
Fructose and the Liver


1
Laboratory of Experimental Hepatology, Department of Pharmacology, Cinvestav-IPN, Apartado Postal 14-740, Mexico City 07300, Mexico
2
Postgraduate Studies and Research Section, School of Higher Education in Medicine-IPN, Plan de San Luis y Díaz Mirón s/n, Casco de Santo Tomás, Mexico City 11340, Mexico
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(13), 6969; https://doi.org/10.3390/ijms22136969
Submission received: 26 May 2021
/
Revised: 22 June 2021
/
Accepted: 25 June 2021
/
Published: 28 June 2021
(This article belongs to the Special Issue Molecular Link between Steatosis and Obesity)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Chronic diseases represent a major challenge in world health. Metabolic syndrome is a constellation of disturbances affecting several organs, and it has been proposed to be a liver-centered condition. Fructose overconsumption may result in insulin resistance, oxidative stress, inflammation, elevated uric acid levels, increased blood pressure, and increased triglyceride concentrations in both the blood and liver. Non-alcoholic fatty liver disease (NAFLD) is a term widely used to describe excessive fatty infiltration in the liver in the absence of alcohol, autoimmune disorders, or viral hepatitis; it is attributed to obesity, high sugar and fat consumption, and sedentarism. If untreated, NAFLD can progress to nonalcoholic steatohepatitis (NASH), characterized by inflammation and mild fibrosis in addition to fat infiltration and, eventually, advanced scar tissue deposition, cirrhosis, and finally liver cancer, which constitutes the culmination of the disease. Notably, fructose is recognized as a major mediator of NAFLD, as a significant correlation between fructose intake and the degree of inflammation and fibrosis has been found in preclinical and clinical studies. Moreover, fructose is a risk factor for liver cancer development. Interestingly, fructose induces a number of proinflammatory, fibrogenic, and oncogenic signaling pathways that explain its deleterious effects in the body, especially in the liver.
1. Introduction
Chronic diseases represent a major challenge in world health. Metabolic syndrome is a constellation of disturbances that includes dyslipidemia, type II diabetes, insulin resistance, visceral obesity, microalbuminuria, and hypertension [1,2]. The prevalence of metabolic syndrome is difficult to establish because there is no consensus on its definition [1], but estimations are 27.93% in North America, 27.65% in South America, 21.27% in Asia, 16.04% in Africa, and 10.47% in Europe [3], affecting a quarter of the world’s population [4]. The most important risk factors for developing metabolic syndrome are related to obesity, a complex disease associated with an imbalance between physical activity and calorie intake, and excessive consumption of fats and simple carbohydrates; the obesogenic environment also plays an important role [5]. Approximately one-third of adults, children, or adolescents worldwide are obese or overweight [1,2,6].
Metabolic syndrome affects several organs, and it has been proposed to be a liver-centered condition [7]. Non-alcoholic fatty liver disease (NAFLD) is a term widely used to describe excessive fat infiltration in the liver in the absence of alcohol, autoimmune disorders, and viral hepatitis [6]. NAFLD now constitutes the main cause of hepatic disorders. It is usually asymptomatic, bidirectionally linked with metabolic syndrome, and difficult to diagnose, affecting about a third of the global population, and it is the prevailing cause of hepatocellular carcinoma (HCC) development [8,9]. Thirty percent of NAFLD patients develop necroinflammation and fibrosis, indicating the presence of nonalcoholic steatohepatitis (NASH), which in turn may predispose patients to HCC [10,11,12,13]. Moreover, NASH is a risk factor for liver cancer development [2,14,15,16,17,18]. HCC is the dominant form of primary liver cancer, which represents 75–90% of the total liver cancer burden [19,20]. In the early stages, HCC lacks symptoms and exhibits a rapid growth of malignant cells, resulting in the late diagnosis of the disease, and, therefore, at least one-quarter of HCC cases remain idiopathic and can be attributed to NAFLD [21,22,23]. Unfortunately, no effective therapy for NAFLD is currently available, and there is no scientific evidence to recommend specific diets for this group of patients. This paper provides a review of fructose deleterious effects on the liver and describes the molecular mechanisms involved.
Based on animal model experiments and clinical studies, fructose is recognized as a major mediator of NAFLD [24,25,26]. A significant correlation between fructose intake and the degree of fibrosis has been found [24,27]. Fructose is present in fruits and table sugar (sucrose, which is 50% glucose and 50% fructose), and has a sweeter taste and lower glycemic index than glucose. Its consumption has recently increased in many parts of the world because of the growing use of high-fructose corn syrup in beverages and processed food [21]. Studies on ancestral diets have shown that the average intake of fructose per capita was around 2 kg per year, while the current global average consumption of fructose per capita is 25 kg per year [28]. High-fructose corn syrup is made from corn using caustic soda, hydrochloric acid, and enzymes and is classified according to the percentage of fructose content (90, 55, and 42%). This powerful and cheap sweetener provides a long shelf life and maintains long-lasting hydration in industrial bakeries [29]. In developing countries, such as Mexico, its importation from the United States has significantly increased recently, indicating a larger demand for added sugars in these emerging markets. Sugar-sweetened beverages provide 60% of the daily sugar intake in the United States. Mexico has the largest number of sugar-sweetened beverage consumers worldwide, where such beverages provide 69% of the total added sugar in the daily diet [30,31]. Additionally, NAFLD patients consume twice as many calories from beverages sweetened with high-fructose corn syrup as healthy patients [32]. The World Health Organization recommends reducing the intake of free sugars to less than 10% of the total daily energy intake because of its association with metabolic diseases and cancer [32,33,34]. Furthermore, elevated consumption of fructose represents a great metabolic risk for not only obese but also lean individuals who have a high consumption of fructose-sweetened beverages [7,24,35].
2. Deleterious Metabolic Effects of Fructose
2.1. The Initial Physiological Impact of High Fructose Consumption
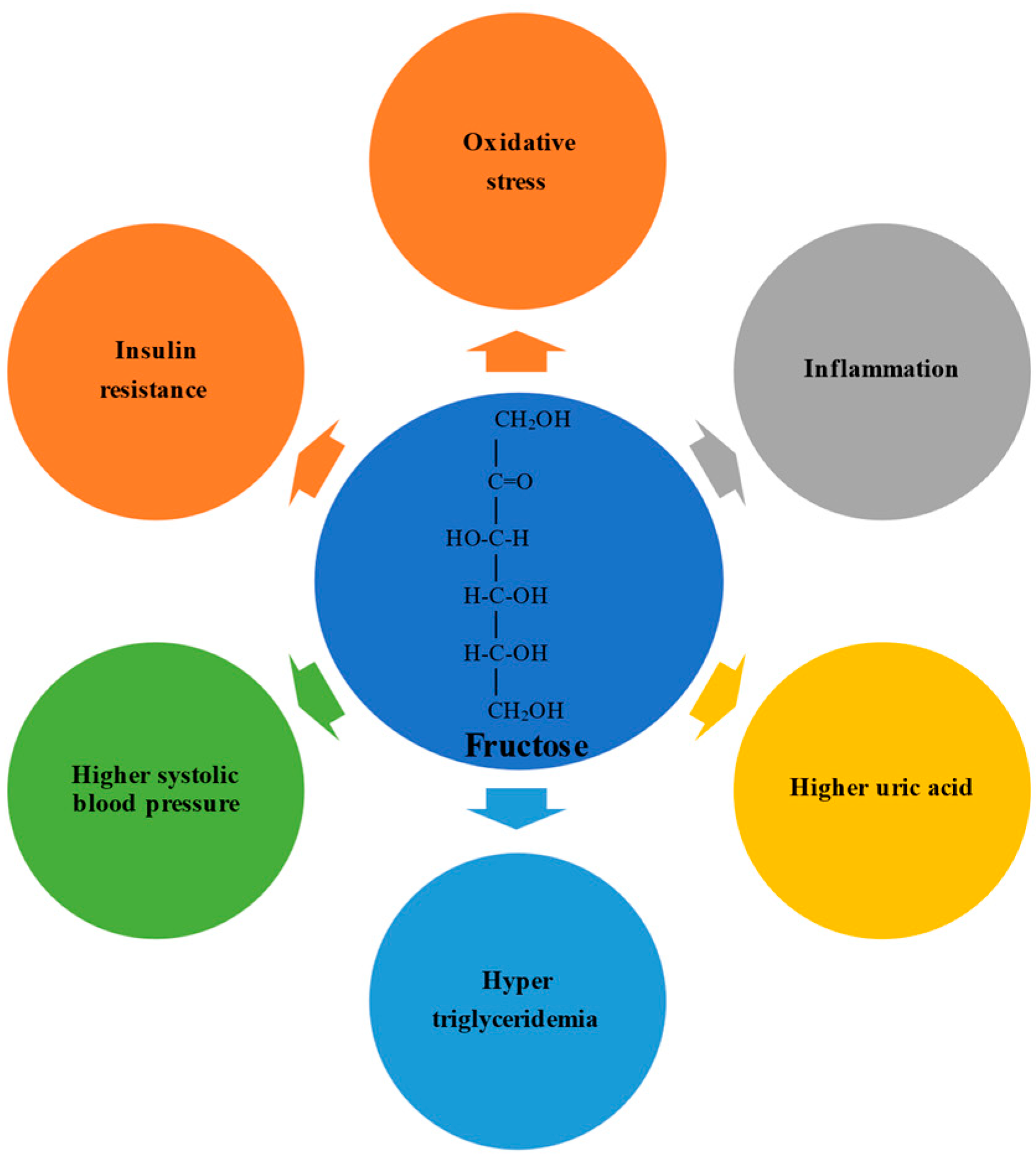
Fructose possesses an open-chain chemical conformation and is therefore much more reactive than glucose [36]. Experimental studies have shown that a high fructose intake promotes oxidative stress, inflammation, higher serum uric acid levels, hypertriglyceridemia, higher systolic blood pressure, and insulin resistance [37,38] (Figure 1). In humans, the physiological impact depends on the formulation in which the fructose is consumed; consumption via solids and liquids differently affects microbiota composition, gut integrity, and liver toxicity [39,40].
