
Pantoea Bacteriophage vB_PagS_MED16—A Siphovirus Containing a 2′-Deoxy-7-amido-7-deazaguanosine-Modified DNA

 , , , , , , , and
, , , , , , , and 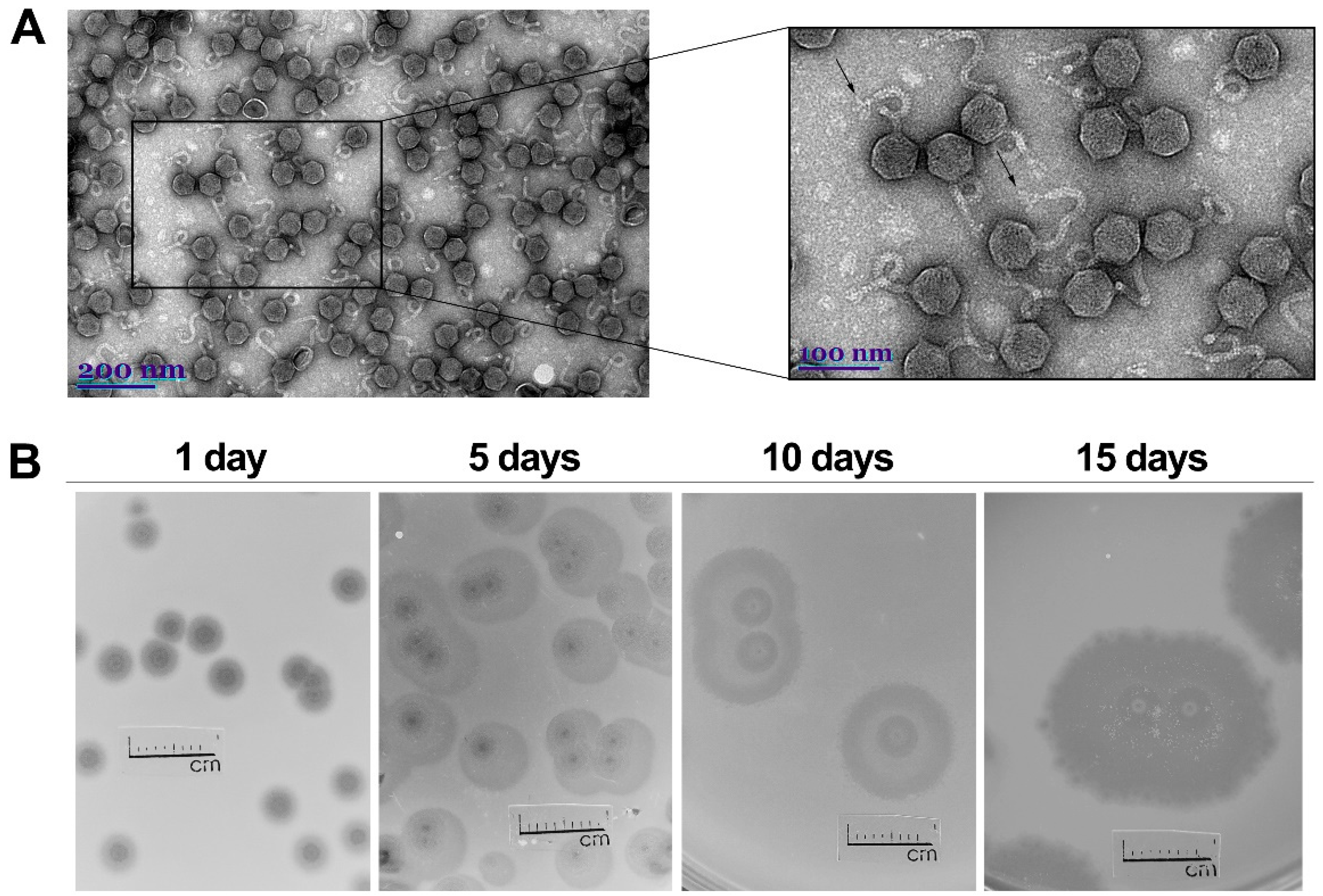{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Phage Morphology, Host Range and Physiological Characteristics
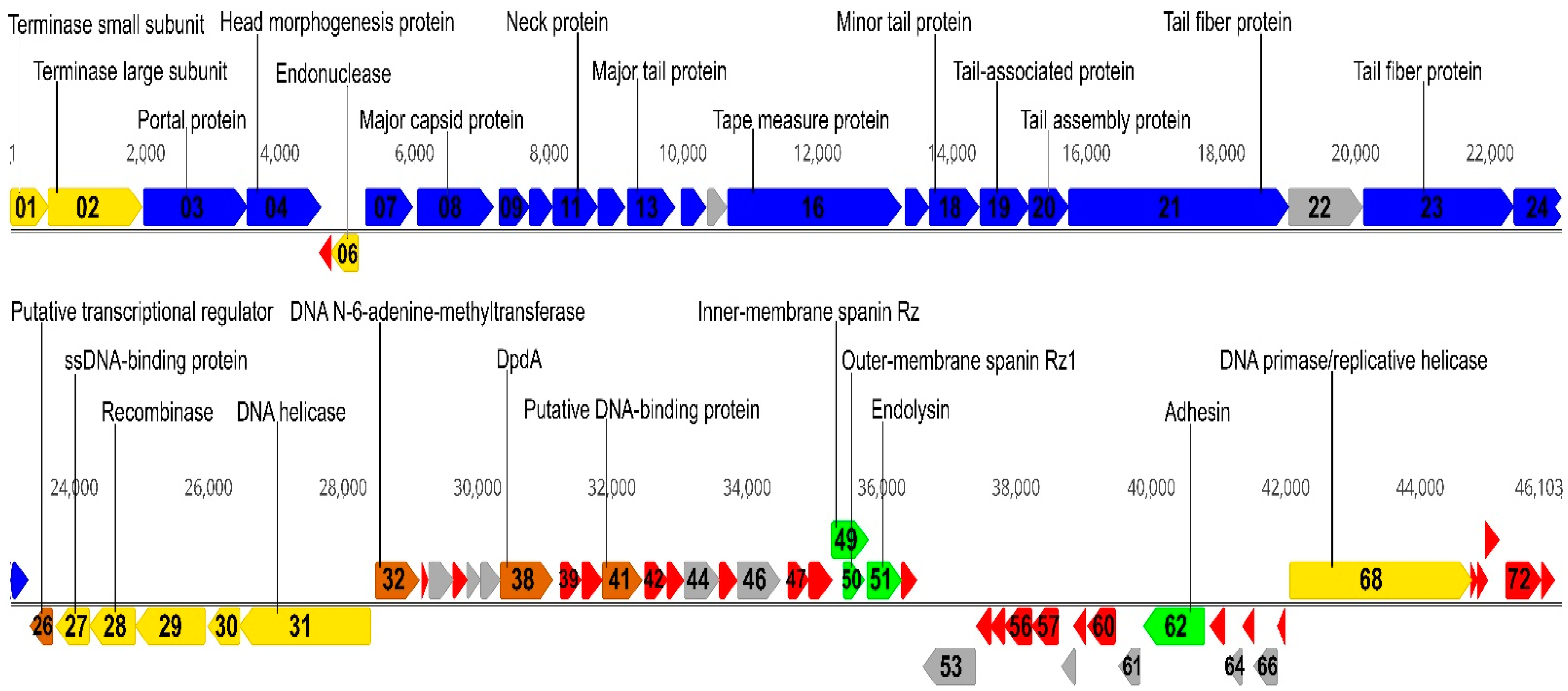
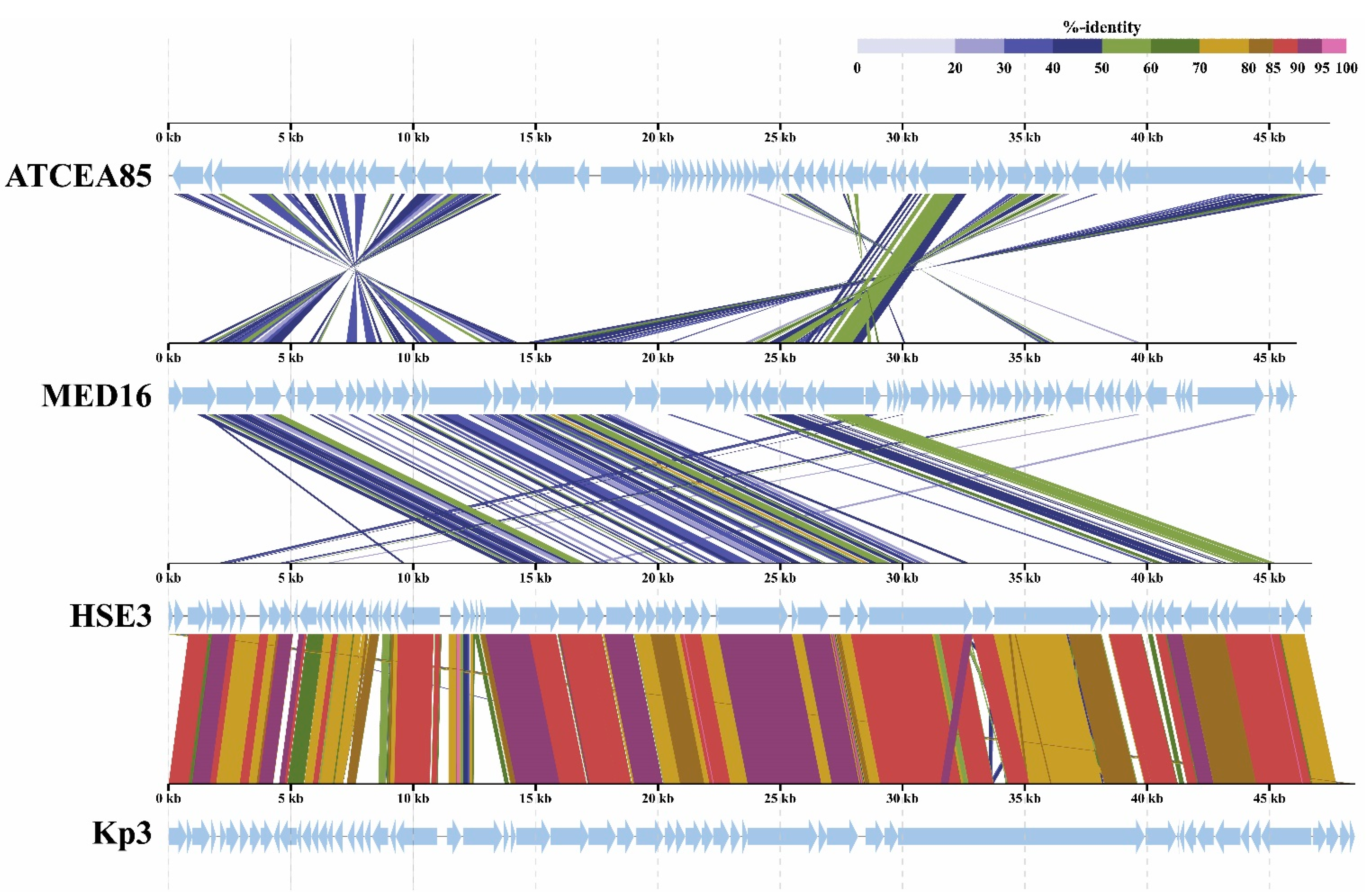
2.2. Overview of Genome
2.3. Structural Proteins
2.4. Packaging
2.5. DNA RRR
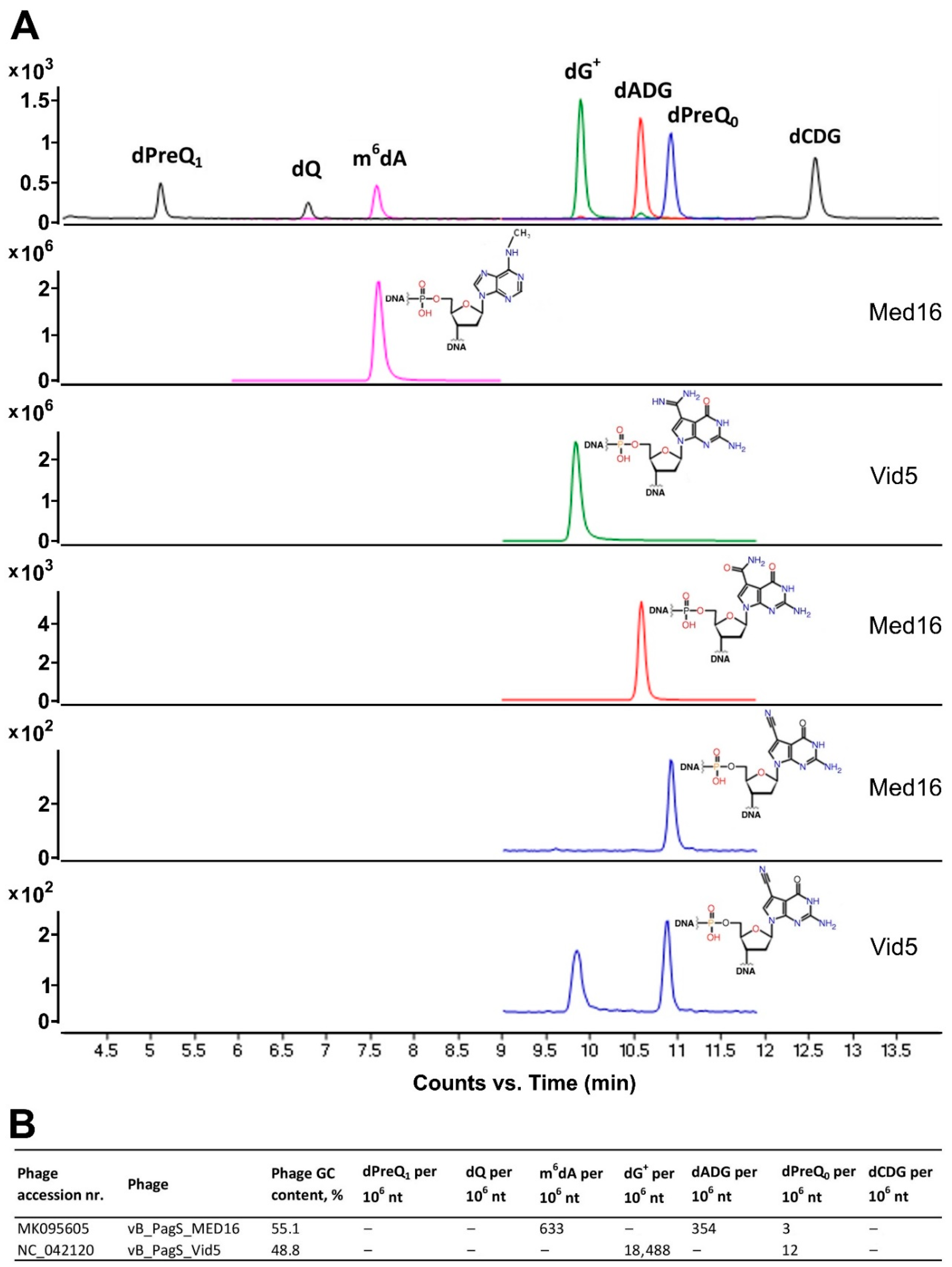
2.6. Transcription, Translation, Nucleotide Metabolism and DNA Modification
2.7. Lysis Cassette
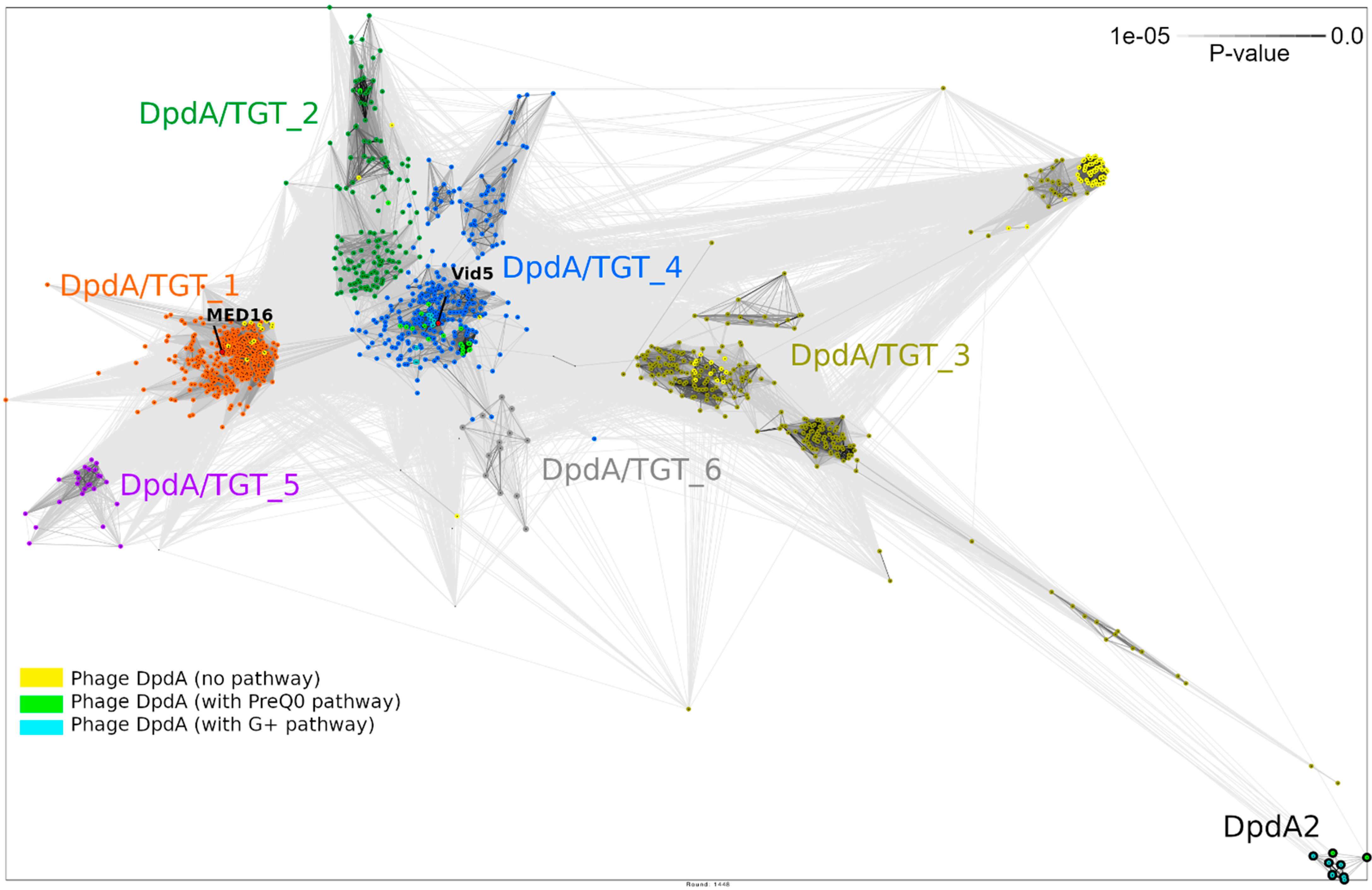
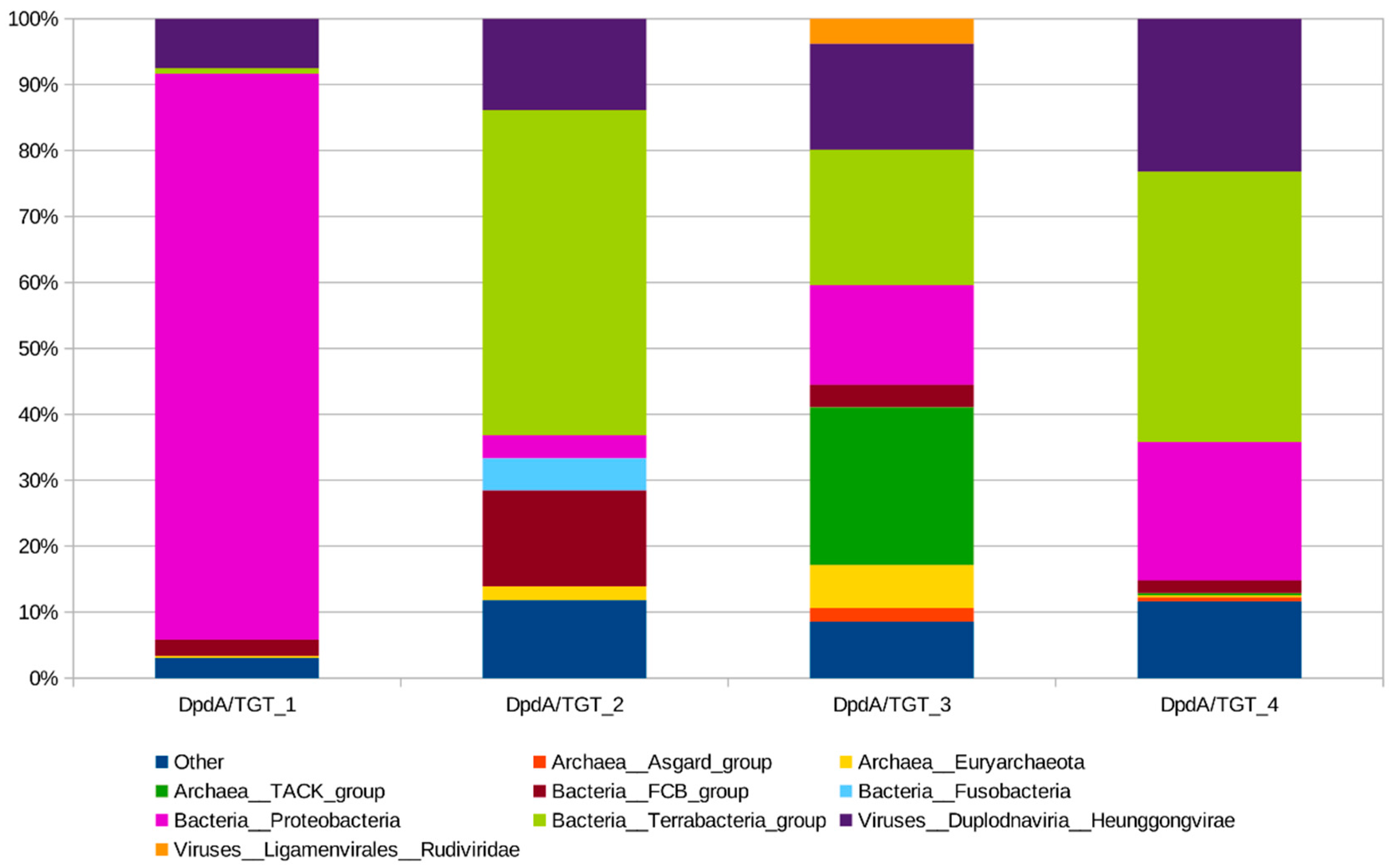
2.8. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Phages and Bacterial Strains
4.2. Phage Isolation, Propagation and Purification Techniques
4.3. Transmission Electron Microscopy
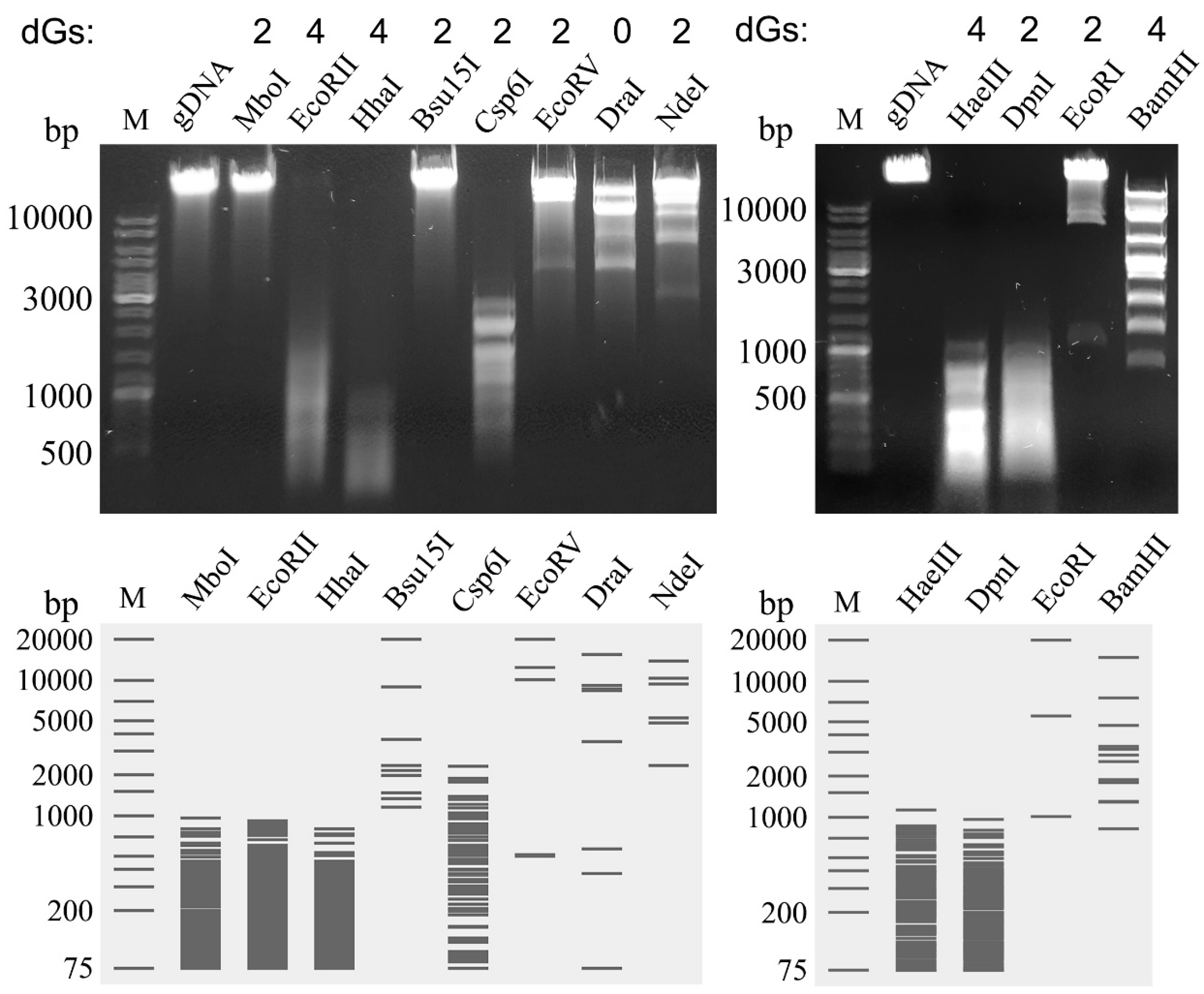
4.4. DNA Isolation and Restriction Analysis
4.5. Genome Sequencing and Analysis
4.6. Analysis of Structural Proteins
4.7. LC–MS/MS Analysis of Phage DNA Modifications
4.8. Bioinformatic Analysis of DpdA-Like Proteins
4.9. Nucleotide Sequence Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, R.A.; Rohwer, F. Opinion: Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Westra, E.R.; Dowling, A.J.; Broniewski, J.M.; van Houte, S. Evolution and ecology of CRISPR. Annu. Rev. Ecol. Evol. Syst. 2016, 47, 307–331. [Google Scholar] [CrossRef] [Green Version]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef]
- Malone, L.M.; Birkholz, N.; Fineran, P.C. Conquering CRISPR: How phages overcome bacterial adaptive immunity. Curr. Opin. Biotechnol. 2020, 68, 30–36. [Google Scholar] [CrossRef]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Weigele, P.; Raleigh, E.A. Biosynthesis and function of modified bases in bacteria and their viruses. Chem. Rev. 2016, 116, 12655–12687. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Turner, D.; Nash, J.H.E.; Ackermann, H.W.; Lingohr, E.J.; Warren, R.A.; Ehrlich, K.C.; Ehrlich, M. The sequence of two bacteriophages with hypermodified bases reveals novel phage-host interactions. Viruses 2018, 10, 217. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Dai, N.; Walsh, S.E.; Muller, S.; Fraser, M.E.; Kauffman, K.M.; Guan, C.; Correa, I.R., Jr.; Weigele, P.R. Identification and biosynthesis of thymidine hypermodifications in the genomic DNA of widespread bacterial viruses. Proc. Natl. Acad. Sci. USA 2018, 115, E3116–E3125. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Weigele, P.R. Detection of modified bases in bacteriophage genomic DNA. In DNA Modifications. Methods in Molecular Biology; Ruzov, A., Gering, M., Eds.; Humana: New York, NY, USA, 2021; Volume 2198, pp. 53–66. [Google Scholar]
