
Integrative Transcriptomic and Proteomic Analysis Reveals an Alternative Molecular Network of Glutamine Synthetase 2 Corresponding to Nitrogen Deficiency in Rice (Oryza sativa L.)

Abstract
:1. Introduction
2. Results
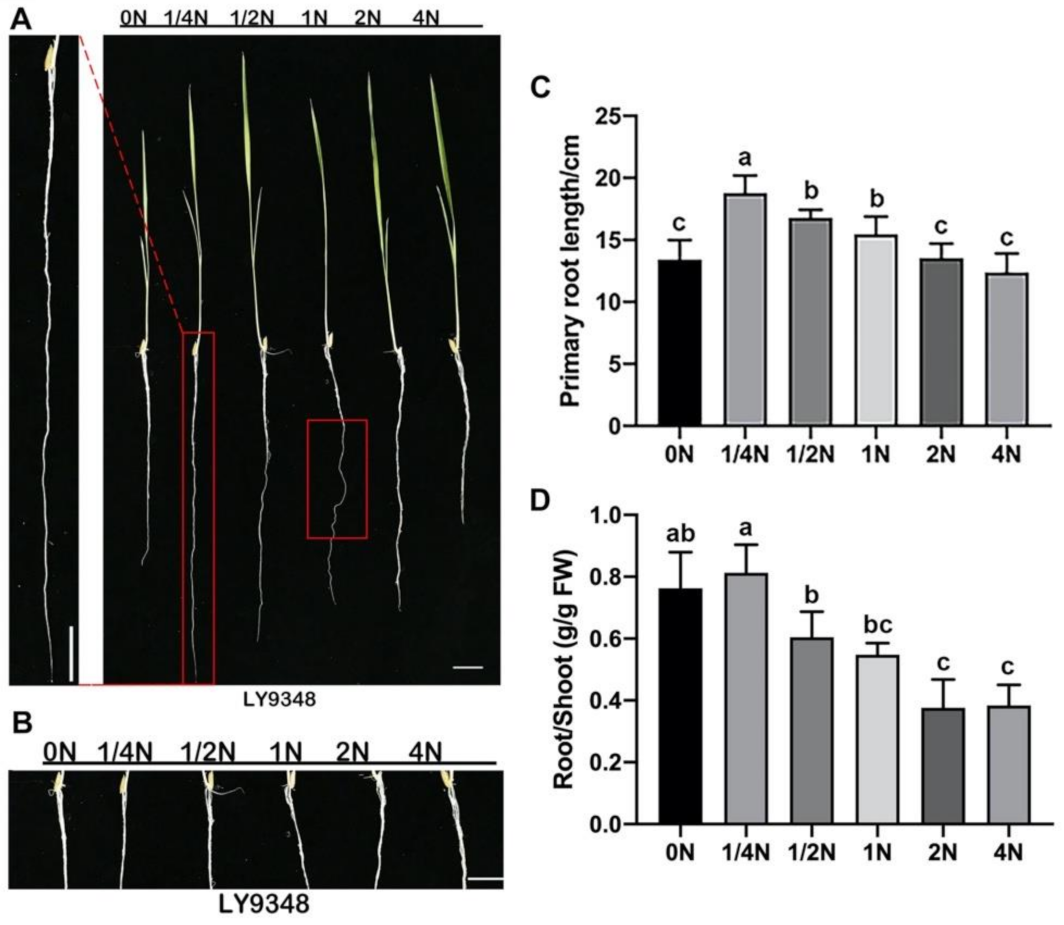
2.1. Root Phenotypes Identified in Different Nitrogen Dosage Treatments
2.2. N-Deficiency Responses in Proteome Profiling in LY9348 Roots
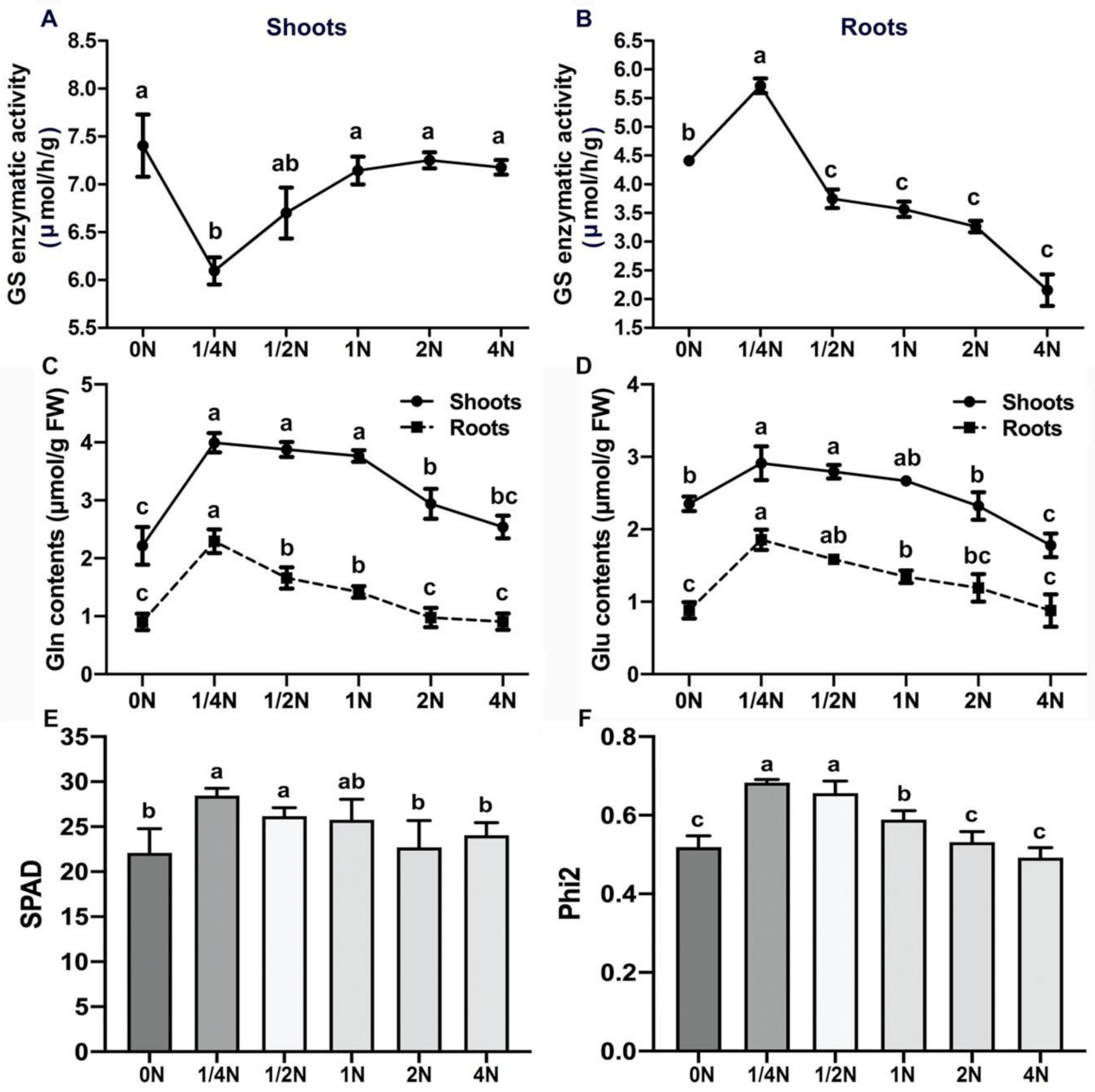
2.3. GS2 Unexpected Concentration in the Roots after 1/4N Treatment
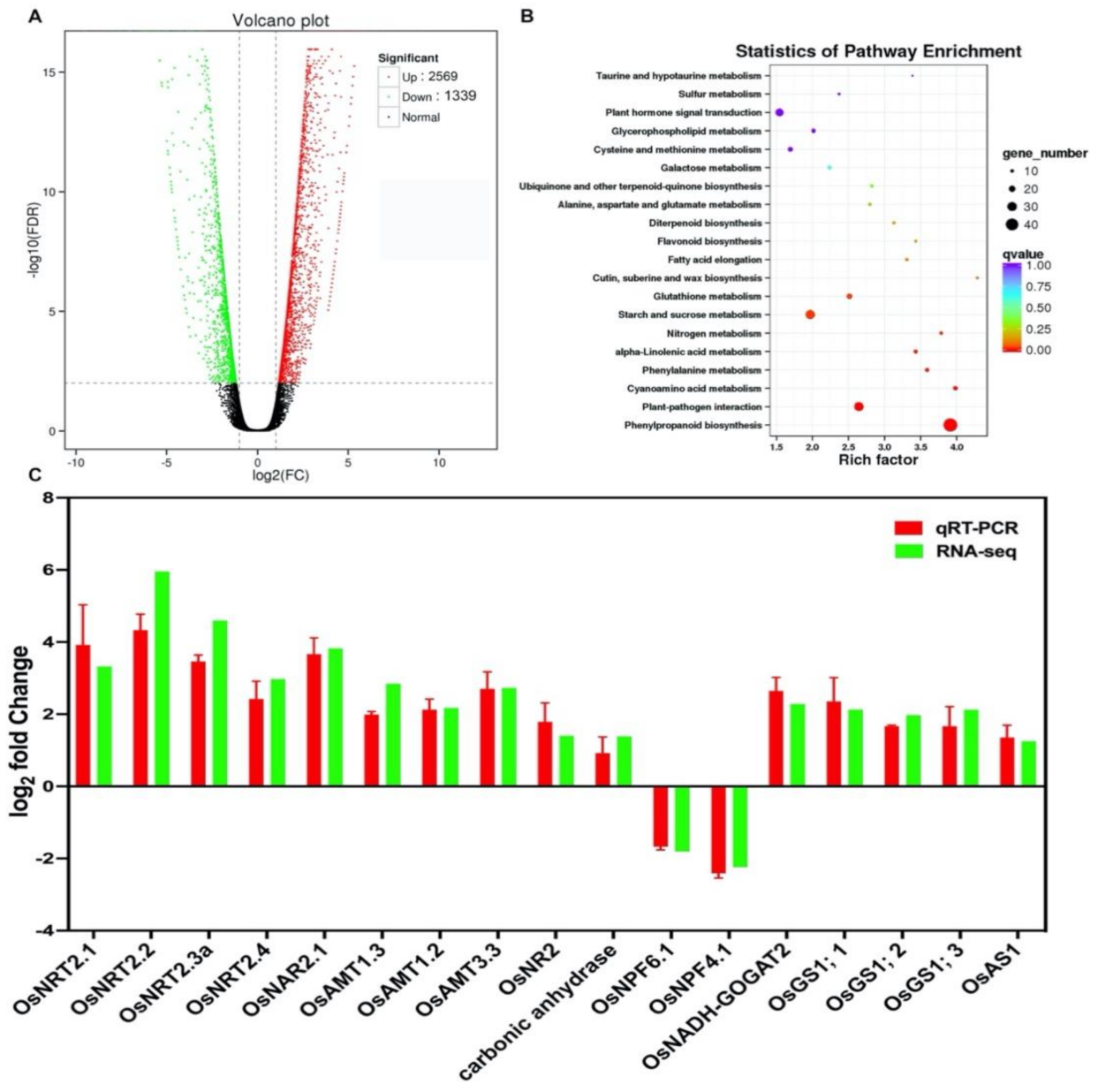
2.4. N-Deficiency Responses in Transcriptome Profiling in LY9348 Roots
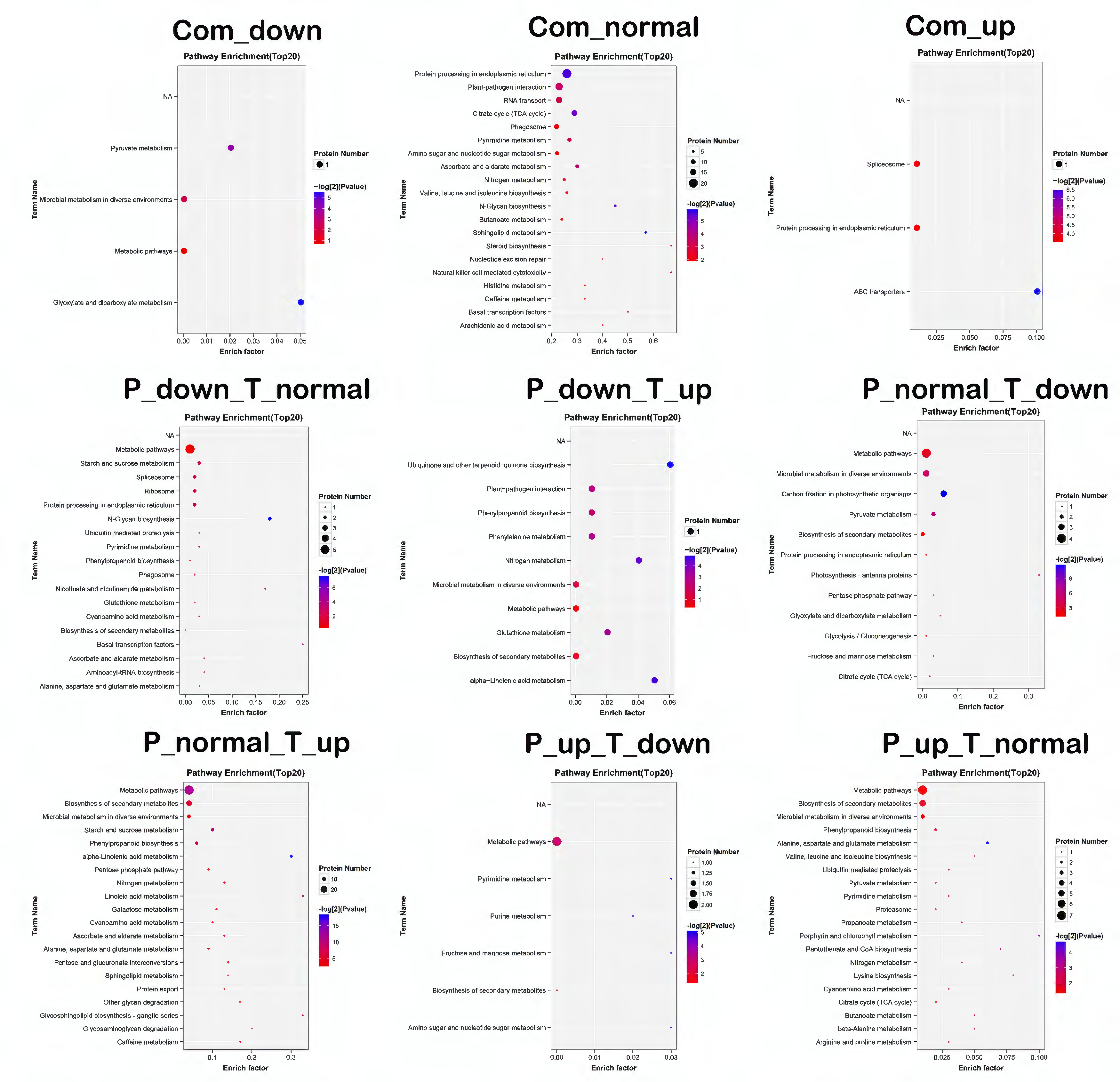
2.5. Correlation Analysis between Proteome and Transcriptome
2.6. Modulation of Phenylpropanoid Biosynthesis Pathways under N-Deficiency Stress
2.7. Modulation of Glutathione Metabolism in N-Deficiency Stress
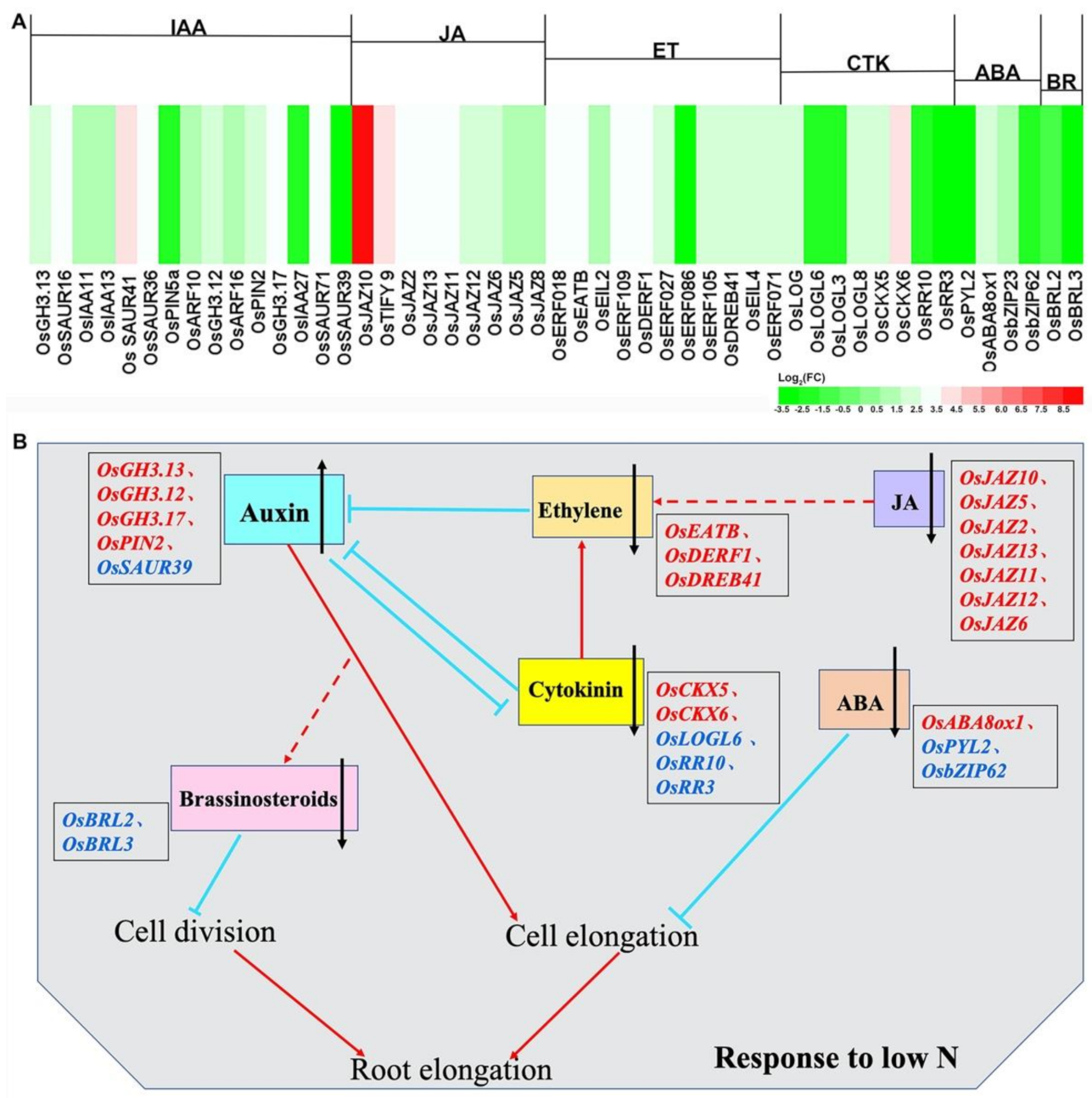
2.8. Transcription Factors (TFs) and Plant Hormones Involved in N-Deficiency Stress
3. Discussion
3.1. Root Morphological Changes in N-Deficiency Stress
3.2. New GS Pathway for N Assimilation under N-Deficiency Stress
3.3. Glutamine as a Signal Molecule for N Assimilation and Transportation
3.4. Relatively Normal Shoot Growth in N-Deficiency Stress
3.5. Prolonged N-Deficiency Responses
4. Materials and Methods
4.1. Plants and Materials
4.2. Phenotypic Analysis
4.3. RNA Extraction and Sequencing
4.4. qRT-PCR Validation
4.5. Protein Extraction, Digestion, and Identification by Using Label-Free Technology
4.6. Protein and RNA Correlation Analysis
4.7. GO, KEGG Enrichment Analysis, and Functional Annotation of DEGs and DAPs
4.8. GS Enzymatic Activity Assays
4.9. Determination of Glu and Gln Contents
4.10. Determination of SPAD and Phi2 Values
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, S.; Fang, J.; Xu, F.; Wang, W.; Chu, C. Rice HOX12 Regulates Panicle Exsertion by Directly Modulating the Expression of ELONGATED UPPERMOST INTERNODE1. Plant Cell 2016, 28, 680–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.H.; Zhang, Y.L.; Shen, Q.R.; Wang, S.-W. College, Nitrate Effect on Rice Growth and Nitrogen Absorption and Assimilation at Different Growth Stages. Pedosphere 2006, 16, 707–717. [Google Scholar]
- Tatsumi, K.; Abiko, T.; Kinose, Y.; Inagaki, S.; Izuta, T. Effects of ozone on the growth and yield of rice (Oryza sativa L.) under different nitrogen fertilization regimes. Environ. Sci. Pollut. Res. 2019, 26, 32103–32113. [Google Scholar] [CrossRef]
- Chen, M.; Chen, G.; Di, D.; Kronzucker, H.J.; Shi, W. Higher nitrogen use efficiency (NUE) in hybrid “super rice” links toimproved morphological and physiological traits in seedling roots. J. Plant Physiol. 2020, 251, 153191. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shi, J.; Fan, T.; Wang, C.; Liu, L.; Jin, H.; Foba, C.N.; Wang, M. The control of the brown planthopper by the rice Bph14 gene is affected by nitrogen. Pest Manag. Sci. 2020, 76, 3649–3656. [Google Scholar] [CrossRef]
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to Green Super Rice. Nat. Rev. Genet. 2018, 19, 505–517. [Google Scholar] [CrossRef]
- Lochab, S.; Kumar, P.A.; Raghuram, N. Molecular characterization of nitrate uptake and assimilatory pathway in Arthrospira platensis reveals nitrate induction and differential regulation. Arch. Microbiol. 2014, 196, 385–394. [Google Scholar] [CrossRef]
- Perchlik, M.; Tegeder, M. Leaf Amino Acid Supply Affects Photosynthetic and Plant Nitrogen Use Efficiency under Nitrogen Stress. Plant Physiol. 2018, 178, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Crawford, N.M.; Forde, B. Molecular and Developmental Biology of Inorganic Nitrogen Nutrition. Arab. Book 2002, 1, e0011. [Google Scholar] [CrossRef] [Green Version]
- Wallach, T.; Daniel, W. EvaluatingDecision Rules for Nitrogen Fertilization. Biometrics 2000, 56, 420–426. [Google Scholar]
- Yi, J.; Gao, J.; Zhang, W.; Zhao, C.; Wang, Y.; Zhen, X. Differential Uptake and Utilization of Two Forms of Nitrogen in Japonica Rice Cultivars From North-Eastern China. Front. Plant Sci. 2019, 10, 1061. [Google Scholar] [CrossRef]
- Temple, S.J.; Vance, C.P.; Gantt, J.S. Glutamate synthase and nitrogen assimilation. Trends Plant Sci. 1998, 3, 51–56. [Google Scholar] [CrossRef]
- Forde, B.G.; Lea, P.J. Glutamate in plants: Metabolism, regulation, and signalling. J. Exp. Bot. 2007, 58, 2339–2358. [Google Scholar] [CrossRef]