Sensory stimulation is the adaptive response to food intake through rapid physiological processes, and one of the most studied is the cephalic-phase insulin response. Oral fructose stimulates autonomic and endocrine responses, which downregulate the cephalic phase of the insulin pathway in taste cells, reducing pancreatic insulin production [41]. Additionally, eating fructose, in contrast to glucose consumption, leads to increased hunger and desire to eat because fructose decreases leptin and glucagon-like peptide 1 and increases ghrelin levels in the serum [42]. Ghrelin activates the neuronal activity of neuropeptide Y, increasing food intake, and glucagon-like peptide 1 inhibition causes a decrease in insulin secretion [43]. Increased dietary fructose intake significantly accelerated the half-emptying time in the stomach compared to a similar intake of glucose [44]. Fructose, in the mouth and gut, may impact eating behavior by sweet-tasting mechanisms [45]. Sweet foods have powerful reinforcing effects mediated, in part, by dopamine receptors and, on vulnerable individuals, may overwhelm the homeostatic control mechanisms of the brain, possibly inducing behavioral alterations observed in addiction, such as anxiety or craving [46,47,48]. Regarding the hedonic value of fructose and the sum of all these events that affect appetite control, more studies are required to understand the role of fructose in the reward system.
2.2. Fructose in the Intestine
The intestinal epithelium is the cell layer closest to the intestinal lumen and is composed by 70–80% of enterocytes, Paneth cells, goblet cells, and intestinal stem cells. Studies attribute the metabolic effects of fructose to enterocytes, cells specialized in absorption [49].
2.2.1. Intestinal Absorption of Fructose
The human small intestine expresses all the fructose-metabolizing enzymes; glucose transporter protein member 5 (Glut5) is the main protein responsible for the absorption of fructose into the cytosol [50,51]. Glut5, which mediates the active transport of fructose in mice, is mainly found in the small intestine [52]. Glut2 (SLC2A2) is a non-specific glucose transporter expressed in the intestinal basolateral membrane, which transports fructose by a facilitated passive mechanism from the gut into the hepatic portal circulation [53]. In humans, fructose is converted to glucose when the intake is moderate (≤1 g/kg of body weight), while high fructose consumption leads to the strong induction of Glut5 but not Glut2, thus increasing the fructose concentration and catabolism in the cytosol of intestinal epithelial cells [36,54,55,56]. The deletion of Glut5 in mice has been shown to reduce fructose absorption [57]. Glut5 is a transceptor, a transporter that binds to its substrate and activates intracellular signaling that triggers multiple responses [58]. Thioredoxin-interacting protein (TXNIP) is a multifunctional intracellular protein that coordinates signaling pathways during oxidative stress and inflammation [59]. TXNIP is also a regulator of carbohydrate metabolism [60]. Glut5 binds to TXNIP, which leads to increased Glut5 gene expression and protein synthesis, facilitating its migration to the apical membrane, thus improving fructose absorption [59]. In the cytoplasm of intestinal cells, the ketohexokinase (KHK) enzyme, also called fructokinase, which has a high affinity for fructose, phosphorylates fructose to fructose-1-phosphate, a toxic metabolite [61]. Excess phosphorylated fructose is conducted to the hexosamine pathway, which increases O-glucosamine-N-acetyl transferase activity and consequently upregulates the expression of transforming growth factor-beta (TGF-β) [62]. There are two isoforms of KHK, KHK-C and KHK-A; the latter has a ten-times-lower affinity for fructose than KHK-C and therefore consumes ATP more slowly [7,63]. KHK-A may decrease fructose metabolism in the liver and, thus, may inhibit the development of metabolic syndrome [36]. By contrast, KHK-C overexpression promotes intestinal fructose clearance and increases fructose-induced lipogenesis in the liver [61]. However, when the capacity for intestinal fructose clearance is exceeded, the increased activity of KHK-C exhausts adenosine triphosphate (ATP) and induces adenosine monophosphate deaminase activation, which results in marked ATP depletion, leading to the accumulation of adenosine monophosphate and uric acid production [64]. Preclinical evidence using human livers, KHK inhibition to improve steatosis, inflammation and fibrosis in NAFLD [65].
2.2.2. Intestinal Production of Uric Acid by Fructose
Uric acid, a weak organic acid end-product of purine catabolism in humans, is an antioxidant molecule that plays an essential role in the cardiac, vascular, and central nervous systems because it can neutralize pro-oxidant free radicals, such as hydroxyl radicals, hydrogen peroxide, and peroxynitrite [64]. Uric acid is produced in the liver and gut and excreted through the urine and feces [64]. According to Yun et al., the duodenum plays an important role in the synthesis and elimination of uric acid; one-third of the total uric acid is excreted through the gut [66]. Thirty percent of uric acid is excreted via ATP-binding cassette subfamily 2 (breast cancer resistance protein) on the luminal surface of the intestine, but an imbalance in its production or excretion can increase uric acid levels, favoring nicotinamide adenine dinucleotide (NADPH) oxidase (NOX) activation in the liver, acting as a damage-associated molecular pattern (DAMP) [67,68].
2.2.3. Fructose Induces Lipogenesis and Oxidative Stress in the Intestine
Furthermore, a high fructose intake in an experimental model can activate carbohydrate-responsive element-binding protein (ChREBP) and sterol-responsive element-binding protein (SREBP), which induce fructolytic and lipogenic enzymes, respectively [69]. ChREBP is a transcription factor activated by a high-fructose diet, improving the KHK and Glut5 capacity for fructose absorption [70]. SREBP is a family of transcription factors consisting of three isoforms that regulate the homeostasis of lipids. In enterocytes, apolipoprotein induces the transcription of SREBP1c, which improves the stability of ApoB-48, the structural protein for chylomicrons, enhances microsomal triglyceride transfer protein, and augments lipogenesis [69]. This uncontrolled lipid metabolism and lower clearance of chylomicrons in the intestinal cells, together with uric acid overproduction, is responsible for increased cardiometabolic risk and leads to the development of NASH [70,71,72].
NASH models showed that cytochrome P450 2E1 activity is linked to increased intestinal inflammation during fructose consumption [73]. Cytochrome P450 2E1 plays a critical role in the metabolism of fatty acids. Furthermore, NASH patients have increased cytochrome P450-2E1 and inducible nitric oxide synthase, which cause the nitration of intestinal tight and adherent junction proteins [74]. The disruption of tight junction proteins and elevated apoptosis of enterocytes, evidenced by the upregulation of caspase 3 and p-JNK after fructose exposure, contributes to endoplasmic reticulum stress, the accumulation of unfolded or misfolded proteins, and the dysfunction of the epithelial barrier, which result in increased gut permeability, allowing lipopolysaccharides (LPS) to translocate from the gut lumen to the portal tract, triggering an inflammatory response in the liver [74]. Ca2+ absorption is one of the most important intestinal functions, and glutathione (GSH) is essential for this process [75]. γ-L-glutamyl-L-cysteinylglycine, or GSH, is the main intracellular cofactor protecting against oxidative stress in the gut, and its biosynthesis occurs in the cytosol through ATP-dependent reactions [76]. The antioxidant activity of GSH is catalyzed by GSH peroxidase (GPx), which reduces hydrogen peroxide and lipid peroxides as GSH is oxidized to GSSG [77]. In animal models that use fructose-rich diets, the intestinal absorption of Ca2+ is decreased, and Ca2+ receptors are depleted, which leads to decreased antioxidant defenses (GPx, catalase, superoxide dismutase, etc., are exhausted), and endoplasmic reticulum stress occurs [75]. Similarly, increased fructose phosphorylation triggers ATP depletion, as mentioned earlier, inhibiting GSH restoration.
2.2.4. Fructose and the Microbiota
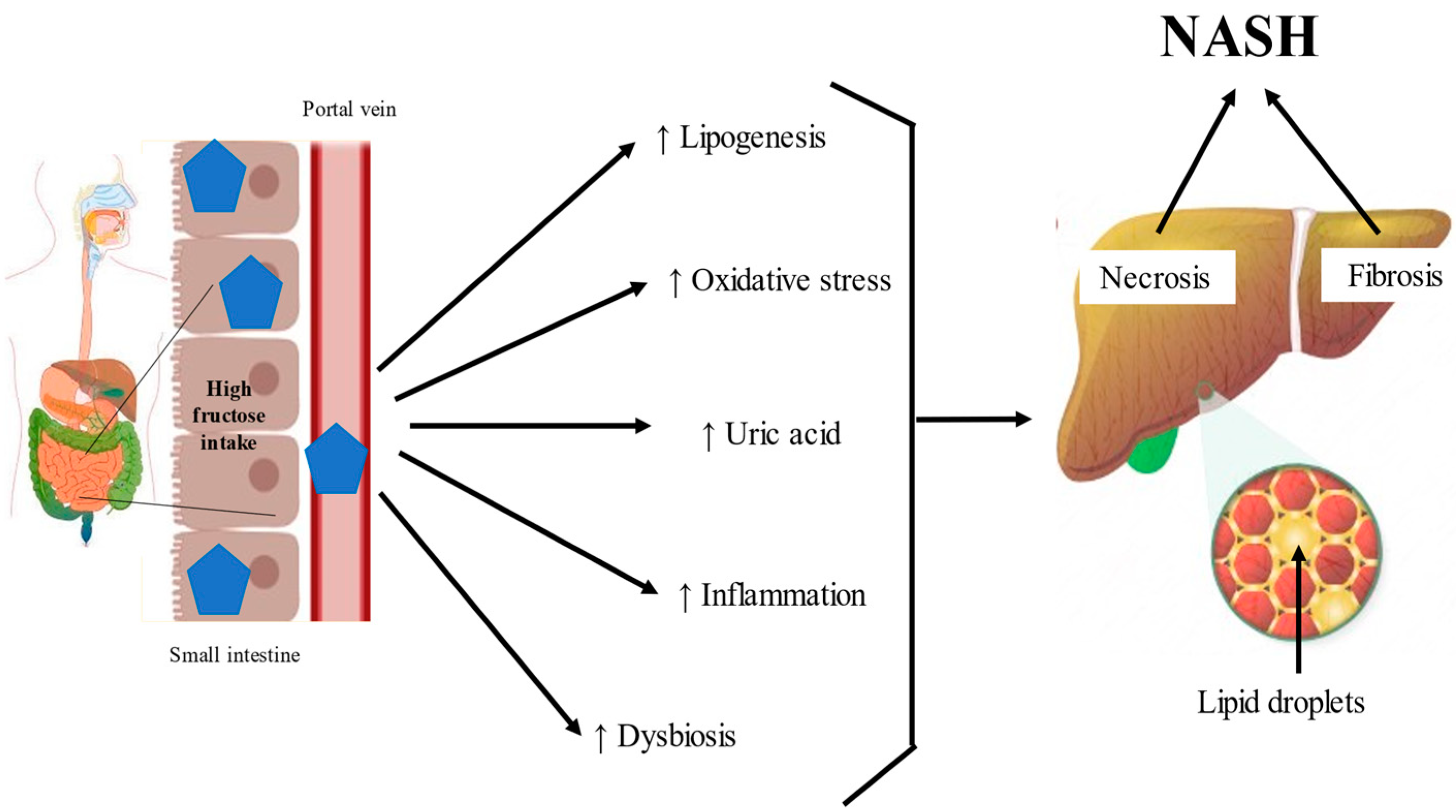
The composition and function of the microbiota are regulated by multiple factors, such as diet and physical activity. Recent reports show that fructose consumption alters the gut microbiota and their bacterial metabolites, in a manner that promotes the development and progression of NASH [78]. Excessive fructose consumption decreases the expression of intestinal tight junction proteins, such as zonula occludens 1, junctional adhesion molecule A, occludin, claudin, β-catenin, and E-cadherin [74,79]. This environment generates dysbiosis by increasing Bacteroides, Proteobacteria, Enterobacteria, Escherichia, Blautia producta, and Bacteroides fragilis while decreasing Actinobacteria, Akkermansia, Verrucomicrobia, Coprococcus eutactus, and Lactobacillus, increasing the loss and blebbing of the laminar propria, which triggers inflammation in the small intestine, and, due to the increase in gut permeability, toxic bacterial metabolites may reach the liver, contributing to inflammation in NASH [29,36,74,80,81]. Similarly, diets enriched with fructose alter the composition of the short-chain fatty acids in the gut, inducing a high microbial production of butyrate, acetate or propionate by the intestinal microbiota, therefore increasing the production of acetyl-CoA from acetate, which contributes to lipogenesis [82]. Ethanol is also an important fructose metabolite that has been associated with NAFLD. Patients suffering from NAFLD who abuse alcohol exhibit more severe liver injury than those with any of these factors individually [83]. It is noteworthy that Escherichia, Bacteroides, and Clostridium bacteria can produce ethanol. In patients with NAFLD, the activity of alcohol-metabolizing enzymes, such as alcohol dehydrogenase, and the microbiota are dysregulated [84]. As a consequence, increased blood ethanol concentrations and/or ethanol metabolites can alter the host’s metabolism, generate reactive oxygen species, and active inflammatory pathways, suggesting that microbiota that produce alcohol can have important effects on the evolution of NAFLD [85,86,87]. Moreover, gut dysbiosis triggered by excessive fructose intake leads to intestinal bacterial overgrowth, a strong decrease in microbial diversity, and increased translocation of bacterial products and cytotoxins, stimulating inflammatory pathways in experimental and human NAFLD [88,89] (Figure 2). These results indicate that high fructose in the intestine plays a major role in NAFLD development. The dysregulated microbiota, disruption of intestinal tight junction proteins, elevated uric acid production, and toxic bacterial metabolites accelerate NASH progression. The deleterious effects of fructose in the intestine could be ameliorated by the development of selective inhibitors of KHK-C, the limiting enzyme in fructose metabolism.