- Ehrlich, M.; Ehrlich, K.; Mayo, J.A. Unusual properties of the DNA from Xanthomonas phage XP-12 in which 5-methylcytosine completely replaces cytosine. Biochim. Biophys. Acta 1975, 395, 109–119. [Google Scholar] [CrossRef]
- Kuo, T.T.; Tu, J. Enzymatic synthesis of deoxy-5-methyl-cytidylic acid replacing deoxycytidylic acid in Xanthomonas oryzae phage Xp12 DNA. Nature 1976, 263, 615. [Google Scholar] [CrossRef]
- Thiaville, J.J.; Kellner, S.M.; Yuan, Y.; Hutinet, G.; Thiaville, P.C.; Jumpathong, W.; Mohapatra, S.; Brochier-Armanet, C.; Letarov, A.V.; Hillebrand, R.; et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, E1452–E1459. [Google Scholar] [CrossRef] [Green Version]
- Tsai, R.; Correa, I.R.; Xu, M.Y.; Xu, S.Y. Restriction and modification of deoxyarchaeosine (dGC)-containing phage 9 g DNA. Sci. Rep. 2017, 7, 8348. [Google Scholar] [CrossRef]
- Vanyushin, B.F.; Buryanov, Y.I.; Belozersky, A.N. Distribution of N6-methyladenine in DNA of T2 phage and its host Escherichia coli B. Nature N. Biol. 1971, 230, 25–27. [Google Scholar] [CrossRef]
- Kelln, R.A.; Warren, R.A. Studies on the biosynthesis of alpha-putrescinylthymine in bacteriophage phi W-14-infected Pseudomonas acidovorans. J. Virol. 1973, 12, 1427–1433. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Bose, R.J.; Warren, R.A. 5-(4-Aminobutylaminomethyl)uracil, an unusual pyrimidine from the deoxyribonucleic acid of bacteriophage phiW-14. Biochemistry 1973, 12, 151–157. [Google Scholar] [CrossRef]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442. [Google Scholar] [CrossRef]
- Crippen, C.S.; Lee, Y.J.; Hutinet, G.; Shajahan, A.; Sacher, J.C.; Azadi, P.; de Crécy-Lagard, V.; Weigele, P.R.; Szymanski, C.M. Deoxyinosine and 7-deaza-2-deoxyguanosine as carriers of genetic information in the DNA of Campylobacter viruses. J. Virol. 2019, 93, e01111-19. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, D.; Garcia, P.S.; Lagune, M.; Loc’h, J.; Haouz, A.; Taib, N.; Röthlisberger, P.; Gribaldo, S.; Marlière, P.; Kaminski, P.A. A third purine biosynthetic pathway encoded by aminoadenine-based viral DNA genomes. Science 2021, 372, 516–520. [Google Scholar] [CrossRef]
- Rihtman, B.; Puxty, R.J.; Hapeshi, A.; Lee, Y.J.; Zhan, Y.; Michniewski, S.; Waterfield, N.R.; Chen, F.; Weigele, P.; Millard, A.D.; et al. A new family of globally distributed lytic roseophages with unusual deoxythymidine to deoxyuridine substitution. Curr. Biol. 2021, S0960-9822, 00673–00674. [Google Scholar]
