
Neither a Novel Tau Proteinopathy nor an Expansion of a Phenotype: Reappraising Clinicopathology-Based Nosology

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
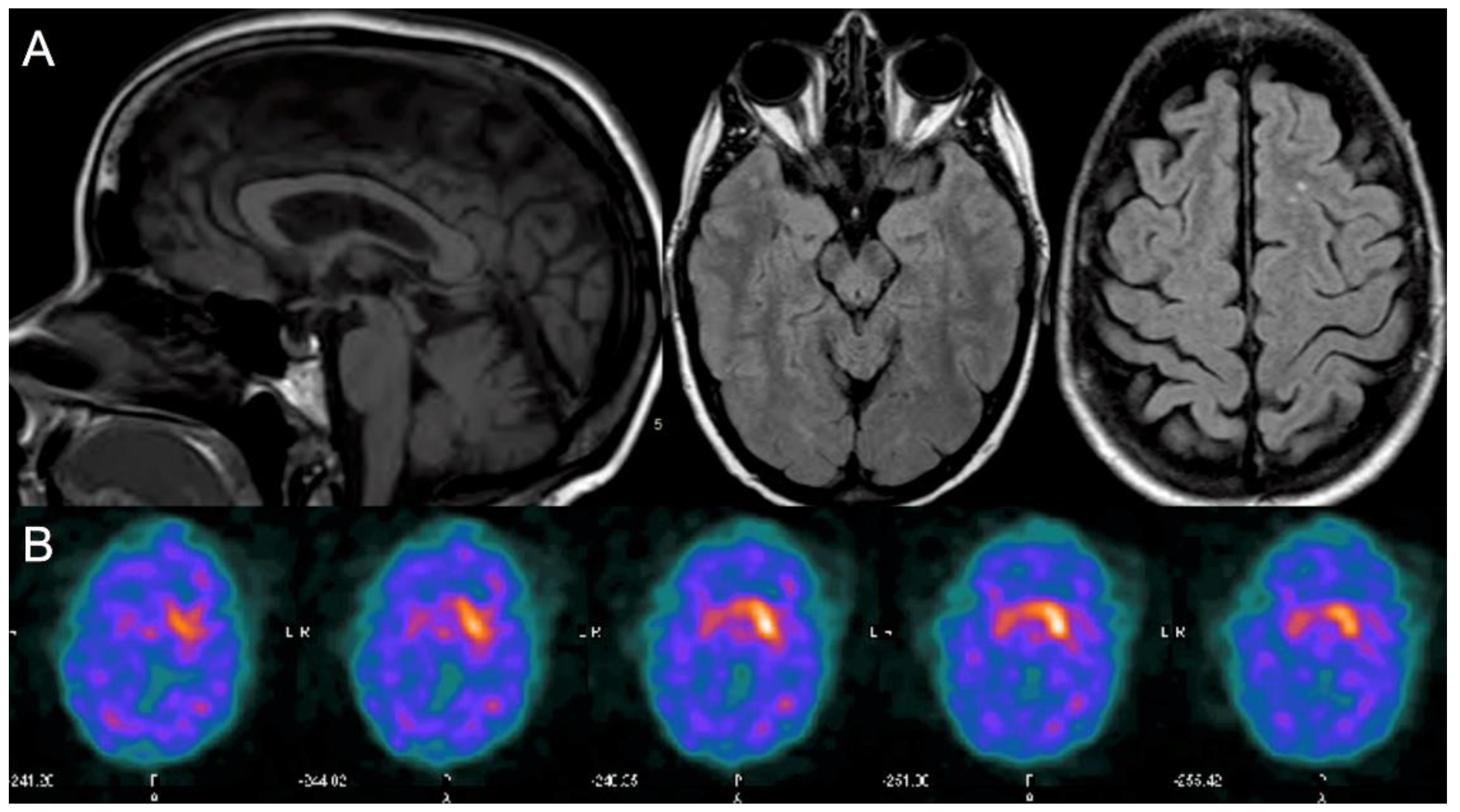
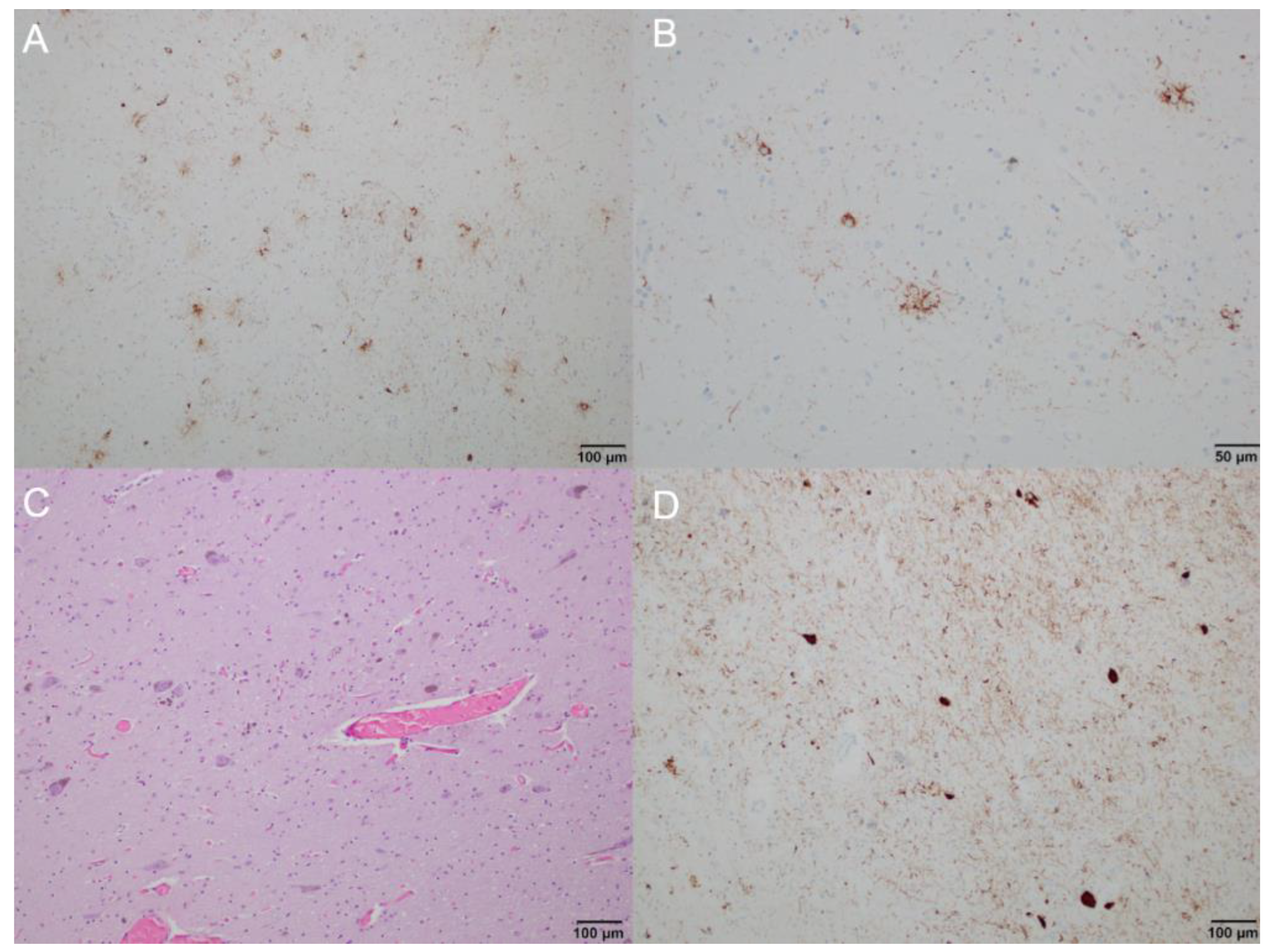
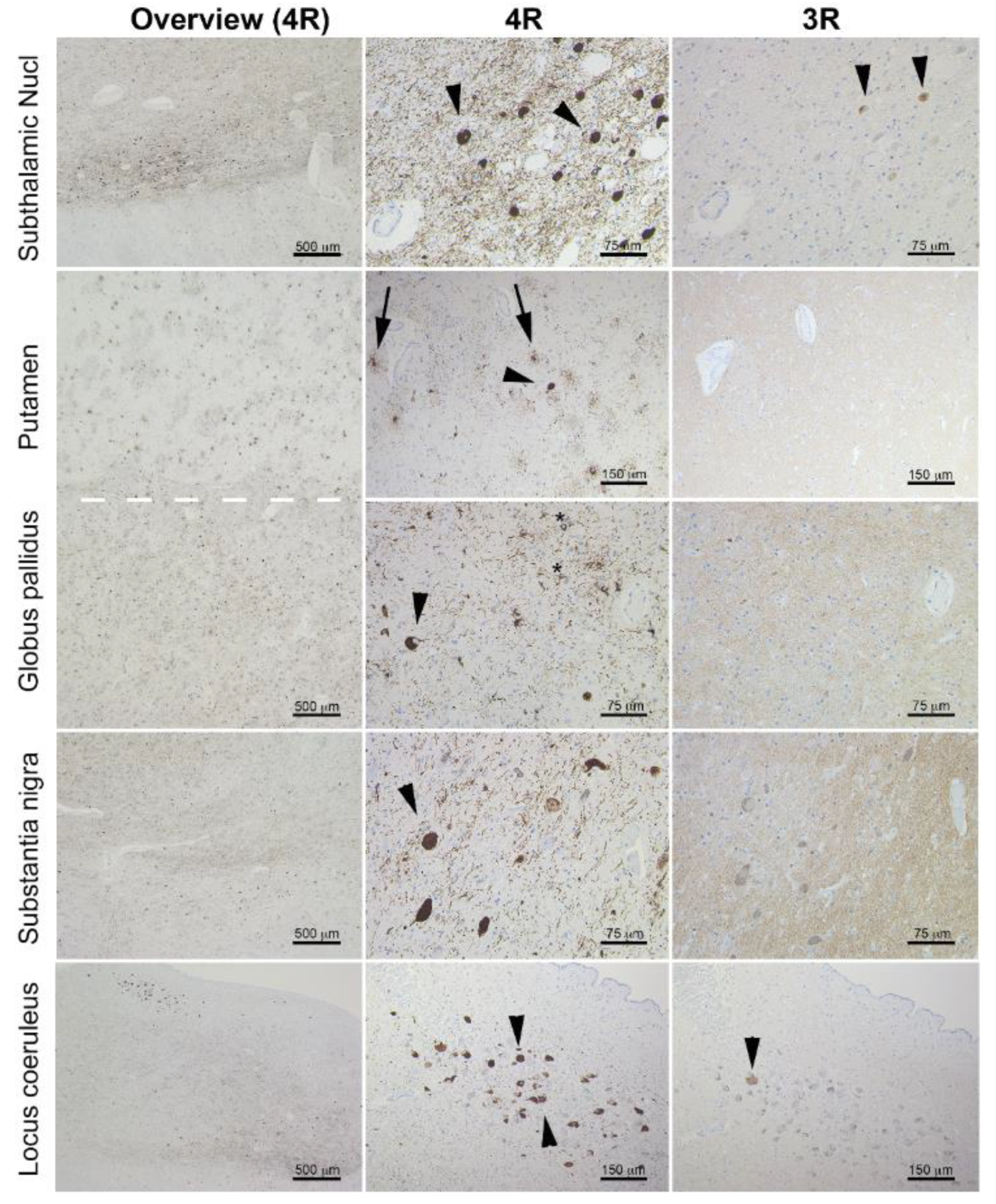
2. Case Report
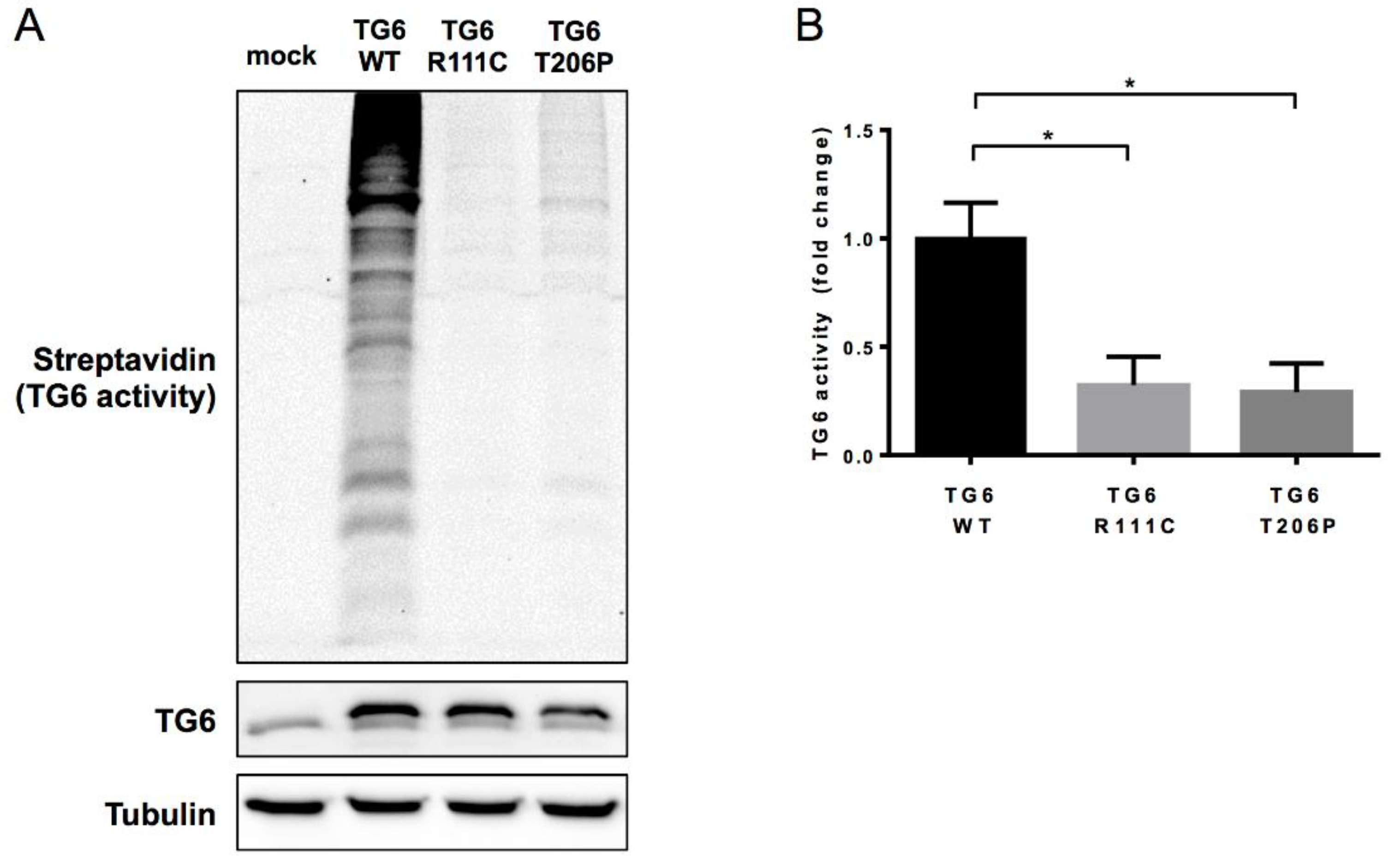
3. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–123. [Google Scholar] [CrossRef] [Green Version]
- Jeitner, T.M.; Battaile, K.; Cooper, A.J. γ-Glutamylamines and neurodegenerative diseases. Amino Acids 2013, 44, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Jeitner, T.M.; Pinto, J.T.; Krasnikov, B.F.; Horswill, M.; Cooper, A.J. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109 (Suppl. 1), 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Mulroy, E.; Jaunmuktane, Z.; Balint, B.; Erro, R.; Latorre, A.; Bhatia, K.P. Some New and Unexpected Tauopathies in Movement Disorders. Mov. Disord. Clin. Pract. 2020, 7, 616–626. [Google Scholar] [CrossRef]