2.3. Fructose in the Liver
In humans, 70% of fructose is metabolized by the liver [90]. A diet rich in fructose induces the hepatic de novo synthesis of fatty acids and triglyceride accumulation [7,38,90]. Therefore, fructose has been postulated as a key factor for the development of NASH. Once fructose exceeds the intestinal clearance capacity, it is driven to the portal vein, where a fructosemic state strongly and quickly induces mechanisms involved in its overflow to the liver, which is the principal organ for fructose metabolism [7,38]. However, the mechanisms of the hepatic cell types (hepatocytes, hepatic stellate cells (HSCs), and Kupffer cells) that are involved in the metabolism of fructose consumed in large quantities are poorly understood [69]. In the liver, fructose is catabolized faster and is more lipogenic than glucose. In particular, chronic high fructose consumption induces the aldolase B enzyme, which breaks down fructose to dihydroxyacetone phosphate and D-glyceraldehyde. Then, triokinase stimulates the phosphorylation of D-glyceraldehyde to produce pyruvate and acetyl-CoA, promoting lipid dysregulation [36,54,91] (Figure 3).
2.3.1. Ketohexokinase and Fructose
The liver plays the most important role in carbohydrate metabolism. The principal isoform of KHK in the liver is KHK-C, which phosphorylates fructose rapidly and without any negative feedback control. Similar to in mice, KHK expression is elevated in obese patients with advanced liver disease compared to in obese subjects without fatty liver [81]. In humans, KHK inhibition has been demonstrated to improve steatosis, ballooning degeneration, inflammation, and fibrosis in the liver [92]. In KHK-knockout mice, ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC)-1, and fatty acid synthase (FASN) are decreased by fructose administration [81]. ACLY is an enzyme that links carbohydrate to lipid metabolism by converting citrate to acetyl-CoA for fatty acid and cholesterol biosynthesis. ACLY inhibition protects against hepatic steatosis, dyslipidemia, and associated complications such as atherosclerosis [93]. ACC-1 coordinates the synthesis of fatty acids in the liver and generates a pool of malonyl-CoA used by FASN to generate palmitate [94]. ACC-1 inhibition reduces lipotoxicity in hepatocytes and prevents HSC activation, which significantly reduces fibrosis in NASH [94] (Figure 3).
2.3.2. Toll-Like Receptor-4 and Fructose
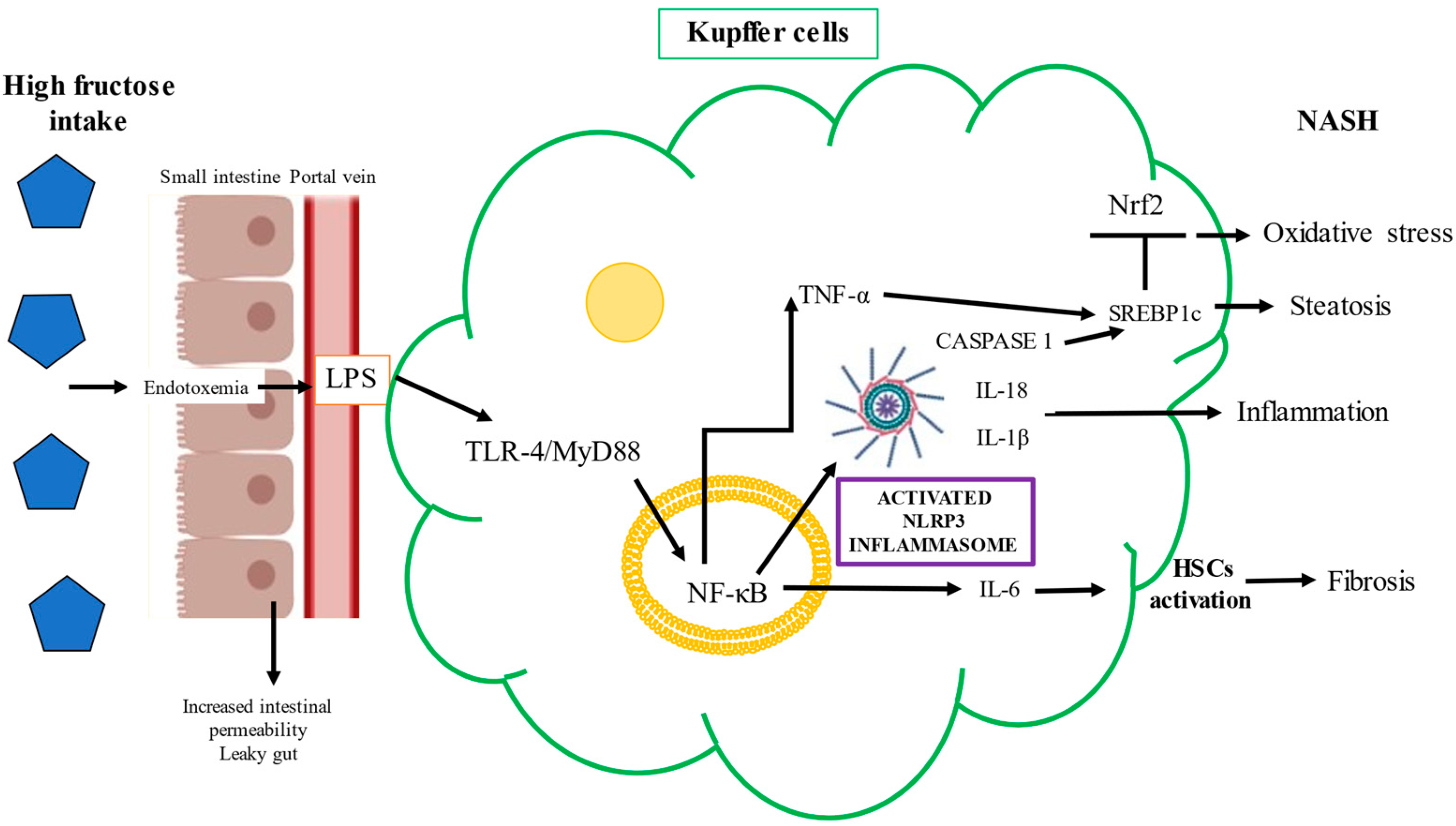
Kupffer cells play a central role in liver damage induced by fructose. The elevated endotoxemia and oxidative stress produced by fructose intake promote hepatic Toll-like receptor (TLR)-4 activation. The TLR-4/MyD88 signaling pathway in liver parenchymal cells plays a pivotal role during NASH development [85,87]. As previously mentioned, fructose causes gut-barrier deterioration through the disruption of tight-junction proteins. Endotoxins produced by Gram-negative bacteria alter intestinal permeability and cause bacterial translocation. Mice chronically fed fructose were found to have increased levels of endotoxins in portal blood and unregulated inflammatory mediators in Kupffer cells [95]. LPS and other bacterial toxins cross the gut barrier and bind to TLR-4 on the macrophages or Kupffer cells’ plasma membranes, which activates the proinflammatory signaling pathway, with a consequent increase in the expression of proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1beta (β) [78,96]. Specifically, the binding of LPS to TLR-4 on macrophages activates the nuclear factor kappa B (NF-κB) signaling pathway via the adaptor protein MyD88 and induces TNF-α expression and secretion [97]. TNF-α also stimulates fructose-driven steatosis in the human liver and induces the expression of the SREBP1-regulated enzymes ACC-1, FASN, and SREBP1c at the mRNA level [97]. The NF-κB/MyD88 pathway drives inflammasome activation, which is a cytosolic regulator of inflammation that, through the caspase-2 pathway, activates SREBP1c to induce ACC-1 and FASN, contributing to the exacerbation of hepatic steatosis and inflammation in NAFLD [97]. The deleterious mechanism induced by the binding of cytotoxic bacterial metabolites to TLR-4 is shown in Figure 4.