- Šimoliūnas, E.; Šimoliūnienė, M.; Kaliniene, L.; Zajančkauskaitė, A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaitė, L. Pantoea bacteriophage vB_PagS_Vid5: A low-temperature siphovirus that harbors a cluster of genes involved in the biosynthesis of archaeosin. Viruses 2018, 10, 583. [Google Scholar] [CrossRef] [Green Version]
- Bradley, D.E. Ultrastructure of bacteriophages and bacteriocins. Bacteriol. Rev. 1967, 31, 230–314. [Google Scholar] [CrossRef]
- Ackermann, H.W.; Eisenstark, A. The present state of phage taxonomy. Intervirology 1974, 3, 201–219. [Google Scholar] [CrossRef]
- Seeley, N.D.; Primrose, S.B. The effect of temperature on the ecology of aquatic bacteriophages. J. Gen. Virol. 1980, 46, 87–95. [Google Scholar] [CrossRef]
- Pires, D.P.; Oliveira, H.; Melo, L.D.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokine, A.; Rossmann, M.G. Molecular architecture of tailed double stranded DNA phages. Bacteriophage 2014, 4, e28281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef]
- Rao, V.B.; Feiss, M. Mechanisms of DNA packaging by large double stranded DNA viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, R.; Choudhury, M.; Zaman, S.; Singh, S.; Santosh, V.; Bastia, D.; Escalante, C.R. Functional architecture of the Reb1-Ter complex of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 2016, 11316, E2267–E2276. [Google Scholar] [CrossRef] [Green Version]
- Žukauskienė, E.; Šimoliūnienė, M.; Truncaitė, L.; Skapas, M.; Kaupinis, A.; Valius, M.; Meškys, R.; Šimoliūnas, E. Pantoea bacteriophage vB_PagS_AAS23: A singleton of the genus Sauletekiovirus. Microorganisms 2021, 9, 668. [Google Scholar] [CrossRef]
- Kot, W.; Olsen, N.S.; Nielsen, T.K.; Hutinet, G.; de Crécy-Lagard, V.; Cui, L.; Dedon, P.C.; Carstens, A.B.; Moineau, S.; Swairjo, M.A.; et al. Detection of preQ0 deazaguanine modifications in bacteriophage CAjan DNA using Nanopore sequencing reveals same hypermodification at two distinct DNA motifs. Nucleic Acids Res. 2020, 48, 10383–10396. [Google Scholar] [CrossRef]
- Young, I.; Wang, I.; Roof, W.D. Phages will out: Strategies of host cell lysis. Trends. Microbiol. 2000, 8, 120–128. [Google Scholar] [CrossRef]