- Tabuchi, M.; Abiko, T.; Yamaya, T. Assimilation of ammonium ions and reutilization of nitrogen in rice (Oryza sativa L.). J. Exp. Bot. 2007, 58, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Forde, B.G. Glutamate signalling in roots. J. Exp. Bot. 2013, 65, 779–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaya, T.; Kusano, M. Evidence supporting distinct functions of three cytosolic glutamine synthetases and two NADH-glutamate synthases in rice. J. Exp. Bot. 2014, 65, 5519–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellamuthu, V.R.; Ermilova, E.; Lapina, T.; Lüddecke, J.; Minaeva, E.; Herrmann, C.; Hartmann, M.D.; Forchhammer, K. Hartmann and Karl Forchhammer, A widespread gluatmine-sensing mechanism in the plant kingdom. Cell 2014, 159, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Linka, M.; Weber, A.P. Shuffling ammonia between mitochondria and plastids during photorespiration. Trends Plant Sci. 2005, 10, 461–465. [Google Scholar] [CrossRef]
- Funayama, K.; Kojima, S.; Tabuchi-Kobayashi, M.; Sawa, Y.; Nakayama, Y.; Hayakawa, T.; Yamaya, T. Cytosolic Glutamine Synthetase1;2 is Responsible for the Primary Assimilation of Ammonium in Rice Roots. Plant Cell Physiol. 2013, 54, 934–943. [Google Scholar] [CrossRef]
- Ohashi, M.; Ishiyama, K.; Kojima, S.; Konishi, N.; Nakano, K.; Kanno, K.; Hayakawa, T.; Yamaya, T. Asparagine Synthetase1, but not Asparagine Synthetase2, is Responsible for the Biosynthesis of Asparagine Following the Supply of Ammonium to Rice Roots. Plant Cell Physiol. 2015, 56, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Y.; Cheng, Y.H.; Chen, K.-E.; Tsay, Y.-F. Nitrate Transport, Signaling, and Use Efficiency. Annu. Rev. Plant Biol. 2018, 69, 85–122. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef] [Green Version]
- Hirel, B.; Le Gouis, J.; Ney, B.; Gallais, A. The challenge of improving nitrogen use efficiency in crop plants: Towards a more central role for genetic variability and quantitative genetics within integrated approaches. J. Exp. Bot. 2007, 58, 2369–2387. [Google Scholar] [CrossRef]
- Hu, B.; Wang, W.; Doug, S.J.B.; Tang, J.Y.; Li, H.; Che, R.H.; Zhang, Z.H.; Chai, X.Y.; Wang, H.R.; Wang, Y.Q.; et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef]
- Wang, W.; Hu, B.; Yuan, D.; Liu, Y.; Che, R.; Hu, Y.; Ou, S.; Liu, Y.; Zhang, Z.; Wang, H.; et al. Expression of the Nitrate Transporter Gene OsNRT1.1A/OsNPF6.3 Confers High Yield and Early Maturation in Rice. Plant Cell 2018, 30, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Fan, X.; Feng, H.; Miller, T.; Shen, Q.; Xu, G. Rice OsNAR2.1 interacts with OsNRT2.1, OsNRT2.2 and OsNRT2.3a nitrate transporters to provide uptake over high and low concentration ranges. Plant Cell Environ. 2011, 34, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Yan, M.; Fan, X.; Li, B.; Shen, Q.; Miller, A.; Xu, G. Spatial expression and regulation of rice high-affinity nitrate transporters by nitrogen and carbon status. J. Exp. Bot. 2011, 62, 2319–2332. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Tang, Z.; Tan, Y.; Zhang, Y.; Luo, B.; Yang, M.; Lian, X.; Shen, Q.; Miller, A.; Xu, G. Overexpression of a pH-sensitive nitrate transporter in rice increases crop yields. Proc. Natl. Acad. Sci. USA 2016, 113, 7118–7123. [Google Scholar] [CrossRef] [Green Version]
- Gaur, V.S.; Singh, U.S.; Gupta, A.K.; Kumar, A. Influence of different nitrogen inputs on the members of ammonium transporter and glutamine synthetase genes in two rice genotypes having differential responsiveness to nitrogen. Mol. Biol. Rep. 2012, 39, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Wang, Y.; Chen, G.; Zhang, A.; Yang, S.; Shang, L.; Wang, D.; Ruan, B.; Liu, C.; Jiang, H.; et al. The indica nitrate reductase gene OsNR2 allele enhances rice yield potential and nitrogen use efficiency. Nat. Commun. 2019, 10, 5207. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xuan, W.; Tian, Y.; Fan, L.; Sun, J.; Tang, W.; Chen, G.; Wang, B.; Liu, Y.; Wu, W.; et al. Enhanced OsNLP4-OsNiR cascade confers nitrogen use efficiency by promoting tiller number in rice. Plant Biotechnol. J. 2021, 19, 167–176. [Google Scholar] [CrossRef]
- Kusano, M.; Fukushima, A.; Tabuchi-Kobayashi, M.; Funayama, K.; Kojima, S.; Maruyama, K.; Yamamoto, Y.Y.; Nishizawa, T.; Kobayashi, M.; Wakazaki, M.; et al. Cytosolic GLUTAMINE SYNTHETASE1;1 Modulates Metabolism and Chloroplast Development in Roots. Plant Physiol. 2020, 182, 1894–1909. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Xiao, J.; Zhang, Q.; Lian, X. Co-suppressed glutamine synthetase2 gene modifies nitrogen metabolism and plant growth in rice. Chin. Sci. Bull. 2010, 55, 823–833. [Google Scholar] [CrossRef]