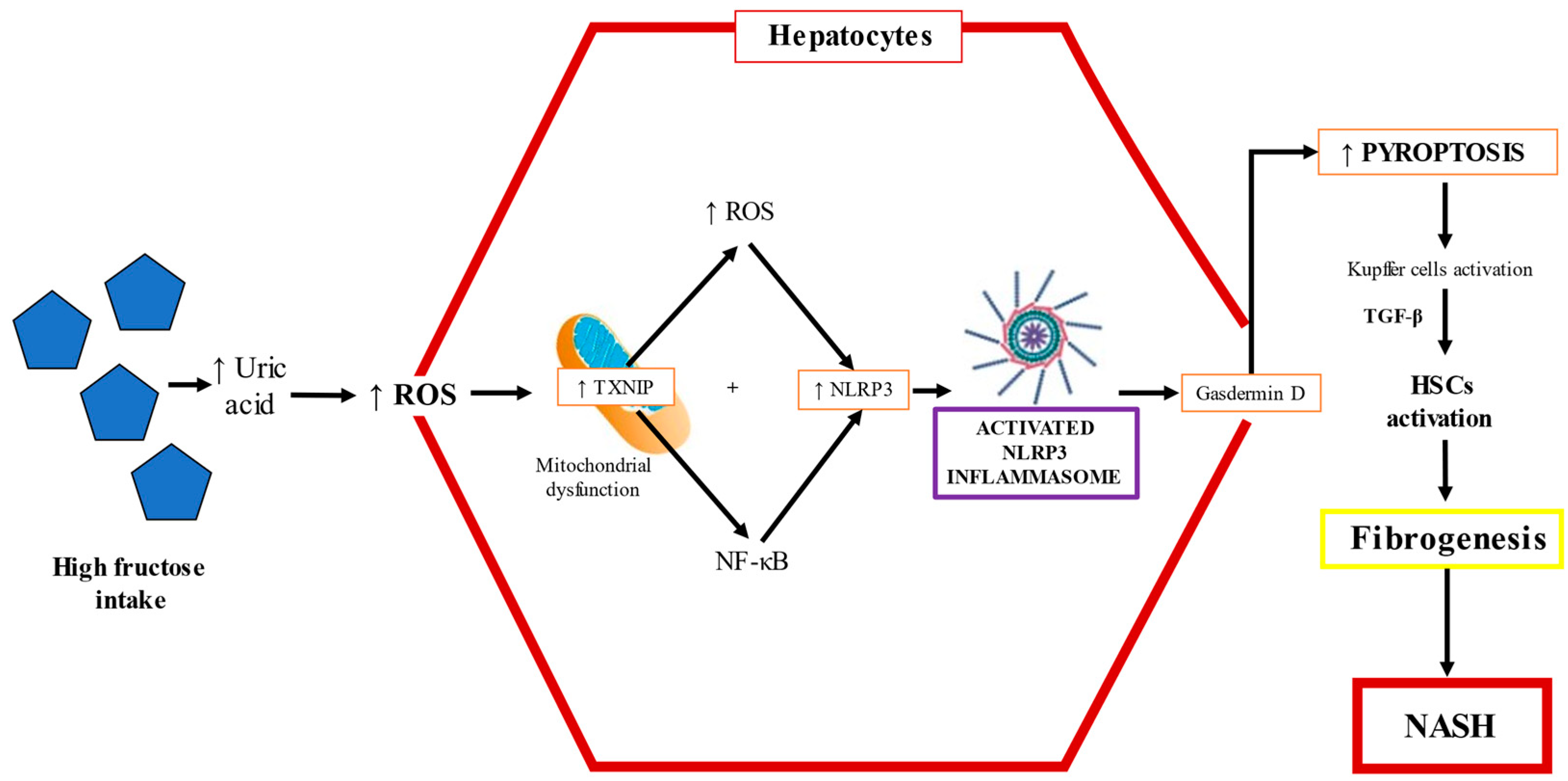
TLR-4 promotes NF-κB signaling, and this pathway upregulates the transcription of the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and proinflammatory cytokines such as IL-1β and TNF-α [96,98]. Studies performed in mice models have shown that fructose triggers the infiltration/activation of macrophages/Kupffer cells, causing increased levels of ROS, and induces the necrosis of hepatocytes through TNF-α and IL-6 upregulation (90). The factors underlying the progression from NAFLD to NASH are multifactorial, but NLRP3 inflammasome activation is critically important. Cytokine release causes hepatocyte death along with activation of HSCs and Kupffer cells. The NLRP3 inflammasome, upregulated by fructose overfeeding, is a sensor of danger signals, DAMPs, uric acid crystals, or derivatives that act like DAMP molecules and induce inflammation [99,100,101]. The NLRP3 inflammasome recruits apoptosis-associated speck-like protein and pro-caspase 1, leading to the maturation and secretion of IL-1β and IL-18 [102,103]. Caspase 1 is necessary for the activation of the NLRP3 inflammasome, as an executioner molecule; then, IL-1β is matured, triggering HSC activation, and thus, fibrogenesis ensues [104]. Indeed, the levels of IL-1β correlate with the mRNA of collagen 1, a key profibrogenic gene [105,106]. The activation of the NLRP3 inflammasome is a synchronized interaction between hepatocytes and Kupffer cells that results in dyslipidemia and lipid accumulation in hepatocytes [107]. High fructose administration to rodents increases TXNIP levels and malondialdehyde and decreases superoxide dismutase, triggering oxidative stress, which is sensed by TXNIP, therefore inducing NLRP3 inflammasome activation [103]. The fructose–ROS–TXNIP–NLRP3 inflammasome axis is crucial in the pathogenesis of uric-acid-induced inflammatory responses [108] (Figure 5).
Wree et al. found that NLRP3 inflammasome activation results in severe liver inflammation and fibrosis via the pyroptotic signaling pathway in hepatocytes [109]. Pyroptosis is a unique form of programed cell death where a plasma membrane pore formed by gasdermin D allows the release of the cellular content, leading to the upregulation of proinflammatory cytokines and profibrogenic factors such as IL-1β, connective tissue growth factor, and TGF-β, triggering the activation of HSCs, leading to the increased production and secretion of scar tissue proteins [109]; as a result, inflammation is exacerbated and liver fibrosis ensues [103,110]. Inflammasome activation by fructose could also be the result of increased Glut5 activity, which induces TXNIP to form the activated complex of ASC with NLRP3, consequently inducing dyslipidemia, hepatic inflammation, and lipid accumulation [111]. In addition, there is evidence indicating that TXNIP is upregulated in the liver by the master nutritional regulator ChREBP [112].
2.3.3. Nuclear Factor E2-Related Factor 2 and Fructose
Increasing evidence indicates that nuclear factor E2-related factor 2 (Nrf2) plays a complex, multicellular role within the processes of liver inflammation and fibrosis through the induction of its target genes [113,114]. Nrf2 is considered to act as the first line of defense against cellular damage due to oxidative stress [115]. It upregulates the expression of protective and antioxidant genes, upregulating the GSH biosynthesis and thioredoxin systems, to maintain cellular redox homeostasis in response to oxidative stress and other insults; therefore, its inactivation can exacerbate oxidant, inflammatory, and profibrotic processes [113,116,117]. Interestingly, oxidative stress, inflammation, and fibrosis are linked by several molecular signaling pathways that have been recently reviewed elsewhere [110]. The cytoplasmic protein repressor Kelch-like ECH-associated protein-1 (Keap1) regulates Nrf2′s function [110]. Keap1 acts as a sensor for oxidative stress, and under stress conditions, the sequestration complex dissociates, allowing Nrf2 to translocate to the nucleus, where it binds to the antioxidant response element and induces the expression of a battery of antioxidant genes [110]. In the liver, the activation of Nrf2 attenuates injuries of diverse etiologies, including chronic diseases such as NAFLD, by inducing heme oxygenase-1 (HO-1) expression and improving GSH efficacy [116,117]. Nrf2 activation prevents metabolic dysregulation and insulin resistance in mice through the repression of hepatic enzymes such as FASN and ACC and protects against hypertriglyceridemia and fatty liver disease; this protection is abolished when Nrf2 is deleted [118]. Acute fructose intake upregulates the expression of Nrf2 pathways, but excessive consumption through high-fructose diets increases reactive species and oxidative damage and downregulates Nrf2 and GSH [119,120]. MiRNAs are non-coding RNAs that regulate genes, silencing or promoting their expression through modulating mRNA transcription. MicroRNA (miRNA)-200a is reported to target Keap1, thereby activating Nrf2, and high fructose decreases miRNA-200a, which inhibits the Nrf2 antioxidant response [121]. The inhibition of KHK in the presence of fructose is accompanied by an increase in Nrf2 and the cytoprotective expression of HO-1, NAD(P)H dehydrogenase (quinone) 1 (NQO-1), and thioredoxin reductase 1 [92,117]. Mice deficient in Glut8 (SLC2A8), a member of the facilitated hexose transporter superfamily, have impaired hepatic first-pass fructose metabolism [122]. Transcriptomic analysis reveals that the excessive consumption of fructose induces mechanisms that increase oxidative stress, such as aryl hydrocarbon receptor downregulation. The aryl hydrocarbon receptor modulates the expression of various biotransformation enzymes classified as phase I and II enzymes; this receptor also has crosstalk with NF-κB [123]. Therefore, fructose intake, which causes the downregulation of xenobiotic-metabolizing enzymes and Nrf2 transcription, also leads to the upregulation of NF-κB [124,125,126].
2.3.4. Carbohydrate Responsive Element-Binding Protein and Fructose
ChREBP is an essential transcription factor involved in hepatic stress that upregulates the ACLY, ACC-1, and FASN enzymes involved in hepatic de novo lipogenesis and, therefore, is a central factor in NAFLD [127,128]. However, the liver-specific deletion of ACLY fails to suppress fructose-induced lipogenesis [82]. By contrast, ACC-1 inhibition was associated with a decrease in hepatic de novo lipogenesis and insulin resistance and increased fatty acid β-oxidation [94]. Moreover, the inhibition of ACC-1 reduced the activation of TGF-β and fibrogenesis because HSC activation requires this factor and de novo lipogenesis [94]. The liver-specific ablation of ChREBP in rodents fed an elevated-fructose diet causes severe transaminitis and hepatomegaly with glycogen accumulation [129]. In addition, ChREBP induces the expression of fibroblast growth factor 21 (FGF21), which ameliorates dyslipidemia in humans [129]. FGF21 activates lipolysis and increases fatty acid oxidation in the liver through the activation of peroxisome proliferator-activated receptor alpha (PPAR-α). At the molecular level, these changes were associated with increases in the liver X receptor, which increases SREBP and decreases PPAR-α activation [130]. In humans, the expression of PPAR-α negatively correlates with the presence of NAFLD and the severity of steatosis [131]. PPAR-α, which is mainly activated during the fasted state and regulates the metabolism of lipids and inflammation, is primarily found in hepatocytes, and fatty acids resulting from the metabolism of fructose are oxidized to produce acetyl-CoA by peroxisomes and mitochondria through PPAR-α [76]. PPAR-α also stimulates the mitochondrial β-oxidation pathway and induces inhibitor kappa B (IκB)α in hepatocytes, which prevents the translocation of nuclear transcription factor kappa B (NF-κB) to the nucleus, a well-known proinflammatory signaler [78,96]. IκBα upregulates lipid metabolism and reduces inflammation, which improves NASH pathology [132]. By contrast, in FGF21-knockout mice, the activation of HSCs and fibrogenesis were increased, evidenced by increased levels of TGF-β, matrix metalloproteinases, and tissue inhibitors of metalloproteinases [129].