- Summer, E.J.; Berry, J.; Tran, T.A.T.; Niu, L.; Struck, D.K.; Young, R. Rz/Rz1 lysis gene equivalents in phages of gram-negative hosts. J. Mol. Biol. 2007, 373, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Cowan, D.A. Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 2014, 80, 4470–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.M.; Weiss, N.; Rainey, F.; Salkinoja-Salonen, M.S. Dust-borne bacteria in animal sheds, schools and children’s day care centres. J. Appl. Microbiol. 1999, 86, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Nadarasah, G.; Stavrinides, J. Quantitative evaluation of the host colonizing capabilities of the enteric bacterium Pantoea using plant and insect hosts. Microbiology 2014, 160, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, J.; Mackiewicz, B.; Lemieszek, M.K.; Golec, M.; Milanowski, J. Pantoea agglomerans: A mysterious bacterium of evil and good. Part IV. Beneficial effects. Ann. Agric. Environ. Med. 2016, 23, 206–222. [Google Scholar] [CrossRef]
- Pileggi, M.; Pileggi, S.; Olchanheski, L.; de Silva, P.A.G.; Gonzalez, A.M.M.; Koskinen, W.C.; Barber, B.; Sadowsky, M.J. Isolation of mesotrione degrading bacteria from aquatic environments in Brazil. Chemosphere 2012, 86, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Nakata, K.; Inagawa, H.; Soma, G. Lipopolysaccharide IPPA1 from Pantoea agglomerans prevents suppression of macrophage function in stress-induced diseases. Anticancer Res. 2011, 31, 2437–2440. [Google Scholar] [PubMed]
- Dutkiewicz, J.; Mackiewicz, B.; Kinga Lemieszek, M.; Golec, M.; Milanowski, J. Pantoea agglomerans: A mysterious bacterium of evil and good. Part III. Deleterious effects: Infections of humans, animals and plants. Ann. Agric. Environ. Med. 2016, 23, 197–205. [Google Scholar] [CrossRef]
- Laanto, E.; Hoikkala, V.; Ravantti, J.; Sundberg, L.R. Long-term genomic coevolution of host-parasite interaction in the natural environment. Nat. Commun. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górski, A.; Międzybrodzki, R.; Łobocka, M.; Głowacka-Rutkowska, A.; Bednarek, A.; Borysowski, J.; Jończyk-Matysiak, E.; Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Bagińska, N. Phage therapy: What have we learned? Viruses 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymaekers, K.; Ponet, L.; Holtappels, D.; Berckmans, B.; Cammue, B.P.A. Screening for novel biocontrol agents applicable in plant disease management—A review. Biol. Control 2020, 144, 104240. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIME-light and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truncaitė, L.; Šimoliūnienė, M.; Alijošius, L.; Petrauskaitė, E.; Kiaušaitė, L.; Meškys, R.; Skapas, M.; Šimoliūnas, E. Complete genome analysis of Pantoea agglomerans-infecting bacteriophage vB_PagM_AAM22. Arch. Virol. 2020, 165, 2111–2114. [Google Scholar] [CrossRef] [PubMed]