- Yang, X.; Nian, J.; Xie, Q.; Feng, J.; Zhang, F.; Jing, H.; Zhang, J.; Dong, G.; Liang, Y.; Peng, J.; et al. Rice Ferredoxin-Dependent Glutamate Synthase Regulates Nitrogen–Carbon Metabolomes and Is Genetically Differentiated between japonica and indica Subspecies. Mol. Plant 2016, 9, 1520–1534. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xu, Z.; Zhang, L.; Li, S.; Wang, S.; Yang, H.; Liu, X.; Zeng, D.; Liu, Q.; Qian, Q.; et al. MYB61 is regulated by GRF4 and promotes nitrogen utilization and biomass production in rice. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ye, J.; Yao, X.; Zhao, P.; Xuan, W.; Tian, Y.; Zhang, Y.; Xu, S.; An, H.; Chen, G.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, H.; Jiang, Z.; Wang, W.; Xu, R.; Wang, Q.; Zhang, Z.; Li, A.; Liang, Y.; Ou, S.; et al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 2021, 590, 600–605. [Google Scholar] [CrossRef]
- Brauer, E.K.; Rochon, A.; Bi, Y.M.; Bozzo, G.G.; Rothstein, S.J.; Shelp, B.J. Reappraisal of nitrogen use efficiency in rice overexpressingglutamine synthetase1. Physiol. Plant. 2011, 141, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Kusano, M.; Tabuchi, M.; Fukushima, A.; Funayama, K.; Diaz, C.; Kobayashi, M.; Hayashi, N.; Tsuchiya, Y.N.; Takahashi, H.; Kamata, A.; et al. Metabolomics data reveal a crucial role of cytosolic glutaminesynthetase 1;1 in coordinating metabolic balance in rice. Plant J. 2011, 66, 456–466. [Google Scholar] [CrossRef] [PubMed]
- James, D.; Borphukan, B.; Fartyal, D.; Ram, B.; Singh, J.; Manna, M.; Sheri, V.; Panditi, V.; Yadav, R.; Achary, V.M.; et al. Concurrent Overexpression of OsGS1;1 and OsGS2 Genes in Transgenic Rice (Oryza sativa L.): Impact on Tolerance to Abiotic Stresses. Front. Plant Sci. 2018, 9, 786. [Google Scholar] [CrossRef]
- Thomsen, H.C.; Eriksson, D.; Møller, I.S.; Schjoerring, J.K. Cytosolic glutamine synthetase: A target for improvement of crop nitrogen use efficiency? Trends Plant Sci. 2014, 19, 656–663. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, C.; Zhang, Y.; Wang, C. Nitrogen Use Efficiency in Rice. Nitrogen Agric.-Updates 2018, 10, 187–208. [Google Scholar] [CrossRef] [Green Version]
- Mi, G.; Chen, F.; Zhang, F. Multiple signaling pathways controls nitrogen-mediated root elongation in maize. Plant Signal. Behav. 2008, 3, 1030–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, T.; Conn, V.; Kaiser, B.N. Root based approaches to improving nitrogen use efficiency in plants. Plant Cell Environ. 2009, 32, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Tobin, A.; Yamaya, T. Cellular compartmentation of ammonium assimilation in rice and barley. J. Exp. Bot. 2001, 52, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Gong, B.; Sun, S.; Liu, S.; Wang, X.; Wei, M.; Yang, F.; Li, Y.; Shi, Q. Promoting Roles of Melatonin in Adventitious Root Development of Solanum lycopersicum L. by Regulating Auxin and Nitric Oxide Signaling. Front. Plant Sci. 2016, 7, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Chen, F.; Yuan, L.; Mi, G. The physiological mechanism underlying root elongation in response to nitrogen deficiency in crop plants. Planta 2020, 251, 84. [Google Scholar] [CrossRef]
- Cormier, F.; Faure, S.; Dubreuil, P.; Heumez, E.; Beauchêne, K.; Lafarge, S.; Praud, S.; Le Gouis, J. A multi-environmental study of recent breeding progress on nitrogen use efficiency in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2013, 126, 3035–3048. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.K.; Sihmar, M.; Santal, A.R.; Singh, N.P. Impact assessment of major abiotic stresses on the proteome profiling of some important crop plants: A current update. Biotechnol. Genet. Eng. Rev. 2019, 35, 126–160. [Google Scholar] [CrossRef]
- Wieczorek, S.; Combes, F.; Borges, H.; Burger, T. Protein-Level Statistical Analysis of Quantitative Label-Free Proteomics Data with ProStaR. Methods Mol. Biol. 2019, 1959, 225–246. [Google Scholar] [CrossRef]
- Fukuoka, S.; Saka, N.; Mizukami, Y.; Koga, H.; Yamanouchi, U.; Yoshioka, Y.; Hayashi, N.; Ebana, K.; Mizobuchi, R.; Yano, M. Gene pyramiding enhances durable blast disease resistance in rice. Sci. Rep. 2015, 5, 7773. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Jin, J.; Zhang, H.; Wang, Y.; Li, Q.; Zou, Y.; Huang, X.; Zhou, B.; Zhou, R.; Ding, Y. Comparative Analysis of Proteomics and Transcriptomics during Fertility Transition in a Two-Line Hybrid Rice Line Wuxiang S. Int. J. Mol. Sci. 2019, 20, 4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvez, J.H.; Tai, H.H.; Lague, M.; Zebarth, B.J.; Stromvik, M.V. The nitrogen responsive transcriptome in potato (Solanum tuberosum L.) reveals significant gene regulatory motifs. Sci. Rep. 2016, 6, 26090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Bucio, J.; Cruz-Ramírez, A.; Herrera-Estrella, L.R. The role of nutrient availability in regulating root architecture. Curr. Opin. Plant Biol. 2003, 6, 280–287. [Google Scholar] [CrossRef]
- Jia, L.; Xie, Y.; Wang, Z.; Luo, L.; Zhang, C.; Pélissier, P.; Parizot, B.; Qi, W.; Zhang, J.; Hu, Z.; et al. Rice plants respond to ammonium stress by adopting a helical root growth pattern. Plant J. 2020, 104, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passardi, F.; Longet, D.; Penel, C.; Dunand, C. The class III peroxidase multigenic family in rice and its evolution in land plants. Phytochemistry 2004, 65, 1879–1893. [Google Scholar] [CrossRef]