- Massey, L.A.; Jäger, H.R.; Paviour, D.C.; O’Sullivan, S.S.; Ling, H.; Williams, D.R.; Kallis, C.; Holton, J.; Revesz, T.; Burn, D.J.; et al. The midbrain to pons ratio: A simple and specific MRI sign of progressive supranuclear palsy. Neurology 2013, 80, 1856–1861. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.L.; Yang, X.; Xia, K.; Hu, Z.M.; Weng, L.; Jin, X.; Jiang, H.; Zhang, P.; Shen, L.; Guo, J.F.; et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010, 133, 3510–3518. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Vignoli, B.; Ramesh, N.; Polanco, M.J.; Coutelier, M.; Stephen, C.D.; Canossa, M.; Monin, M.L.; Aeschlimann, P.; Turberville, S.; et al. Mutations in TGM6 induce the unfolded protein response in SCA35. Hum. Mol. Genet. 2017, 26, 3749–3762. [Google Scholar] [CrossRef]
- Chen, K.; Lu, Y.; Peng, F.; Yu, H.L.; Wu, J.Y.; Tan, Y.; Zhao, Y.X. TGM6 variants in Parkinson’s disease: Clinical findings and functional evidence. J. Integr. Neurosci. 2020, 19, 51–64. [Google Scholar] [CrossRef]
- Fung, J.L.F.; Tsang, M.H.Y.; Leung, G.K.C.; Yeung, K.S.; Mak, C.C.Y.; Fung, C.W.; Chan, S.H.S.; Yu, M.H.C.; Chung, B.H.Y. A significant inflation in TGM6 genetic risk casts doubt in its causation in spinocerebellar ataxia type 35. Parkinsonism Relat. Disord. 2019, 63, 42–45. [Google Scholar] [CrossRef]