- Šimoliūnienė, M.; Truncaitė, L.; Petrauskaitė, E.; Zajančkauskaitė, A.; Meškys, R.; Skapas, M.; Kaupinis, A.; Valius, M.; Šimoliūnas, E. Pantoea agglomerans-infecting bacteriophage vB_PagS_AAS21: A cold-adapted virus representing a novel genus within the family Siphoviridae. Viruses 2020, 12, 479. [Google Scholar] [CrossRef] [Green Version]
- McDougall, D.L.; Soutar, C.D.; Perry, B.J.; Brown, C.; Alexander, D.; Yost, C.K.; Stavrinides, J. Isolation and characterization of vB_PagP-SK1, a T7-like phage infecting Pantoea agglomerans. J. PHAGE 2020, 1, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.; Lurz, R.; Kube, M.; Quedenau, C.; Jelkmann, W.; Geider, K. Molecular and physiological properties of bacteriophages from North America and Germany affecting the fire blight pathogen Erwinia amylovora. Microb. Biotechnol. 2011, 4, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Born, Y.; Fiesler, L.; Marazzi, J.; Lurz, R.; Duffy, B.; Loessner, M.J. Novel virulent and broad host range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and relationship to Enterobacteriacea phages. Appl. Environ. Microbiol. 2011, 77, 5945–5954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulé, J.; Sholberg, P.L.; Lehman, S.M.; O’gorman, D.T.; Svircev, A.M. Isolation and characterization of eight bacteriophages infecting Erwinia amylovora and their potential as biological control agents in British Columbia, Canada. Can. J. Plant Pathol. 2011, 33, 308–317. [Google Scholar] [CrossRef]
- Lagonenko, A.L.; Sadovskaya, O.; Valentovich, L.N.; Evtushenkov, A.N. Characterization of a new Vil-like Erwinia amylovora bacteriophage phiEa2809. FEMS Microbiol. Lett. 2015, 362, fnv031. [Google Scholar] [CrossRef] [PubMed]
- Schwarczinger, I.; Kolozsváriné Nagy, J.; Künstler, A.; Szabó, L.; Geider, K.; Király, L.; Pogány, M. Characterization of Myoviridae and Podoviridae family bacteriophages of Erwinia amylovora from Hungary—potential of application in biological control of fire blight. Eur. J. Plant Pathol. 2017, 149, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Arens, D.K.; Brady, T.S.; Carter, J.L.; Pape, J.A.; Robinson, D.M.; Russell, K.A.; Staley, L.A.; Stettler, J.M.; Tateoka, O.B.; Townsend, M.H.; et al. Characterization of two related Erwinia myoviruses that are distant relatives of the PhiKZ-like Jumbo phages. PLoS ONE 2018, 13, e0200202. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Born, Y.; Lucid, A.; Loessner, M.J.; Fieseler, L.; Coffey, A. Erwinia amylovora phage vB_EamM_Y3 represents another lineage of hairy Myoviridae. Res. Microbiol. 2018, 169, 505–514. [Google Scholar] [CrossRef]