- Opassiri, R.; Maneesan, J.; Akiyama, T.; Pomthong, B.; Jin, S.; Kimura, A.; Cairns, J.K. Rice Os4BGlu12 is a wound-induced β-glucosidase that hydrolyzes cell wall-β-glucan-derived oligosaccharides and glycosides. Plant Sci. 2010, 179, 273–280. [Google Scholar] [CrossRef]
- Esen, A. β-Glucosidase overview. In β-Glucosidase: Biochemistry and Molecular Biology; Esen, A., Ed.; ACS Symposium Series 533 American Chemical Society: Washinton, DC, USA, 1993. [Google Scholar]
- Seshadri, S.; Akiyama, T.; Opassiri, R.; Kuaprasert, B.; Cairns, J.K. Structural and enymatic characterization of Os3BGlu6, a rice β-glucosidase hydrolyzing hydrophobic glycosides and (1-3)- and (1-2)-linked disaccharides. Plant Physiol. 2009, 151, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, J.; Shen, J.; Li, L. Functional Characterization of Evolutionarily Divergent 4-Coumarate:Coenzyme A Ligases in Rice. Plant Physiol. 2011, 157, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Aya, K.; Kondo, M.; Okuno, A.; Morinaka, Y.; Matsuoka, M. OsCAD2 is the major CAD gene responsible for monolignol biosynthesis in rice culm. Plant Cell Rep. 2012, 31, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, K.; Chern, M.; Liu, Y.; Zhu, Z.; Liu, J.; Zhu, X.; Yin, J.; Ran, L.; Xiong, J.; et al. Sclerenchyma cell thickening through enhanced lignification induced by OsMYB30 prevents fungal penetration of rice leaves. New Phytol. 2020, 226, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen, B.W.; Manosalva, P.; Lang, J.; Baraoidan, M.; Bordeos, A.; Mauleon, R.; Oard, J.; Hulbert, S.; Leung, H.; Leach, J.E. Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol. Biol. 2014, 87, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Mhamdi, A.; Chaouch, S.; Han, Y.; Neukermans, J.; Garcia, B.M.; Queval, G.; Foyer, C. Glutathione in plants: An integrated overview. Plant Cell Environ. 2011, 35, 454–484. [Google Scholar] [CrossRef]
- Yamazaki, S.; Ochiai, K.; Matoh, T. Rice plants have three homologs of glutathione synthetase genes, one of which, OsGS2, codes for hydroxymethyl-glutathione synthetase. Plant Direct 2019, 3, e00119. [Google Scholar] [CrossRef] [Green Version]
- Esteban, R.; Ariz, I.; Cruz, C.; Moran, J.F. Mechanisms of ammonium toxicity and the quest for tolerance. Plant Sci. 2016, 248, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Valdivia, M.D.; Aparicio-Tejo, P.M.; Lamsfus, C.; Cruz, C.; Martins-Loução, M.A.; Moran, J.F. Nitrogen nutrition and antioxidant metabolism in ammonium-tolerant and -sensitive plants. Physiol. Plant. 2008, 132, 359–369. [Google Scholar] [CrossRef]
- Hong, C.-Y.; Hsu, Y.T.; Tsai, Y.-C.; Kao, C.H. Expression of ASCORBATE PEROXIDASE 8 in roots of rice (Oryza sativa L.) seedlings in response to NaCl. J. Exp. Bot. 2007, 58, 3273–3283. [Google Scholar] [CrossRef]
- Chou, T.S.; Chao, Y.Y.; Kao, C.H. Involvement of hydrogen peroxide in heat shock- and cadmium-induced expression of ascorbate peroxidase and glutathione reductase in leaves of rice seedlings. J. Plant Physiol. 2012, 169, 478–486. [Google Scholar] [CrossRef]
- Sharma, R.; Sahoo, A.; Devendran, R.; Jain, M. Over-Expression of a Rice Tau Class Glutathione S-Transferase Gene Improves Tolerance to Salinity and Oxidative Stresses in Arabidopsis. PLoS ONE 2014, 9, e92900. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Ghanashyam, C.; Bhattacharjee, A. Comprehensive expression analysis suggests overlapping and specific roles of rice glutathione S-transferase genes during development and stress responses. BMC Genom. 2010, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passaia, G.; Caverzan, A.; Fonini, L.S.; Carvalho, F.; Silveira, J.A.G.; Margis-Pinheiro, M. Chloroplastic and mitochondrial GPX genes play a critical role in rice development. Biol. Plant. 2014, 58, 375–378. [Google Scholar] [CrossRef]
- Lima-Melo, Y.; Carvalho, F.E.L.; Martins, M.O.; Passaia, G.; Sousa, R.H.V.; Neto, M.C.L.; Margis-Pinheiro, M.; Silveira, J.A.G. Mitochondrial GPX1 silencing triggers differential photosynthesis impairment in response to salinity in rice plants. J. Integr. Plant Biol. 2016, 58, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, S.; Akiyama, A.; Kisaka, H.; Uchimiya, H.; Miwa, T. Metabolic engineering with Dof1 transcription factor in plants: Improved nitrogen assimilation and growth under low-nitrogen conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Liu, Y.; Shen, Q.J. The WRKY Gene Family in Rice (Oryza sativa). J. Integr. Plant Biol. 2007, 49, 827–842. [Google Scholar] [CrossRef]