The respiratory chain of the mitochondria produces ROS, but ROS are decreased by antioxidant enzymes to prevent the deleterious effects of free radicals on important biological molecules. Long-term elevated fructose intake produces oxidative alterations in liver cells, particularly in the lipid components of mitochondria, and diminished superoxide dismutase and catalase activities, which are important enzymes for counteracting mitochondrially produced ROS [133,134]. Fructose intake diminishes the antioxidant machinery of mitochondria, increasing oxidative stress, which causes the lipid peroxidation of polyunsaturated fatty acids, and allows the attack of free radicals on mitochondrial DNA; as a result, mitochondrial biogenesis is also affected [133]. Mitochondrial dysfunction results in low fatty acid oxidation, decreased hepatic ATP levels, and increased hepatic oxidative stress [135,136]. All these effects are, at least in part, regulated through PPAR-α inhibition. On the other hand, fructose oxidation also produces carbonyl compounds such as glycolaldehyde, a metabolite of glyceraldehyde, and glyoxal, the major product of glycolaldehyde oxidation, which is associated with cellular injury and dysfunction, including the inhibition of mitochondrial respiration and induction of mitochondrial permeability transition, leading to cell death [33,67,137].
Additionally, the consumption of fructose but not glucose increases apolipoprotein CIII through the ChREBP pathway, increasing triglyceride and low-density lipoprotein levels upon fructose metabolism, and represents a significant contributor to cardiometabolic risk [138,139]. These observations suggest that ChREBP plays an important role in the pathogenesis of NASH; however, the suggested protective role of ChREBP deserves further investigation [127].
2.3.5. Sterol-Responsive Element-Binding Protein and Fructose
The SREBP protein is generated in the endoplasmic reticulum as a complex with SREBP cleavage-activating protein (SCAP). SREBP1c is mainly produced in the liver and is activated by changes in nutritional status [140]. As in the intestine, fructose in the liver also contributes to increasing SREBP1c expression, which plays a pivotal role in lipid metabolism [138,141]. The deleterious effects on lipid metabolism of excessive fructose consumption are fasting and postprandial hypertriglyceridemia, and increased hepatic synthesis of lipids, very-low-density lipoproteins (VLDLs), and cholesterol [138,139,142,143]. It has been shown that the elevated levels of plasma triacylglycerol during high fructose feeding may be due to the overproduction and impaired clearance of VLDL, and chronic oxidative stress potentiates the effects of high fructose on the export of newly synthesized VLDL [144]. Moreover, in humans diets high in fructose have been observed to reduce postprandial serum insulin concentration; therefore, there is less stimulation of lipoprotein lipase, which causes a greater accumulation of chylomicrons and VLDL because lipoprotein lipase is an enzyme that hydrolyzes triglycerides in plasma lipoproteins [145]. High fructose consumption induces the hepatic transcription of hepatocyte nuclear factor 1, which upregulates aldolase B and cholesterol esterification 2, triggering the assembly and secretion of VLDL, resulting in the overproduction of free fatty acids [146]. These free fatty acids increase acetyl-CoA formation and maintain NADPH levels and NOX activation [146]. NOX, which uses NADPH to oxidize molecular oxygen to the superoxide anion [140], and xanthine oxidoreductase (XO), which catalyzes the oxidative hydroxylation of hypoxanthine to xanthine and xanthine to uric acid, are the main intracellular sources of ROS in the liver [147,148]. NOX reduces the bioavailability of nitric oxide and thus impairs the hepatic microcirculation and promotes the proliferation of HSCs, accelerating the development of liver fibrosis [147,148]. ROS derived from NOX lead to the accumulation of unfolded proteins in the endoplasmic reticulum lumen, which increases oxidative stress [146].
In hepatocytes, cytoplasmic Ca2+ is an important regulator of lipid metabolism. An increased Ca2+ concentration stimulates exacerbated lipid synthesis [145]. A high fructose intake induces lipid accumulation, leading to protein kinase C phosphorylation, stressing the endoplasmic reticulum [149]. Elevated activity of the protein kinase C pathway has been reported to stimulate ROS-generating enzymes such as lipoxygenases. A prolonged endoplasmic reticulum stress response activates SREBP1c and leads to insulin resistance [140,150]. Calcium signaling is also important for liver regeneration, and increased intracellular calcium homeostasis is known to be involved in tumor initiation, progression, and metastasis; therefore, the alteration of calcium homeostasis by high fructose consumption could be an important mechanism in the development of cancer [151,152].
Some evidence indicates that there is a synergy between SREBP activation with the stimulation of the inflammatory pathway mediated by NF-κB and cholesterol homeostasis. Activated NF-κB increases SCAP expression, resulting in the activation of the SCAP–SREBP complex, triggering an exacerbated inflammatory response and cholesterol accumulation [153]. Furthermore, some reports indicate that fructose supplementation leads to insulin receptor downregulation because protein-tyrosine phosphatase 1B activity decreases the phosphorylation of the insulin receptor and induces protein phosphatase 2A, increasing SREBP1c, aggravating hepatic insulin resistance via intricate metabolic pathways [88]. Extensive reviews have been published on the lipogenic effect of fructose [70,135,154]; however, the deleterious effects of fructose in the liver go beyond the steatotic effect. Hepatic cholesterol accumulation is associated with inflammatory cell infiltration [155]. Dietary fructose induces strong SREBP1c activation, and the consequent palmitate production causes lipotoxicity in the endoplasmic reticulum; these events are the leading factors responsible for the greater Nrf2 inhibition and more intense hepatic inflammatory response driven by NLRP3 inflammasome activation [107,156].
Some authors have proposed “multiple parallel hit” theories to explain the development of the metabolic disease NAFLD, the first hit being the accumulation of fat in the liver (mainly triglycerides), followed by multifactorial processes that involve oxidative stress, inflammation, and hyperuricemia as the main factors [157,158]. DNA methylation is an epigenetic mechanism that decreases gene expression. Accumulating evidence suggests that excessive fructose intake drives epigenetic alterations, including the hypermethylation of the carnitine palmitoyl transferase 1A and PPAR-α genes [159,160]. Increased malonyl-CoA, which is synthesized by the enzyme acetyl-CoA carboxylase, inhibits carnitine palmitoyl transferase 1A, which is the rate-limiting step of the oxidation of lipids in the mitochondria, leading to the disruption of β-oxidation and accumulation of hepatic lipids, particularly fatty acids such as diacylglycerols and ceramides, which inhibit the insulin signaling pathway through protein kinase C activation and the inhibition of the protein kinase AKT, respectively [102,160] (Figure 3).
This scenario can be worsened because high-glycemic diets induce the conversion of glucose to fructose by the aldose reductase enzyme. Fructose can be endogenously synthesized in the body via the polyol pathway, a two-step conversion of glucose to fructose, which is relatively inactive under physiological conditions [161,162]. In addition, in high-glycemic diets, glucose is metabolized by fructose-3-phosphokinase to a highly reactive molecule, fructose-3-phosphate, causing the formation of advanced glycation end products, which can trigger inflammatory pathways through the activation of signaling pathways such as NF-κB and mitogen-activated protein kinases, aside from increasing lipogenesis and the disruption of β-oxidation, independently of caloric intake and weight gain [135,163,164]. On the other hand, fructose can be released from the liver to the systemic circulation and filtered and excreted by the kidneys, a decisive organ for fructose disposal, increasing metabolic abnormalities [165,166].
2.3.6. Uric Acid and Fructose
KHK utilizes ATP to phosphorylate fructose to form fructose-1-phosphate, leading to intracellular phosphate exhaustion, which in turn activates the enzyme adenosine monophosphate (AMP) deaminase, which converts AMP to inosine monophosphate (IMP) (Figure 3). Consequently, xanthine is formed by the XO enzyme, which is associated with the production of large amounts of highly cytotoxic ROS as well as hydrogen peroxide [72]. Chronic fructose consumption stimulates purine nucleotide turnover, which culminates in the synthesis of uric acid from xanthine by XO, leading to uric acid accumulation within hepatic cells [167]. 4-Hydroxynonenal, which is formed by the attack of ROS on biological membranes, induces XO, a key enzyme in purine and free radical metabolism; in turn, high activity of XO may further promote oxidative stress in the liver [168,169]. Increased systemic oxidative stress is recognized as an essential cause of elevated uric acid and inflammation [170]. Uric acid is a potent inducer of the inflammatory response by activating NF-κB through inducing the phosphorylation of IKK and IκBα, followed by the subsequent stimulation of NF-κB activity [108,171,172]. The systemic effects of uric acid are related to endothelial injury and dysfunction. Uric acid directly inhibits endothelial nitric oxide synthase; the impairment of nitric oxide synthesis decreases vascular smooth muscle relaxation and increases systolic blood pressure, leading to hypertension [173].
Uric acid also promotes fat synthesis within hepatocytes through the translocation of the NADPH oxidase subunit 4 to the mitochondria, increasing superoxide formation [174]. In turn, this increase in ROS inhibits the enzyme aconitase, which catalyzes the conversion of citrate to isocitrate in the mitochondrial matrix in the Krebs cycle and promotes citrate accumulation; then, citrate is converted to acetyl-CoA for de novo lipogenesis by FASN [174]. Oxidative stress and uric acid are amplifying actors that activate the nuclear factor of activated T cells, which plays a role in the regulation of inflammation and upregulates aldose reductase via the polyol pathway, leading to hepatic steatosis [162]. Oral and coworkers (2019) found a positive correlation between the degree of liver damage and uric acid concentration in non-obese and young patients with NAFLD, who had higher uric acid concentrations than the healthy control group [175]. Some reports showed that uric acid in the liver promotes oxidative stress and inflammation through the inhibition of Nrf2 and overproduction of thioredoxin, which results in NLRP3 inflammasome activation [103,176,177]. In NAFLD patients who have fatty liver but no inflammation, the expression of NLRP3 components is increased, but the inflammasome is not activated; however, in patients with fatty liver and inflammation, the NLRP3 inflammasome complex is assembled and functional [106]. Therefore, the activation of the NLRP3 inflammasome is associated with liver disease progression from simple fatty liver to NASH with inflammation and fibrosis [106]. Additionally, increased serum uric acid levels are indicative of a greater probability of elevated serum alanine aminotransferase and gamma-glutamyl transferase, two markers of hepatic necroinflammation, and are associated with a greater risk of cirrhosis-related hospitalization or death [64]. These studies support the role of uric acid as a risk marker of liver damage via NLRP3 inflammasome activation; moreover, it represents a non-invasive marker and a possible predictor of NASH. These findings suggest that activation of NLRP3 inflammasome induces a fibrogenic micro-environment in the liver. Therefore, the inhibition of NLRP3 inflammasome is a promising therapeutic tool to ameliorate hepatic fibrosis. In addition, some antioxidants have been shown to block the NRLP3 inflammasome signaling pathway and thus may be helpful to decrease NASH development.