- Thompson, D.W.; Casjens, S.R.; Sharma, R.; Grose, J.H. Genomic comparison of 60 completely sequenced bacteriophages that infect Erwinia and/or Pantoea bacteria. Virology 2019, 535, 59–73. [Google Scholar] [CrossRef]
- Evans, T.J.; Crow, M.A.; Williamson, N.R.; Orme, W.; Thomson, N.R.; Komitopoulou, E.; Salmond, G.P.C. Characterization of a broad-host-range flagellum-dependent phage that mediates high-efficiency generalized transduction in, and between, Serratia and Pantoea. Microbiology 2010, 156, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the “Enterobacteriales”: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468–470, 421–443. [Google Scholar] [CrossRef] [Green Version]
- Bryson, A.L.; Hwang, Y.; Sherrill-Mix, S.; Wu, G.D.; Lewis, J.D.; Black, L.; Clark, T.A.; Bushman, F.D. Covalent modification of bacteriophage T4 DNA inhibits CRISPR-Cas9. MBio 2015, 6, e00648. [Google Scholar] [CrossRef] [Green Version]
- Vlot, M.; Houkes, J.; Lochs, S.J.A.; Swarts, D.C.; Zheng, P.; Kunne, T.; Mohanraju, P.; Anders, C.; Jinek, M.; van der Oost, J.; et al. Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR-Cas effector complexes. Nucleic Acids Res. 2018, 46, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Hattman, S. Unusual transcriptional and translational regulation of the bacteriophage Mu mom operon. Pharmacol. Ther. 1999, 84, 367–388. [Google Scholar] [CrossRef]
- Greene, J.R.; Morrissey, L.M.; Geiduschek, E.P. DNA binding by the bacteriophage SPO1-encoded type II DNA-binding protein, transcription factor 1. Site-specific binding requires 5-hydroxymethyluracil-containing DNA. J. Biol. Chem. 1986, 261, 12828–12833. [Google Scholar] [CrossRef]
- Sternberg, N.; Coulby, J. Cleavage of the bacteriophage P1 packaging site (pac) is regulated by adenine methylation. Proc. Natl. Acad. Sci. USA 1990, 87, 8070–8074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scraba, D.G.; Bradley, R.D.; Leyritz-Wills, M.; Warren, R.A. Bacteriophage phi W-14: The contribution of covalently bound putrescine to DNA packing in the phage head. Virology 1983, 124, 152–160. [Google Scholar] [CrossRef]
- Yuan, Y.; Hutinet, G.; Valera, J.G.; Hillebrand, R.; Gustafson, A.; Iwata-Reuyl, D.; Dedon, P.C.; de Crécy-Lagard, V. Identification of the minimal bacterial 2′-deoxy-7-amido-7-deazaguanine synthesis machinery. Mol. Microbiol. 2018, 110, 469–483. [Google Scholar] [CrossRef]
- Xu, D.; Ma, M.; Liu, Y.; Zhou, T.; Wang, K.; Deng, Z.; Hong, K. PreQ0 base, an unusual metabolite with anti-cancer activity from Streptomyces qinglanensis 172205. Anticancer Agents Med. Chem. 2015, 15, 285–290. [Google Scholar] [CrossRef]