- Yamada, S.; Kano, A.; Tamaoki, D.; Miyamoto, A.; Shishido, H.; Miyoshi, S.; Taniguchi, S.; Akimitsu, K.; Gomi, K. Involvement of OsJAZ8 in Jasmonate-Induced Resistance to Bacterial Blight in Rice. Plant Cell Physiol. 2012, 53, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol. Biol. 2009, 71, 291–305. [Google Scholar] [CrossRef]
- Asim, M.; Ullah, Z.; Xu, F.; An, L.; Aluko, O.O.; Wang, Q.; Liu, H. Nitrate signaling, function, and regulation of root system architechture:insights from Arabidopsis thaliana. Gene 2020, 11, 633. [Google Scholar] [CrossRef]
- Forde, B.G. Nitrogen signalling pathways shaping root system architecture: An update. Curr. Opin. Plant Biol. 2014, 21, 30–36. [Google Scholar] [CrossRef]
- Giehl, R.F.; Gruber, B.D.; von Wirén, N. It’s time to make changes: Modulation of root system architecture by nutrient signals. J. Exp. Bot. 2014, 65, 769–778. [Google Scholar] [CrossRef]
- Nacry, P.; Bouguyon, E.; Gojon, A. Nitrogen acquisition by roots: Physiological and developmental mechanisms ensuring plant adaptation to a fluctuating resource. Plant Soil 2013, 370, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Osmont, K.S.; Sibout, R.; Hardtke, C.S. Hidden branches: Developments in root system architecture. Annu. Rev. Plant Biol. 2007, 58, 93–113. [Google Scholar] [CrossRef]
- Motte, H.; Beeckman, T. The evolution of root branching: Increasing the level of plasticity. J. Exp. Bot. 2019, 70, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Nemoto, K. Morphology and anatomy of rice roots with special reference to coordination in organo- and histogenesis. In Structure and Function of Roots; Springer: Berlin/Heidelberg, Germany, 1995; pp. 76–86. [Google Scholar]
- Hochholdinger, F.; Park, W.J.; Sauer, M.; Woll, K. From weeds to crops: Genetic analysis of root development in cereals. Trends Plant Sci. 2004, 9, 42–48. [Google Scholar] [CrossRef]
- Zhang, Q.; Wing, R. (Eds.) Root development. In Genetics and Genomics of Rice, Plant Genetics and Genomics: Crops and Models 5; Springer Science + Business Media: New York, NY, USA, 2013; pp. 297–316. [Google Scholar]
- Banda, J.; Bellande, K.; von Wangenheim, D.; Goh, T.; Guyomarc’H, S.; Laplaze, L.; Bennett, M.J. Lateral Root Formation in Arabidopsis: A Well-Ordered LRexit. Trends Plant Sci. 2019, 24, 826–839. [Google Scholar] [CrossRef]
- Coskun, D.; Britto, D.T.; Shi, W.; Kronzucker, H.J. How Plant Root Exudates Shape the Nitrogen Cycle. Trends Plant Sci. 2017, 22, 661–673. [Google Scholar] [CrossRef]
- Liu, B.; Wu, J.; Yang, S.; Schiefelbein, J.; Gan, Y. Nitrate regulation of lateral root and root hair development in plants. J. Exp. Bot. 2020, 71, 4405–4414. [Google Scholar] [CrossRef]
- Muller, B.; Guédon, Y.; Passot, S.; Lobet, G.; Nacry, P.; Pagès, L.; Wissuwa, M.; Draye, X. Lateral roots:random diversity in adversity. Trends Plant Sci. 2019, 24, 810–825. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Krapp, A. Plant nitrogen acquisition under low availability:regulation of uptake and root architecture. Plant Cell Physiol. 2016, 57, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Forde, B.G.; Roberts, M.R. Glutamate receptor-like channels in plants: A role as amino acid sensors in plants. F1000Prime Rep. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, S.; Lancien, M.; Hodges, M. Are isocitrate dehydrogenases and 2-oxoglutarate invovled in the regulation of glutamate synthesis? Trends Plant Sci. 1999, 4, 484–490. [Google Scholar] [CrossRef]
- Lancien, M.; Gadal, P.; Hodges, M. Enzyme Redundancy and the Importance of 2-Oxoglutarate in Higher Plant Ammonium Assimilation. Plant Physiol. 2000, 123, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.K.; Ghosh, S. Regulation of glutamine synthetase isoforms in two differentially drought-tolerant rice (Oryza sativa L.) cultivars under water deficit conditions. Plant Cell Rep. 2013, 32, 183–193. [Google Scholar] [CrossRef]
- Prabhakar, V.; Löttgert, T.; Gigolashvili, T.; Bell, K.; Flügge, U.-I.; Häusler, R.E. Molecular and functional characterization of the plastid-localized Phosphoenolpyruvate enolase (ENO1) fromArabidopsis thaliana. FEBS Lett. 2009, 583, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ye, Q.; He, D.; Bai, H.; Wen, J. The ubiquity and coexistence of two FBPases in chloroplasts of photosynthetic eukaryotes and its evolutionary and functional implications. Plant Divers. 2020, 42, 120–125. [Google Scholar] [CrossRef]
- Von Wirén, N.; Gazzarrini, S.; Gojon, A.; Frommer, W.B. The molecular physiology of ammonium uptake and retrieval. Curr. Opin. Plant Biol. 2000, 3, 254–261. [Google Scholar] [CrossRef]
