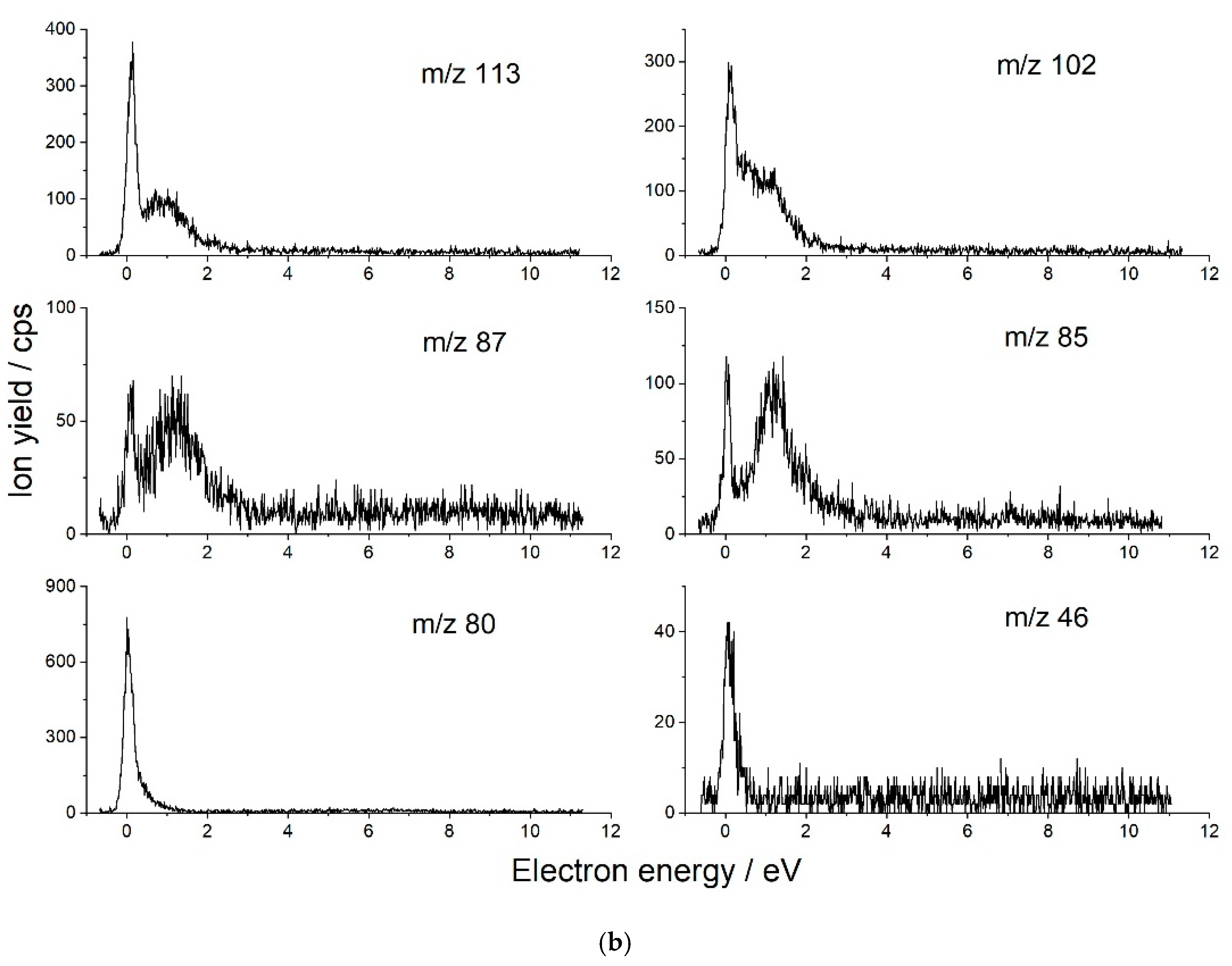
| Anion Fragment from OAA m/z | Resonance Energies (eV) | Anion Fragment from CA m/z | Resonance Energies (eV) |
|---|---|---|---|
| 131 [C4H3O5]− [OAA-H]− | 0.14 0.48 0.88 2.78 * | 191 [C6H7O7]− [CA-H]− | 0.01 0.99 |
| 113 [C4HO4]− [OAA-H2O-H]− | 0.15 0.44 0.71 1.13 2.75 * 4.8 * | 175 [C6H7O6]− [CA-OH]− | 0.0 0.85 |
| 86 [C3H2O3]− [OAA-HCOOH]− | 0.14 1.3 * 3.1 * 6.4 * | 146 [C5H6O5]− [CA-HCOOH]− | 0.0 |
| 80 [C4O2]− [OAA-H2O-H2O2]− | 0.16 0.56 1.4 * | 133 [C4H5O5]− [CA-C2H3O2]− | 0.0 0.94 |
| 70 [C2H2O2]− [OAA-HOCOOH]− | 0.17 0.53 * 1.3 2.83 * 4.4 * | 113 [C5H5O3]− [CA-H2O2-HCO2]− | 0.12 0.89 |
| 46 [HCOOH]− | 0.17 | 102 [C4H6O3]− [CA-2(HCO2)]− | 0.11 0.41 0.98 |
| 45 [HCOO]− | 3.7 5.0 6.3 7.0 | 87 [C3H3O3]− | 0.09 1.22 |
| 41 [C2OH]− | 3.7 4.95 6.6 ~8.0 | 85 [C3HO3]− | 0.04 1.20 |
| 25 [C2H]− | 6.5 8.0 | 80 [C4O2,]− | 0.05 |
| 17 [OH]− | 5.3(4) * 6.3 8.1 | 46 [HCOOH]− | 0.09 |
| 16 [O]− | 5.2 6.3 7.0 |
| M = OAA * | M = CA | |||
|---|---|---|---|---|
| Enthalpy | Gibbs Free Energy | Enthalpy | Gibbs Free Energy | |
| [M-H]− | 0.95 (0.84) | 0.19 (0.05) 0.31 (0.17) | 0.89 | 0.17 0.27 |