- McCarty, R.M.; Bandarian, V. Biosynthesis of pyrrolopyrimidines. Bioorg. Chem. 2012, 43, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Matsuo, M.; Tanaka, S.; Akimoto, H.; Asahi, S.; Nishimura, S.; Katze, J.R.; Hashizume, T.; Crain, P.F.; McCloskey, J.A.; et al. Biosynthesis of archaeosine, a novel derivative of 7-deazaguanosine specific to archaeal tRNA, proceeds via a pathway involving base replacement on the tRNA polynucleotide chain. J. Biol. Chem. 1997, 272, 20146–20151. [Google Scholar] [CrossRef] [Green Version]
- Phillips, G.; Chikwana, V.M.; Maxwell, A.; El-Yacoubi, B.; Swairjo, M.A.; Iwata-Reuyl, D.; de Crécy-Lagard, V. Discovery and characterization of an amidinotransferase involved in the modification of archaeal tRNA. J. Biol. Chem. 2010, 285, 12706–12713. [Google Scholar] [CrossRef] [Green Version]
- Phillips, G.; Swairjo, M.A.; Gaston, K.W.; Bailly, M.; Limbach, P.A.; Iwata-Reuyl, D.; de Crécy-Lagard, V. Diversity of archaeosine synthesis in crenarchaeota. ACS Chem. Biol. 2012, 7, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Bon Ramos, A.; Bao, L.; Turner, B.; de Crécy-Lagard, V.; Iwata-Reuyl, D. QueF-Like, a Non-homologous archaeosine synthase from the crenarchaeota. Biomolecules 2017, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Van Lanen, S.G.; Reader, J.S.; Swairjo, M.A.; de Crecy-Lagard, V.; Lee, B.; Iwata-Reuyl, D. From cyclohydrolase to oxidoreductase: Discovery of nitrile reductase activity in a common fold. Proc. Natl. Acad. Sci. USA 2005, 102, 4264–4269. [Google Scholar] [CrossRef] [Green Version]
- Stengl, B.; Reuter, K.; Klebe, G. Mechanism and substrate specificity of tRNA-guanine transglycosylases (TGTs): tRNA-modifying enzymes from the three different kingdoms of life share a common catalytic mechanism. Chembiochem 2005, 6, 1926–1939. [Google Scholar] [CrossRef]
- Van Lanen, S.G.; Iwata-Reuyl, D. Kinetic mechanism of the tRNAmodifying enzyme S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA). Biochemistry 2003, 42, 5312–5320. [Google Scholar] [CrossRef] [PubMed]
- Miles, Z.D.; McCarty, R.M.; Molnar, G.; Bandarian, V. Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci. USA 2011, 108, 7368–7372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zallot, R.; Ross, R.; Chen, W.H.; Bruner, S.D.; Limbach, P.A.; de Crécy-Lagard, V. Identification of a novel epoxyqueuosine reductase family by comparative genomics. ACS Chem. Biol. 2017, 12, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Sambrook, J.; Russel, D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Šimoliūnas, E.; Kaliniene, L.