2.3.7. MicroRNAs and Fructose
Novel evidence suggests that miRNAs play an important role in liver health and disease. The expression of miRNAs can be modified by increasing fructose intake and/or uric acid production. Rats fed a high-fructose diet have decreased miRNA-122, miRNA-451, and miRNA-27a compared to control-fed rats [178]. Additionally, miRNAs in mice such as miRNA-34a, miRNA-335, miRNA-221, and miRNA-9 are upregulated in the liver by high fructose intake [179]. There is cumulative evidence that some miRNAs regulate several signaling pathways, leading to oxidative stress and inflammation in the liver. For example, in humans the elevation of miR-214 levels decreases glutathione reductase and cytochrome P450 activities; consequently, hepatic oxidative stress is augmented [180]. The attenuation of miRNA-199a-5p produces apoptosis associated with endoplasmic reticulum stress [181]. miRNA-223 is expressed in the liver and prevents inflammation, the activation of HSCs, and fibrosis through disrupting the activation of the NLRP3 inflammasome [182]. In addition, it has been observed through an in vitro transfection assay that miRNA-33 is responsible for the regulation of SREBP1 after fructose ingestion [183]. Mice with miRNA-29a overexpression show decreased DNA oxidative damage in an NAFLD model, suggesting its role in neutralizing oxidative stress [184]. Furthermore, miRNA-29a contributes to a reduction in NF-κB activity, which leads to a decrease in the inflammatory process and provides protection against fibrosis by suppressing TGF-β and SMAD3, the canonical signaling pathway for HSC activation. MiRNA-149-5p is induced by uric acid in hepatocytes, causing lipid accumulation via the upregulation of FGF21, a protein implicated in lipid metabolism that is considered an anti-metabolic-syndrome hormone, therefore playing an important role in the prevention of NAFLD development [57].
2.3.8. Cancer and Fructose
In 1924, Otto Warburg described that cancer cells could obtain energy by fermenting glucose into lactate, and this is called the “Warburg effect” [185]. Fructose promotes the Warburg effect, increasing glycolysis and suppressing fat oxidation, which may promote mitochondrial dysfunction, tumor growth, and metastasis [185]. Fructose-rich diets can increase HCC incidence because it was found that fructokinase and Glut5 are highly expressed in diverse types of cancer, and that the upregulation of Glut5 correlates with a poor prognosis in HCC [186,187]. Importantly, several investigators have suggested that high fructose intake not only promotes cancer development in various tissues but also proposed that endogenously produced fructose in cancer cells could potentially stimulate cancer growth [185]. The key enzyme that stimulates endogenous fructose production is aldose reductase in the polyol pathway. Fructose also induces metabolic changes via KHK-A, promoting the pentose phosphate pathway, the development of HCC [188], and the serine-to-glycine synthesis pathway for HCC growth [189]. Notably, fructose can be utilized by cancer cells as an energy source and, subsequently, for the synthesis of nucleic acids through the pentose phosphate pathway. Fructose also promotes colon cancer metastasis to the liver via the KHK–aldolase B pathway, and a high-fructose diet increases colorectal liver metastasis [190]. The silencing of aldolase B or the restriction of fructose in the diet suppresses liver metastasis from colorectal cancer [190,191]. Moreover, as mentioned above, uric acid is a by-product of fructose metabolism that stimulates the production of mitochondrial ROS and aldolase. In clinical studies, high uric acid is considered a significant risk factor for active hepatocarcinogenesis [191]. Fructose metabolism during carcinogenesis elevates oxidative stress and inflammation [192]. However, the effects of endogenous or exogenous fructose in cancer need to be investigated in more detail.
3. Conclusions and Perspectives
Research on the impact of human nutrition on health and disease is vast. However, the molecular mechanisms involved in nutrition’s effects on human diseases are far from being fully understood. Plenty of evidence indicates that fructose and its metabolites play a significant role in the development of liver disease. The multiple mechanisms that fructose triggers have placed it in the eye of the hurricane in metabolic disorders of the liver. Although direct extrapolation from animal findings to humans is not recommended, basic research has illuminated some of the cellular and molecular mechanisms that are involved in the deleterious effects of the overconsumption of fructose, including oxidative stress, inflammation, higher serum uric acid levels, hypertriglyceridemia, higher systolic blood pressure, insulin resistance, fibrosis, cirrhosis, and HCC. Fructose-induced hepatic injury depends strongly on the activation of lipogenesis and inflammatory signaling pathways, which, in turn, trigger fibrosis and HCC development. Free radical and uric acid overproduction induced by excessive fructose consumption also play pivotal roles in fatty liver, inflammation, fibrosis, and HCC progression through a variety of signaling pathways. These observations provide mechanistic information on NASH development and may be used for the development of new drugs and therapies. Several anti-inflammatory, antifibrotic, and anticancer targets are now known in the pathogenic pathways involved in fructose overconsumption. However, more in-depth studies dealing with the involved molecular mechanisms of fructose-driven fibrogenesis are required to find new therapeutic targets for drug development to prevent hepatic fibrosis.
The alarming increase in metabolic syndrome and comorbidities can only be attenuated if the consumption of fructose, mainly in soft beverages, is significantly reduced worldwide. In addition, an active lifestyle incorporating the practice of sports seems to be useful for fighting the sedentarism associated with obesity. Patients suffering from hepatic maladies should be recommended to reduce fructose consumption to prevent aggravation of their condition because fructose may act as a conjoint pathological agent.
Author Contributions
Conceptualization, P.M. and E.R.-T.; writing—review and editing, P.M., P.L.-S. and E.R.-T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by Consejo Nacional de Ciencia y Tecnología through the Ciencia de Frontera funding program, grant number 53358.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Erika Ramos Tovar is thankful for a postdoctoral scholarship from Consejo Nacional de Ciencia y Tecnología.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- Eckel, R.H.; Alberti, K.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Streba, L.A.M. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: An open question. World J. Gastroenterol. 2015, 21, 4103. [Google Scholar] [CrossRef] [PubMed]
- Roomi, M.A.; Mohammadnezhad, M. Prevalence of Metabolic Syndrome Among Apparently Healthy Workforce. J. Ayub Med. Coll. 2019, 31, 252–254. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthoud, H.-R.; Morrison, C.D.; Münzberg, H. The obesity epidemic in the face of homeostatic body weight regulation: What went wrong and how can it be fixed? Physiol. Behav. 2020, 222, 112959. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014. [Google Scholar] [CrossRef] [Green Version]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab. 2020. [Google Scholar] [CrossRef]
- Wai-Sun Wong, V.; Lai-Hung Wong, G.; Woo, J.; Abrigo, J.M.; Ka-Man Chan, C.; She-Ting Shu, S.; Ka-Yu Leung, J.; Mei-Ling Chim, A.; Pik-Shan Kong, A.; Chung-Yan Lui, G.; et al. Impact of the New Definition of Metabolic Associated Fatty Liver Disease on the Epidemiology of the Disease. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Nagaoki, Y.; Hyogo, H.; Aikata, H.; Tanaka, M.; Naeshiro, N.; Nakahara, T.; Honda, Y.; Miyaki, D.; Kawaoka, T.; Takaki, S.; et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol. Res. 2012, 42, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, R. Primary hepatocellular carcinoma and metabolic syndrome: An update. World J. Gastrointest. Oncol. 2013, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.C.; Niederpruem, M.G.; Wallace, J.P.; Lindeman, A.K. Dietary fat, sugar, and fiber predict body fat content. J. Am. Diet. Assoc. 1994, 94, 612–615. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-medicare database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Margini, C.; Dufour, J.F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016, 36, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Muhidin, S.O.; Magan, A.A.; Osman, K.A.; Syed, S.; Ahmed, M.H. The Relationship between Nonalcoholic Fatty Liver Disease and Colorectal Cancer: The Future Challenges and Outcomes of the Metabolic Syndrome. J. Obes. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Wong, M.C.S.; Jiang, J.Y.; Goggins, W.B.; Liang, M.; Fang, Y.; Fung, F.D.H.; Leung, C.; Wang, H.H.X.; Wong, G.L.H.; Wong, V.W.S.; et al. International incidence and mortality trends of liver cancer: A global profile. Sci. Rep. 2017, 7, 45846. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Paliogiannis, P.; Cigliano, A.; Pilo, M.G.; Chen, X.; Calvisi, D.F. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Li, L.; Dai, J.; Natarajan, Y.; Yu, X.; Asch, S.M.; El-Serag, H.B. Effect of Metabolic Traits on the Risk of Cirrhosis and Hepatocellular Cancer in Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 808–819. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucera, O. Experimental models of non-alcoholic fatty liver disease in rats. World J. Gastroenterol. 2014, 20, 8364. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef]
- Kmietowicz, Z. Countries that use large amounts of high fructose corn syrup have higher rates of type 2 diabetes. BMJ 2012, 345, e7994. [Google Scholar] [CrossRef]
- Rosas-Villegas, A.; Sánchez-Tapia, M.; Avila-Nava, A.; Ramírez, V.; Tovar, A.; Torres, N. Differential Effect of Sucrose and Fructose in Combination with a High Fat Diet on Intestinal Microbiota and Kidney Oxidative Stress. Nutrients 2017, 9, 393. [Google Scholar] [CrossRef]
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J. Dietary Sugars Intake and Cardiovascular Health. Circulation 2009, 120, 1011–1020. [Google Scholar] [CrossRef]
- Basto-Abreu, A.; Braverman-Bronstein, A.; Camacho-García-Formentí, D.; Zepeda-Tello, R.; Popkin, B.M.; Rivera-Dommarco, J.; Hernández-Ávila, M.; Barrientos-Gutiérrez, T. Expected changes in obesity after reformulation to reduce added sugars in beverages: A modeling study. PLoS Med. 2018, 15, e1002664. [Google Scholar] [CrossRef] [Green Version]
- Powell, E.S.; Smith-Taillie, L.P.; Popkin, B.M. Added Sugars Intake Across the Distribution of US Children and Adult Consumers: 1977-2012. J. Acad. Nutr. Diet. 2016, 116, 1543–1550.e1. [Google Scholar] [CrossRef] [Green Version]
- Santhekadur, P.K. The dark face of fructose as a tumor promoter. Genes Dis. 2020, 7, 163–165. [Google Scholar] [CrossRef]
- WHO Guideline: Sugars Intake for Adult and Children; WHO: Geneva, Switzerland, 2015.