- Sonoda, Y.; Ikeda, A.; Saiki, S.; Yamaya, T.; Yamaguchi, J. Feedback regulation of the ammonium transporter gene family AMT1 by glutamine in rice. Plant Cell Physiol. 2003, 44, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.Z.; Merrick, M.; Li, S.M.; Li, H.Y.; Zhu, S.W.; Shi, W.M.; Su, Y.H. Molecular Basis and Regulation of Ammonium Transporter in Rice. Rice Sci. 2009, 16, 314–322. [Google Scholar] [CrossRef]
- Krapp, A. Plant nitrogen assimilation and its regulation: A complex puzzle with missing pieces. Curr. Opin. Plant Biol. 2015, 25, 115–122. [Google Scholar] [CrossRef]
- Ohashi, M.; Ishiyama, K.; Kusano, M.; Fukushima, A.; Kojima, S.; Hanada, A.; Kanno, K.; Hayakawa, T.; Seto, Y.; Kyozuka, J. Lack of cytosolic glutamine sythetase1;2 in vascular tissues of axillary buds causes severe reduction in their outgrowth and disorder of metabolic balance in rice seedlings. Plant J. 2015, 81, 347–356. [Google Scholar] [CrossRef]
- Hirano, T.; Satoh, Y.; Ohki, A.; Takada, R.; Arai, T.; Michiyama, H. Inhibition of ammonium assimilation restores elongation of seminal rice roots respressed by high levels of exogenous ammonium. Physiol. Plant. 2008, 134, 183–190. [Google Scholar] [CrossRef]
- Walch-Liu, P.; Liu, L.-H.; Remans, T.; Tester, M.; Forde, B.G. Evidence that l -Glutamate Can Act as an Exogenous Signal to Modulate Root Growth and Branching in Arabidopsis thaliana. Plant Cell Physiol. 2006, 47, 1045–1057. [Google Scholar] [CrossRef] [Green Version]
- Geiger, M.; Haake, V.; Ludewig, F.; Sonnewald, U.; Stitt, M.; Geiger, M.; Haake, V.; Ludewig, F.; Sonnewald, U.; Stitt, M. The nitrate and ammonium nitrate supply have a major influence on the response of photosynthesis, carbon metabolism, nitrogen metabolism and growth to elevated carbon dioxide in tobacco. Plant Cell Environ. 1999, 22, 1177–1199. [Google Scholar] [CrossRef]
- Geiger, M.; Walch-Liu, P.; Engels, C.; Harnecker, J.; Schulze, E.; Ludewig, F.; Sonnewald, U.; Scheible, W.; Stitt, M. Enhanced carbon dioxide leads to a modified diurnal rhythm of nitrate reductase activity in older plants, and a large stimulation of nitrate reductase activity and higher levels of amino acids in young tobacco plants. Plant Cell Environ. 1998, 21, 253–268. [Google Scholar] [CrossRef]
- Sakakibara, H.; Takei, K.; Hirose, N. Interactions between nitrogen and cytokinin in the regulation of metabolism and development. Trends Plant Sci. 2006, 11, 440–448. [Google Scholar] [CrossRef]
- Cai, H.; Lu, Y.; Xie, W.; Zhu, T.; Lian, X. Transcriptome response to nitrogen starvation in rice. J. Biosci. 2012, 37, 731–747. [Google Scholar] [CrossRef]
- Yang, W.; Yoon, J.; Choi, H.; Fan, Y.; Chen, R.; An, G. Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol. 2015, 15, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-C.; Zhou, C.-Z.; Burnap, R.L.; Peng, L. Carbon/Nitrogen Metabolic Balance: Lessons from Cyanobacteria. Trends Plant Sci. 2018, 23, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Sade, N.; Umnajkitikorn, K.; Wilhelmi, M.D.M.R.; Wright, M.; Wang, S.; Blumwald, E. Delaying chloroplast turnover increases water-deficit stress tolerance through the enhancement of nitrogen assimilation in rice. J. Exp. Bot. 2017, 69, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Inaba, J.; Kudo, T.; Ueda, N.; Konishi, M.; Mitsuda, N.; Takiguchi, Y.; Kondou, Y.; Yoshizumi, T.; Ohme-Takagi, M.; et al. Repression of Nitrogen Starvation Responses by Members of the Arabidopsis GARP-Type Transcription Factor NIGT1/HRS1 Subfamily. Plant Cell 2018, 30, 925–945. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xu, J.; Gao, Z.; Tian, H.; Gao, Y.; Kariman, K. Genetically modifed crops are superior in their nitrogen use efciency-A meta-analysis of three major cereals. Sci. Rep. 2020, 10, 8365. [Google Scholar]
- Wang, R.; Qian, J.; Fang, Z.; Tang, J. Transcriptomic and physiological analyses of rice seedlings under different nitrogen supplies provide insight into the regulation involved in axillary bud outgrowth. BMC Plant Biol. 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurai, T.; Wakayama, M.; Abiko, T.; Yanagisawa, S.; Aoki, N.; Ohsugi, R. Introduction of the ZmDof1 gene into rice enhances carbon and nitrogen assimilation under low-nitrogen conditions. Plant Biotechnol. J. 2011, 9, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.N.; Zhou, X.A.; Sha, A.H.; Wang, C.; Zhou, R.; Chen, S.L. Identification of genes associated with nitrogen-use efficiency by genome-wide transcriptional analysis of two soybean genotypes. BMC Genom. 2011, 12, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaings, L.; Camargo, A.; Pocholle, D.; Gaudon, V.; Texier, Y.; Boutet-Mercey, S.; Taconnat, L.; Renou, J.-P.; Daniel-Vedele, F.; Fernandez, E.; et al. The nodule inception-like protein 7 modulates nitrate sensing and metabolism in Arabidopsis. Plant J. 2009, 57, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liang, Z.; Chen, S.; Sun, H.; Fan, X.; Wang, C.; Xu, G.; Zhang, Y. A Transcription Factor, OsMADS57, Regulates Long-Distance Nitrate Transport and Root Elongation. Plant Physiol. 