- Guo, Y.C.; Lin, J.J.; Liao, Y.C.; Tsai, P.C.; Lee, Y.C.; Soong, B.W. Spinocerebellar ataxia 35: Novel mutations in TGM6 with clinical and genetic characterization. Neurology 2014, 83, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Shi, M.M.; Liu, Y.T.; Wang, Y.L.; Luo, H.Y.; Wang, Z.L.; Shi, C.H.; Xu, Y.M. TGM6 gene mutations in undiagnosed cerebellar ataxia patients. Parkinsonism Relat. Disord. 2018, 46, 84–86. [Google Scholar] [CrossRef]
- Li, M.; Pang, S.Y.; Song, Y.; Kung, M.H.; Ho, S.L.; Sham, P.C. Whole exome sequencing identifies a novel mutation in the transglutaminase 6 gene for spinocerebellar ataxia in a Chinese family. Clin. Genet. 2013, 83, 269–273. [Google Scholar] [CrossRef]
- Fasano, A.; Hodaie, M.; Munhoz, R.P.; Rohani, M. SCA 35 presenting as isolated treatment-resistant dystonic hand tremor. Parkinsonism Relat. Disord. 2017, 37, 118–119. [Google Scholar] [CrossRef]
- Manini, A.; Bocci, T.; Migazzi, A.; Monfrini, E.; Ronchi, D.; Franco, G.; De Rosa, A.; Sartucci, F.; Priori, A.; Corti, S.; et al. A case report of late-onset cerebellar ataxia associated with a rare p.R342W TGM6 (SCA35) mutation. BMC Neurol. 2020, 20, 408. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef]