| [M-HCOO-H]− | 4.78 (4.73) | 3.05 (3.02) 3.38 (3.34) | 4.72 | 2.98 3.31 |
| [M-HCOOH]− | 0.22 (0.17) | −0.73 (−0.76) −0.53 (−0.57) | 0.16 | −0.80 −0.60 |
| [M-HCOO-OH]− | 1.06 (1.01) | 0.13 (0.10) 0.68 (0.66) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopyra, J.; Wierzbicka, P.; Tulwin, A.; Thiam, G.; Bald, I.; Rabilloud, F.; Abdoul-Carime, H. Experimental and Theoretical Studies of Dissociative Electron Attachment to Metabolites Oxaloacetic and Citric Acids. Int. J. Mol. Sci. 2021, 22, 7676. https://doi.org/10.3390/ijms22147676
Kopyra J, Wierzbicka P, Tulwin A, Thiam G, Bald I, Rabilloud F, Abdoul-Carime H. Experimental and Theoretical Studies of Dissociative Electron Attachment to Metabolites Oxaloacetic and Citric Acids. International Journal of Molecular Sciences. 2021; 22(14):7676. https://doi.org/10.3390/ijms22147676
Chicago/Turabian StyleKopyra, Janina, Paulina Wierzbicka, Adrian Tulwin, Guillaume Thiam, Ilko Bald, Franck Rabilloud, and Hassan Abdoul-Carime. 2021. "Experimental and Theoretical Studies of Dissociative Electron Attachment to Metabolites Oxaloacetic and Citric Acids" International Journal of Molecular Sciences 22, no. 14: 7676. https://doi.org/10.3390/ijms22147676