- Caliceti, C.; Calabria, D.; Roda, A.; Cicero, A. Fructose Intake, Serum Uric Acid, and Cardiometabolic Disorders: A Critical Review. Nutrients 2017, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [Green Version]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221, jeb164202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Ferrocino, I.; Liberto, E.; Chiazza, F.; Cento, A.S.; Collotta, D.; Querio, G.; Nigro, D.; Bitonto, V.; Cutrin, J.C.; et al. Fructose liquid and solid formulations differently affect gut integrity, microbiota composition and related liver toxicity: A comparative in vivo study. J. Nutr. Biochem. 2018, 55, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Baena, M.; Sánchez, R.M.; Merlos, M.; Alegret, M.; Laguna, J.C. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Glendinning, J.I.; Frim, Y.G.; Hochman, A.; Lubitz, G.S.; Basile, A.J.; Sclafani, A. Glucose elicits cephalic-phase insulin release in mice by activating K ATP channels in taste cells. Am. J. Physiol. Integr. Comp. Physiol. 2017, 312, R597–R610. [Google Scholar] [CrossRef] [Green Version]
- Bodnaruc, A.M.; Prud’homme, D.; Blanchet, R.; Giroux, I. Nutritional modulation of endogenous glucagon-like peptide-1 secretion: A review. Nutr. Metab. 2016, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Kokkinos, A.; Tentolouris, N.; Katsilambros, N. The Effect of Ingested Macronutrients on Postprandial Ghrelin Response: A Critical Review of Existing Literature Data. Int. J. Pept. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yau, A.M.W.; McLaughlin, J.; Maughan, R.J.; Gilmore, W.; Evans, G.H. Short-term dietary supplementation with fructose accelerates gastric emptying of a fructose but not a glucose solution. Nutrition 2014, 30, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, M.; Lallès, J.-P.; Malbert, C.-H.; Val-Laillet, D. Dietary sugars: Their detection by the gut–brain axis and their peripheral and central effects in health and diseases. Eur. J. Nutr. 2015, 54, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustig, R.H. Fructose: Metabolic, Hedonic, and Societal Parallels with Ethanol. J. Am. Diet. Assoc. 2010, 110, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P.; Choe, J.; Patel, C.R. Intestinal Absorption of Fructose. Annu. Rev. Nutr. 2018, 38, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Lowette, K.; Roosen, L.; Tack, J.; Vanden Berghe, P. Effects of High-Fructose Diets on Central Appetite Signaling and Cognitive Function. Front. Nutr. 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Kishida, K.; Pearce, S.C.; Yu, S.; Gao, N.; Ferraris, R.P. Nutrient sensing by absorptive and secretory progenies of small intestinal stem cells. Am. J. Physiol. Liver Physiol. 2017, 312, G592–G605. [Google Scholar] [CrossRef] [Green Version]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) Is Essential for the Absorption of Fructose in the Intestine and Generation of Fructose-induced Hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.A.; Samuel, V.T. The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 2016, 27, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.; Douard, V.; Yu, S.; Tharabenjasin, P.; Gao, N.; Ferraris, R.P. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Integr. Comp. Physiol. 2015, 309, R499–R509. [Google Scholar] [CrossRef] [Green Version]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar Absorption in the Intestine: The Role of GLUT2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Packard, C.J.; Borén, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef] [Green Version]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef]
- Jones, H.F.; Butler, R.N.; Brooks, D.A. Intestinal fructose transport and malabsorption in humans. Am. J. Physiol. Liver Physiol. 2011, 300, G202–G206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Dagdeviren, S.; Lewandowski, J.P.; Schmider, A.B.; Ricci-Blair, E.M.; Natarajan, N.; Hundal, H.; Noh, H.L.; Friedline, R.H.; Vidoudez, C.; et al. Thioredoxin Interacting Protein Is Required for a Chronic Energy-Rich Diet to Promote Intestinal Fructose Absorption. iScience 2020, 23, 101521. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin Interacting Protein: Redox Dependent and Independent Regulatory Mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Jang, C.; Wada, S.; Yang, S.; Gosis, B.; Zeng, X.; Zhang, Z.; Shen, Y.; Lee, G.; Arany, Z.; Rabinowitz, J.D. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab. 2020, 2, 586–593. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and Localization of the Principal Fructose-metabolizing Enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Afzali, A.; Weiss, N.S.; Boyko, E.J.; Ioannou, G.N. Association between serum uric acid level and chronic liver disease in the United States. Hepatology 2010, 52, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, W.; Zhang, J.; Gobejishvili, L.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Elevated Fructose and Uric Acid Through Aldose Reductase Contribute to Experimental and Human Alcoholic Liver Disease. Hepatology 2020, 72, 1617–1637. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.; Yin, H.; Gao, Z.; Li, Y.; Gao, T.; Duan, J.; Yang, R.; Dong, X.; Zhang, L.; Duan, W. Intestinal tract is an important organ for lowering serum uric acid in rats. PLoS ONE 2017, 12, e0190194. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, C.; Ogura, J.; Sasaki, S.; Okamoto, K.; Kobayashi, M.; Kuwayama, K.; Narumi, K.; Iseki, K. Fructose suppresses uric acid excretion to the intestinal lumen as a result of the induction of oxidative stress by NADPH oxidase activation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Al-Jawadi, A.; Patel, C.R.; Shiarella, R.J.; Romelus, E.; Auvinen, M.; Guardia, J.; Pearce, S.C.; Kishida, K.; Yu, S.; Gao, N.; et al. Cell-Type–Specific, Ketohexokinase-Dependent Induction by Fructose of Lipogenic Gene Expression in Mouse Small Intestine. J. Nutr. 2020, 150, 1722–1730. [Google Scholar] [CrossRef]
- Lee, H.-J.; Cha, J.-Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef] [Green Version]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Francis, H.; Alpini, G. Fructose Promotion of Intestinal and Liver Injury: A Sugar by Any Other Name That Isn’t So Sweet. Hepatology 2019, hep.30843. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, D.; Seo, W.; Gao, B.; Yoo, S.; Song, B. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1–Mediated Oxidative and Nitrative Stress. Hepatology 2019, hep.30652. [Google Scholar] [CrossRef] [PubMed]
- Moine, L.; Rivoira, M.; de Barboza, G.D.; Pérez, A.; de Talamoni, N.T. Glutathione depleting drugs, antioxidants and intestinal calcium absorption. World J. Gastroenterol. 2018, 24, 4979–4988. [Google Scholar] [CrossRef]
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann. Hepatol. 2016, 15, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [Green Version]
- Do, M.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [Green Version]
- Softic, S.; Gupta, M.K.; Wang, G.-X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Akbar, M.; Jo, I.; Hardwick, J.P.; Abdelmegeed, M.A. Translational Implications of the Alcohol-Metabolizing Enzymes, Including Cytochrome P450-2E1, in Alcoholic and Nonalcoholic Liver Disease. Adv. Pharmacol. 2015, 74, 303–372. [Google Scholar]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Simeonova, D.; Ivanovska, M.; Murdjeva, M.; Carvalho, A.F.; Maes, M. Recognizing the Leaky Gut as a Trans-diagnostic Target for Neuroimmune Disorders Using Clinical Chemistry and Molecular Immunology Assays. Curr. Top. Med. Chem. 2018, 18, 1641–1655. [Google Scholar] [CrossRef] [PubMed]
- Francey, C.; Cros, J.; Rosset, R.; Crézé, C.; Rey, V.; Stefanoni, N.; Schneiter, P.; Tappy, L.; Seyssel, K. The extra-splanchnic fructose escape after ingestion of a fructose–glucose drink: An exploratory study in healthy humans using a dual fructose isotope method. Clin. Nutr. ESPEN 2019, 29, 125–132. [Google Scholar] [CrossRef]
- Nergiz-Unal, R.; Ulug, E.; Kisioglu, B.; Tamer, F.; Bodur, M.; Yalcimin, H.; Yuruk, A.A. Hepatic cholesterol synthesis and lipoprotein levels impaired by dietary fructose and saturated fatty acids in mice: Insight on PCSK9 and CD36. Nutrition 2020, 110954. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.L.; Saborano, R.; Northall, E.; Matsuda, K.; Ogino, H.; Yashiro, H.; Pickens, J.; Feaver, R.E.; Cole, B.K.; Hoang, S.A.; et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep. 2021, 3, 100217. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhang, L.; Xu, S.; Shen, A. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: An updated review. Prog. Lipid Res. 2020, 77, 101006. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.; Vijayakumar, A.; Ghoshal, S.; Marchand, B.; Yi, S.; Kornyeyev, D.; Zagorska, A.; Hollenback, D.; Walker, K.; Liu, K.; et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 2020, 73, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1–9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear factor-Κb—A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 1. [Google Scholar] [CrossRef]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation—The negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Paschos, P.; Athyros, V.G.; Tsimperidis, A.; Katsoula, A.; Grammatikos, N.; Giouleme, O. Can Serum Uric Acid Lowering Therapy Contribute to the Prevention or Treatment of Nonalcoholic Fatty Liver Disease? Curr. Vasc. Pharmacol. 2018, 16, 269–275. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef]
- Merino, B.; Fernández-Díaz, C.M.; Cózar-Castellano, I.; Perdomo, G. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients 2019, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, J.-H.; Chen, X.-Y.; Hu, Q.-H.; Wang, M.-X.; Jin, R.; Zhang, Q.-Y.; Wang, W.; Wang, R.; Kang, L.-L.; et al. Reactive Oxygen Species-Induced TXNIP Drives Fructose-Mediated Hepatic Inflammation and Lipid Accumulation Through NLRP3 Inflammasome Activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignat, S.-R.; Dinescu, S.; Hermenean, A.; Costache, M. Cellular Interplay as a Consequence of Inflammatory Signals Leading to Liver Fibrosis Development. Cells 2020, 9, 461. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M. The Role of the NLRP3 Inflammasome in Fibrosis. Open Rheumatol. J. 2012, 6, 80–86. [Google Scholar] [CrossRef]