- Kanazawa, M.; Shimohata, T.; Toyoshima, Y.; Tada, M.; Kakita, A.; Morita, T.; Ozawa, T.; Takahashi, H.; Nishizawa, M. Cerebellar involvement in progressive supranuclear palsy: A clinicopathological study. Mov. Disord. 2009, 24, 1312–1318. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Lukic, M.J.; Irwin, D.J.; Arzberger, T.; Respondek, G.; Lee, E.B.; Coughlin, D.; Giese, A.; Grossman, M.; Kurz, C.; et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020, 140, 99–119. [Google Scholar] [CrossRef]
- Guan, W.J.; Wang, J.L.; Liu, Y.T.; Ma, Y.T.; Zhou, Y.; Jiang, H.; Shen, L.; Guo, J.F.; Xia, K.; Li, J.D.; et al. Spinocerebellar ataxia type 35 (SCA35)-associated transglutaminase 6 mutants sensitize cells to apoptosis. Biochem. Biophys. Res. Commun. 2013, 430, 780–786. [Google Scholar] [CrossRef]
- Thomas, H.; Beck, K.; Adamczyk, M.; Aeschlimann, P.; Langley, M.; Oita, R.C.; Thiebach, L.; Hils, M.; Aeschlimann, D. Transglutaminase 6: A protein associated with central nervous system development and motor function. Amino Acids 2013, 44, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, M.; Ratan, R.R. Transglutaminase is a therapeutic target for oxidative stress, excitotoxicity and stroke: A new epigenetic kid on the CNS block. J. Cereb. Blood Flow Metab. 