2019, 180, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yang, N.; Guo, Z.; Qian, M.; Gan, L. An ethylene and ROS-dependent pathway is involved in low ammonium-induced root hair elongation in Arabidopsis seedlings. Plant Physiol. Biochem. 2016, 105, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Kiba, T.; Kudo, T.; Kojima, M.; Sakakibara, H. Hormonal control of nitrogen acquisition: Roles of auxin, abscisic acid, and cytokinin. J. Exp. Bot. 2010, 62, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Di, D.; Li, G.; Li, Y.; Kronzucker, H.; Shi, W. Transcriptome analysis of rice (Oryza sativa L.) in response to ammonium resupply reveals the involvement of phytohormone signaling and the transcription factor OsJAZ9 in reprogramming of nitrogen uptake and metabolism. J. Plant Physiol. 2020, 246, 153137. [Google Scholar] [CrossRef] [PubMed]
- Dhondge, H.V.; Pable, A.A.; Barvkar, V.T.; Dastager, S.G.; Nadaf, A.B. Rhizobacterial consortium mediated aroma and yield enhancement in basmati and non-basmati rice (Oryza sativa L.). J. Biotechnol. 2021, 328, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Knuesting, J.; Scheibe, R. Small molecules govern thiol redox switches. Trends Plant Sci. 2018, 23, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.; Dixon, R.A. Plant phenylalanine/tyrosine ammonia-lyases. Trends Plant Sci. 2020, 25, 66–79. [Google Scholar] [CrossRef]
- Lee, S.; Marmagne, A.; Park, J.; Fabien, C.; Yim, Y.; Kim, S.; Kim, T.; Lim, P.O.; Masclaux-Daubresse, C.; Gil Nam, H.; et al. Concurrent activation of OsAMT1;2 and OsGOGAT1 in rice leads to enhanced nitrogen use efficiency under nitrogen limitation. Plant J. 2020, 103, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Von Wittgenstein, N.J.J.B.; Le, C.H.; Hawkins, B.J.; Ehlting, J. Evolutionary classification of ammonium, nitrate, and peptide transporters in land plants. BMC Evol. Biol. 2014, 14, 11. [Google Scholar] [CrossRef] [Green Version]
- Krouk, G.; Crawford, N.M.; Coruzzi, G.M.; Tsay, Y.-F. Nitrate signaling: Adaptation to fluctuating environments. Curr. Opin. Plant Biol. 2010, 13, 265–272. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Hsu, P.K.; Tsay, Y.F. Uptake, allocation and signaling of nitrate. Trends Plant Sci. 2012, 17, 458–467. [Google Scholar] [CrossRef]
- Krapp, A.; David, L.C.; Chardin, C.; Girin, T.; Marmagne, A.; Leprince, A.S.; Chaillou, S.; Ferrario-Méry, S.; Meyer, C.; Daniel-Vedele, F. Nitrate transport and signalling in Arabidopsis. J. Exp. Bot. 2014, 65, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data using Real-Time Quantitative PCR. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Q.; Zhou, W.; Li, P.; Alolga, R.N.; Qi, L.-W.; Yin, X. A comparative proteomic characterization and nutritional assessment of naturally- and artificially-cultivated Cordyceps sinensis. J. Proteom. 2018, 181, 24–35. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Protein ID | MSU_Locus | Gene Annotation |
|---|---|---|---|
| Com_down | sp|Q6K4E7|APM1D | LOC_Os09g19800 | aminopeptidase |
| sp|Q7XUG1|MASY | LOC_Os04g40990 | glyoxysomal | |
| Com_up | sp|Q5WA72|PDI15 | LOC_Os06g06790 | OsPDIL1-5 protein disulfide isomerase |
| sp|Q7PC80|AB34G | LOC_Os01g42350 | pleiotropic drug resistance protein | |
| P_up_T_down | sp|Q6Z9A3|GMPP3 | LOC_Os08g13930 | GDP-D-mannose pyrophosphorylase |
| sp|B8BH95|ITPA | LOC_Os10g31940 | inosine triphosphate pyrophosphatase | |
| P_down_T_up | sp|Q06398|GSTU6 | LOC_Os01g37750 | glutathione S-transferase |
| sp|Q10LX4|CML27 | LOC_Os03g21380 | OsCML27 | |
| sp|Q10S72|4CLL4 | LOC_Os03g04000 | AMP-binding domain containing protein | |
| sp|O04985|HBL2 | LOC_Os03g12510 | non-symbiotic hemoglobin 2 | |
| sp|Q42997|NIR | LOC_Os02g52730 | ferredoxin--nitrite reductase |
| Protein ID | MSU_Locus | Annotation | FC(1/4N/1N) |
|---|---|---|---|
| Phenylpropanoid Biosynthesis (ko00940) | |||
| upregulated | |||
| A2Y626 | LOC_Os05g41440 | cytochrome P450 98A1 | 3.76 |
| A2ZCW8 | LOC_Os11g12760 | O-methyltransferase ZRP4 | 3.53 |
| B8AND5 | LOC_Os03g05780 | 4-coumarate--CoA ligase-like 7 | 2.42 |
| Q6ZHS4 | LOC_Os02g09490 | cinnamyl alcohol dehydrogenase | 3.37 |
| A0A0N7KK39 | LOC_Os05g04470 | peroxidase precursor | 4.44 |
| A2YHC0 | LOC_Os07g01420 | peroxidase 1 precursor | 2.66 |
| downregulated | |||
| A2XZ41 | LOC_Os04g58710 | peroxisomal-coenzyme A synthetase | 0.10 |
| B8ARF5 | LOC_Os04g57850 | AMP-binding protein gene | 0.11 |
| B8AMG2 | LOC_Os03g19250 | AMP-binding protein | 0.25 |
| Q10S72 | LOC_Os03g04000 | 4-coumarate--CoA ligase-like 4 | 0.31 |
| A2XB41 | LOC_Os02g57480 | anthocyanin 5-aromatic acyltransferase | 0.49 |