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Kaupinis, J.; Ger, M.; Valius, M.; Meškys, R. Klebsiella phage vB_KleM-RaK2—A giant singleton virus of the family Myoviridae. PLoS ONE 2013, 8, e60717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropinski, A. Measurement of the rate of attachment of bacteriophage to cells. In Bacteriophages; Clokie, M.J., Kropinski, A., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 151–155. [Google Scholar]
- Carlson, K.; Miller, E. Experiments in T4 genetics. In Bacteriophage T4; Karam, J.D., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 419–483. [Google Scholar]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics 2013, 14, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Nam, S.Z.; Söding, J.; Lupas, A.N. The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. Viptree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [Green Version]
- Šimoliūnas, E.; Kaliniene, L.; Stasilo, M.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Nainys, J.; Kaupinis, A.; Valius, M.; Meškys, R. Isolation and characterization of vB_ArS-ArV2—First Arthrobacter sp. infecting bacteriophage with completely sequenced genome. PLoS ONE 2014, 9, e111230. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabler, F.; Nam, S.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Frickey, T.; Lupas, A. CLANS: A Java application for visualizing protein families based on pairwise similarity. Bioinformatics 2004, 20, 3702–3704. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimoliūnienė, M.; Žukauskienė, E.; Truncaitė, L.; Cui, L.; Hutinet, G.; Kazlauskas, D.; Kaupinis, A.; Skapas, M.; Crécy-Lagard, V.d.; Dedon, P.C.; et al. Pantoea Bacteriophage vB_PagS_MED16—A Siphovirus Containing a 2′-Deoxy-7-amido-7-deazaguanosine-Modified DNA. Int. J. Mol. Sci. 2021, 22, 7333. https://doi.org/10.3390/ijms22147333
Šimoliūnienė M, Žukauskienė E, Truncaitė L, Cui L, Hutinet G, Kazlauskas D, Kaupinis A, Skapas M, Crécy-Lagard Vd, Dedon PC, et al. Pantoea Bacteriophage vB_PagS_MED16—A Siphovirus Containing a 2′-Deoxy-7-amido-7-deazaguanosine-Modified DNA. International Journal of Molecular Sciences. 2021; 22(14):7333. https://doi.org/10.3390/ijms22147333
Chicago/Turabian StyleŠimoliūnienė, Monika, Emilija Žukauskienė, Lidija Truncaitė, Liang Cui, Geoffrey Hutinet, Darius Kazlauskas, Algirdas Kaupinis, Martynas Skapas, Valérie de Crécy-Lagard, Peter C. Dedon, and et al. 2021. "Pantoea Bacteriophage vB_PagS_MED16—A Siphovirus Containing a 2′-Deoxy-7-amido-7-deazaguanosine-Modified DNA" International Journal of Molecular Sciences 22, no. 14: 7333. https://doi.org/10.3390/ijms22147333