2013, 33, 809–818. [Google Scholar] [CrossRef]
- Zemaitaitis, M.O.; Kim, S.Y.; Halverson, R.A.; Troncoso, J.C.; Lee, J.M.; Muma, N.A. Transglutaminase activity, protein, and mRNA expression are increased in progressive supranuclear palsy. J. Neuropathol. Exp. Neurol. 2003, 62, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Picillo, M.; Amboni, M.; Bruni, A.; Maletta, R.; Barone, P. Prevalence of heterozygous mutations in Niemann-Pick type C genes in a cohort of progressive supranuclear palsy. Parkinsonism Relat. Disord. 2020, 79, 9–10. [Google Scholar] [CrossRef]
- Malmberg, M.; Malm, T.; Gustafsson, O.; Sturchio, A.; Graff, C.; Espay, A.J.; Wright, A.P.; El Andaloussi, S.; Lindén, A.; Ezzat, K. Disentangling the Amyloid Pathways: A Mechanistic Approach to Etiology. Front. Neurosci. 2020, 14, 256. [Google Scholar] [CrossRef] [Green Version]
- Espay, A.J.; Vizcarra, J.A.; Marsili, L.; Lang, A.E.; Simon, D.K.; Merola, A.; Josephs, K.A.; Fasano, A.; Morgante, F.; Savica, R.; et al. Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 2019, 92, 329–337. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsili, L.; Sharma, J.; Espay, A.J.; Migazzi, A.; Abdelghany, E.; Hill, E.J.; Duque, K.R.; Hagen, M.C.; Stephen, C.D.; Kovacs, G.G.; et al. Neither a Novel Tau Proteinopathy nor an Expansion of a Phenotype: Reappraising Clinicopathology-Based Nosology. Int. J. Mol. Sci. 2021, 22, 7292. https://doi.org/10.3390/ijms22147292
Marsili L, Sharma J, Espay AJ, Migazzi A, Abdelghany E, Hill EJ, Duque KR, Hagen MC, Stephen CD, Kovacs GG, et al. Neither a Novel Tau Proteinopathy nor an Expansion of a Phenotype: Reappraising Clinicopathology-Based Nosology. International Journal of Molecular Sciences. 2021; 22(14):7292. https://doi.org/10.3390/ijms22147292
Chicago/Turabian StyleMarsili, Luca, Jennifer Sharma, Alberto J. Espay, Alice Migazzi, Elhusseini Abdelghany, Emily J. Hill, Kevin R. Duque, Matthew C. Hagen, Christopher D. Stephen, Gabor G. Kovacs, and et al. 2021. "Neither a Novel Tau Proteinopathy nor an Expansion of a Phenotype: Reappraising Clinicopathology-Based Nosology" International Journal of Molecular Sciences 22, no. 14: 7292. https://doi.org/10.3390/ijms22147292