| A2Y375 | LOC_Os05g25640 | trans-cinnamate 4-monooxygenase | 0.31 |
| Q60DX8 | LOC_Os05g30350 | Os5bglu22—beta-glucosidase homologue | 0.42 |
| B8AV76 | LOC_Os04g54810 | beta-D-xylosidase | 0.43 |
| B8B5W7 | LOC_Os07g48010 | peroxidase 2 precursor | 0.26 |
| A2WZD6 | LOC_Os01g73170 | peroxidase 12 precursor | 0.42 |
| P0C5D0 | LOC_Os07g44440 | peroxiredoxin | 0.47 |
| Glutathione Metabolism (ko00480) | |||
| upregulated | |||
| B8BM87 | LOC_Os12g34380 | glutathione synthetase | 12.30 |
| A2YL42 | LOC_Os07g28480 | glutathione S-transferase | 4.71 |
| A2Z9K4 | LOC_Os10g38580 | glutathione S-transferase GSTU6 | 2.94 |
| B8BI29 | LOC_Os10g39740 | glutathione S-transferase | 2.42 |
| A6N0E3 | LOC_Os01g27390 | glutathione transferase | 2.24 |
| A1XBB7 | LOC_Os03g17470 | IN2-1 protein | 2.17 |
| A2Z263 | LOC_Os09g29200 | glutathione S-transferase | 2.03 |
| downregulated | |||
| A0A0P0UYM7 | LOC_Os01g05820 | gamma-glutamyltranspeptidase 1 precursor | 0.23 |
| Q06398 | LOC_Os10g38740 | glutathione S-transferase | 0.12 |
| Q65XA0 | LOC_Os05g02530 | cytosolic dehydroascorbate reductase | 0.30 |
| A2XC68 | LOC_Os03g04250 | glutathione S-transferase 6 | 0.46 |
| Q7XJ02 | LOC_Os04g35520 | probable L-ascorbate peroxidase 7 | 0.42 |
| Gene Locus | FDR | log2FC | Annotation |
|---|---|---|---|
| Phenylpropanoid biosynthesis (ko00940) | |||
| upregulated | |||
| LOC_Os06g29470 | 0 | 5.99 | peroxidase 1-like |
| LOC_Os02g56700 | 0 | 4.45 | cinnamoyl-CoA reductase 1 |
| LOC_Os08g34790 | 0 | 2.92 | Os4CL5 |
| LOC_Os06g46799 | 0 | 2.90 | peroxidase 3 |
| LOC_Os03g25360 | 4.53 × 10−12 | 2.88 | peroxidase 2 |
| LOC_Os04g39880 | 4.51 × 10−12 | 2.35 | Os4BGlu12 |
| LOC_Os02g41670 | 2.91 × 10−10 | 2.18 | OsPAL3 |
| LOC_Os05g35290 | 1.14 × 10−5 | 1.65 | OsPAL7 |
| LOC_Os03g11420 | 1.48 × 10−4 | 1.53 | Os3BGlu6 |
| LOC_Os01g40860 | 1.04 × 10−4 | 1.52 | OsALDH2C4 |
| LOC_Os08g14760 | 7.57 × 10−5 | 1.52 | Os4CL1 |
| LOC_Os06g44620 | 9.84 × 10−5 | 1.50 | Os4CL4 |
| LOC_Os04g43800 | 8.16 × 10−4 | 1.35 | OsPAL6 |
| LOC_Os02g41680 | 1.88 × 10−3 | 1.34 | OsPAL4 |
| downregulated | |||
| LOC_Os08g02110 | 3.72 × 10−10 | −2.18 | peroxidase 47 |
| LOC_Os07g02440 | 4.12 × 10−11 | −2.29 | peroxidase 50 |
| Glutathione metabolism (ko00480) | |||
| upregulated | |||
| LOC_Os10g38740 | 2.44 × 10−15 | 2.79 | glutathione S-transferase GSTU6 |
| LOC_Os03g44170 | 5.34 × 10−12 | 2.69 | probable glutathione S-transferase GSTU1 |
| LOC_Os09g20220 | 3.71 × 10−11 | 2.30 | probable glutathione S-transferase GSTU1 |
| LOC_Os04g46960 | 1.55 × 10−8 | 2.00 | OsGPX1 |
| LOC_Os01g72160 | 6.23 × 10−7 | 1.90 | probable glutathione S-transferase |
| LOC_Os04g38450 | 8.48 × 10−7 | 1.82 | gamma-glutamyltranspeptidase 1 precursor |
| LOC_Os10g38580 | 3.43 × 10−6 | 1.77 | probable glutathione S-transferase GSTU6 |
| LOC_Os10g38640 | 1.33 × 10−6 | 1.76 | putative glutathione S-transferase |
| LOC_Os03g57200 | 1.07 × 10−6 | 1.76 | glutathione S-transferase GSTU1 |
| LOC_Os10g38600 | 2.61 × 10−5 | 1.57 | putative glutathione S-transferase |
| LOC_Os01g70770 | 4.75 × 10−4 | 1.42 | glutathione S-transferase 3 |
| LOC_Os10g38314 | 7.88 × 10−4 | 1.37 | glutathione S-transferase |
| LOC_Os09g29200 | 8.16 × 10−4 | 1.36 | glutathione transferase GST23 |
| LOC_Os11g29400 | 2.54 × 10−3 | 1.30 | 6-phosphogluconate dehydrogenase |
| LOC_Os10g38189 | 3.78 × 10−3 | 1.25 | glutathione S-transferase |
| downregulated | |||
| LOC_Os04g14680 | 1.61 × 10−3 | −1.41 | OsAPx3 |
| LOC_Os03g29950 | 1.50 × 10−3 | −2.08 | glucose-6-phosphate 1-dehydrogenase |
| LOC_Os04g51300 | 4.07 × 10−4 | −2.40 | peroxidase precursor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, T.; Yuan, Z.; Fu, L.; Zhu, M.; Luo, X.; Xu, W.; Yuan, H.; Zhu, R.; Hu, Z.; Wu, X. Integrative Transcriptomic and Proteomic Analysis Reveals an Alternative Molecular Network of Glutamine Synthetase 2 Corresponding to Nitrogen Deficiency in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2021, 22, 7674. https://doi.org/10.3390/ijms22147674
Liang T, Yuan Z, Fu L, Zhu M, Luo X, Xu W, Yuan H, Zhu R, Hu Z, Wu X. Integrative Transcriptomic and Proteomic Analysis Reveals an Alternative Molecular Network of Glutamine Synthetase 2 Corresponding to Nitrogen Deficiency in Rice (Oryza sativa L.). International Journal of Molecular Sciences. 2021; 22(14):7674. https://doi.org/10.3390/ijms22147674
Chicago/Turabian StyleLiang, Ting, Zhengqing Yuan, Lu Fu, Menghan Zhu, Xiaoyun Luo, Wuwu Xu, Huanran Yuan, Renshan Zhu, Zhongli Hu, and Xianting Wu. 2021. "Integrative Transcriptomic and Proteomic Analysis Reveals an Alternative Molecular Network of Glutamine Synthetase 2 Corresponding to Nitrogen Deficiency in Rice (Oryza sativa L.)" International Journal of Molecular Sciences 22, no. 14: 7674. https://doi.org/10.3390/ijms22147674