- Wu, X.; Dong, L.; Lin, X.; Li, J. Relevance of the NLRP3 Inflammasome in the Pathogenesis of Chronic Liver Disease. Front. Immunol. 2017, 8, 1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic administration of saturated fats and fructose differently affect SREBP activity resulting in different modulation of Nrf2 and Nlrp3 inflammasome pathways in mice liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef]
- Kim, S.-K.; Choe, J.-Y.; Park, K.-Y. TXNIP-mediated nuclear factor-κB signaling pathway and intracellular shifting of TXNIP in uric acid-induced NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2019, 511, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Tovar, E.; Muriel, P. Molecular Mechanisms That Link Oxidative Stress, Inflammation, and Fibrosis in the Liver. Antioxidants 2020, 9, 1279. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Noblet, B.; Benhamed, F.; O-Sullivan, I.; Zhang, W.; Filhoulaud, G.; Montagner, A.; Polizzi, A.; Marmier, S.; Burnol, A.-F.; Guilmeau, S.; et al. Dual regulation of TxNIP by ChREBP and FoxO1 in liver. iScience 2021, 102218. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Tovar, E.; Muriel, P. Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage. J. Appl. Toxicol. 2020, 40, 151–168. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J. Gastroenterol. Hepatol. 2011, 26, 173–179. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.M.; Zaravinos, A.; Khoo, N.K.H.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 prevents Notch-induced insulin resistance and tumorigenesis in mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Maurya, C.K.; Arha, D.; Avisetti, D.R.; Prathapan, A.; Raj, P.S.; Raghu, K.G.; Kalivendi, S.V.; Tamrakar, A.K. Fructose induces mitochondrial dysfunction and triggers apoptosis in skeletal muscle cells by provoking oxidative stress. Apoptosis 2015, 20, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, S.; Ospina, E.; Shabandri, O.; Lank, D.; Akakpo, J.Y.; Zhao, Z.; Yang, M.; Wu, J.; Jaeschke, H.; et al. Fructose Protects Against Acetaminophen-Induced Hepatotoxicity Mainly by Activating the Carbohydrate-Response Element-Binding Protein α–Fibroblast Growth Factor 21 Axis in Mice. Hepatol. Commun. 2021, hep4.1683. [Google Scholar] [CrossRef]
- Zhao, X.-J.; Yu, H.-W.; Yang, Y.-Z.; Wu, W.-Y.; Chen, T.-Y.; Jia, K.-K.; Kang, L.-L.; Jiao, R.-Q.; Kong, L.-D. Polydatin prevents fructose-induced liver inflammation and lipid deposition through increasing miR-200a to regulate Keap1/Nrf2 pathway. Redox Biol. 2018, 18, 124–137. [Google Scholar] [CrossRef]
- Novelle, M.G.; Bravo, S.B.; Deshons, M.; Iglesias, C.; García-Vence, M.; Annells, R.; da Silva Lima, N.; Nogueiras, R.; Fernández-Rojo, M.A.; Diéguez, C.; et al. Impact of liver-specific GLUT8 silencing on fructose-induced inflammation and omega oxidation. iScience 2021, 24, 102071. [Google Scholar] [CrossRef]
- Ishihara, Y.; Kado, S.Y.; Hoeper, C.; Harel, S.; Vogel, C.F.A. Role of NF-kB RelB in Aryl Hydrocarbon Receptor-Mediated Ligand Specific Effects. Int. J. Mol. Sci. 2019, 20, 2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.H.; Tang, J.; Beane, K.E.; Redding, M.C.; Cho, Y.J.; Kim, Y.J.; Zhao, J.; Shin, E.-C.; Lee, J.H.; Kong, B.C.; et al. Hepatic transcriptomics analysis reveals that fructose intervention down-regulated xenobiotics-metabolising enzymes through aryl hydrocarbon receptor signalling suppression in C57BL/6N mice. Br. J. Nutr. 2019, 122, 769–779. [Google Scholar] [CrossRef]
- Pan, J.H.; Tang, J.; Beane, K.; Redding, M.; Zhao, J.; Lee, J.H.; Kong, B.; Kim, J.K. Hepatic Transcriptome Reveals That Fructose Downregulated Xenobiotics Metabolizing Enzymes Through Aryl Hydrocarbon Receptor Signaling Suppression in C57BL/6 N Mice (P15-011-19). Curr. Dev. Nutr. 2019, 3. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-H.; Lu, J.-Y.; Chen, H.-Y.; Wei, C.-C.; Xu, X.; Li, H.; Bai, Q.; Xia, F.-Z.; Lam, S.M.; Zhang, H.; et al. Liver ChREBP Protects Against Fructose-Induced Glycogenic Hepatotoxicity by Regulating L-Type Pyruvate Kinase. Diabetes 2020, 69, 591–602. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.-R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-Rich Diet Affects Mitochondrial DNA Damage and Repair in Rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoli, A.; Crescenzo, R.; Cigliano, L.; Spagnuolo, M.S.; Cancelliere, R.; Gatto, C.; Iossa, S. Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats. Nutrients 2019, 11, 2670. [Google Scholar] [CrossRef] [Green Version]
- Softic, S.; Stanhope, K.L.; Boucher, J.; Divanovic, S.; Lanaspa, M.A.; Johnson, R.J.; Kahn, C.R. Fructose and hepatic insulin resistance. Crit. Rev. Clin. Lab. Sci. 2020, 1–15. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.-X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.M.M.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Bruce, W.R.; Dong, Q.; Bruce, J.; Mehta, R.; O’Brien, P.J. Fructose and carbonyl metabolites as endogenous toxins. Chem. Biol. Interact. 2009, 178, 332–339. [Google Scholar] [CrossRef]
- Hieronimus, B.; Griffen, S.C.; Keim, N.L.; Bremer, A.A.; Berglund, L.; Nakajima, K.; Havel, P.J.; Stanhope, K.L. Effects of Fructose or Glucose on Circulating ApoCIII and Triglyceride and Cholesterol Content of Lipoprotein Subfractions in Humans. J. Clin. Med. 2019, 8, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichigo, Y.; Takeshita, A.; Hibino, M.; Nakagawa, T.; Hayakawa, T.; Patel, D.; Field, C.J.; Shimada, M. High-Fructose Diet-Induced Hypertriglyceridemia Is Associated with Enhanced Hepatic Expression of ACAT2 in Rats. Physiol. Res. 2019, 1021–1026. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanashi, M.; Kimura, T.; Li, C.; Tanaka, M.; Matsuhashi, A.; Yoshida, H.; Noda, A.; Xu, P.; Takase, S.; Okazaki, S.; et al. Critical Role of SREBP-1c Large-VLDL Pathway in Environment-Induced Hypertriglyceridemia of Apo AV Deficiency. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hieronimus, B.; Stanhope, K.L. Dietary fructose and dyslipidemia. Curr. Opin. Lipidol. 2020, 31, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Milutinović, D.V.; Brkljačić, J.; Teofilović, A.; Bursać, B.; Nikolić, M.; Gligorovska, L.; Kovačević, S.; Djordjevic, A.; Preitner, F.; Tappy, L.; et al. Chronic Stress Potentiates High Fructose–Induced Lipogenesis in Rat Liver and Kidney. Mol. Nutr. Food Res. 2020, 64, 1901141. [Google Scholar] [CrossRef] [PubMed]
- Steenson, S.; Shojaee-Moradie, F.; Whyte, M.B.; Jackson, K.G.; Lovegrove, J.A.; Fielding, B.A.; Umpleby, A.M. The Effect of Fructose Feeding on Intestinal Triacylglycerol Production and De Novo Fatty Acid Synthesis in Humans. Nutrients 2020, 12, 1781. [Google Scholar] [CrossRef]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef]
- Lian, C.-Y.; Zhai, Z.-Z.; Li, Z.-F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef]
- Pereira, R.; Botezelli, J.; da Cruz Rodrigues, K.; Mekary, R.; Cintra, D.; Pauli, J.; da Silva, A.; Ropelle, E.; de Moura, L. Fructose Consumption in the Development of Obesity and the Effects of Different Protocols of Physical Exercise on the Hepatic Metabolism. Nutrients 2017, 9, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, E.S.; Petrovsky, N. Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends Endocrinol. Metab. 2019, 30, 270–281. [Google Scholar] [CrossRef]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellouk, K.Z.; Louchami, E.; Hupkens, A.; Sener, D.A.; Yahia, W.J.M. The metabolic syndrome of fructose-fed rats: Effects of long-chain polyunsaturated ω3 and ω6 fatty acids. V. Post-mortem findings. Mol. Med. Rep. 2012, 6, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-C.; Varghese, Z.; Moorhead, J.F.; Lee, C.-T.; Chen, J.-B.; Ruan, X.Z. Cross-talk between TLR4-MyD88-NF-κB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am. J. Physiol. Circ. Physiol. 2013, 304, H874–H884. [Google Scholar] [CrossRef]
- Jegatheesan, P.; De Bandt, J. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C.; van Rooyen, D. Liver cholesterol: Is it playing possum in NASH? Am. J. Physiol. Liver Physiol. 2012, 303, G9–G11. [Google Scholar] [CrossRef] [Green Version]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Yamazaki, M.; Munetsuna, E.; Yamada, H.; Ando, Y.; Mizuno, G.; Fujii, R.; Nouchi, Y.; Kageyama, I.; Teshigawara, A.; Ishikawa, H.; et al. Maternal fructose consumption down-regulates Lxra expression via miR-206-mediated regulation. J. Nutr. Biochem. 2020, 82, 108386. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Munetsuna, E.; Yamada, H.; Ando, Y.; Yamazaki, M.; Taromaru, N.; Nagura, A.; Ishikawa, H.; Suzuki, K.; Teradaira, R.; et al. High fructose consumption induces DNA methylation at PPARα and CPT1A promoter regions in the rat liver. Biochem. Biophys. Res. Commun. 2015, 468, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Johnson, R.J.; Lanaspa, M.A. Endogenous fructose production. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The Role of Dietary Advanced Glycation End Products in Metabolic Dysfunction. Mol. Nutr. Food Res. 2020, 1900934. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Zhang, Y.; Zeng, C. Recent advances in fructose intake and risk of hyperuricemia. Biomed. Pharmacother. 2020, 131, 110795. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Vicente, A.; Cabral, P.D.; Hong, N.J.; Asirwatham, J.; Saez, F.; Garvin, J.L. Fructose reabsorption by rat proximal tubules: Role of Na + -linked cotransporters and the effect of dietary fructose. Am. J. Physiol. Physiol. 2019, 316, F473–F480. [Google Scholar] [CrossRef]
- Bjornstad, P.; Lanaspa, M.A.; Ishimoto, T.; Kosugi, T.; Kume, S.; Jalal, D.; Maahs, D.M.; Snell-Bergeon, J.K.; Johnson, R.J.; Nakagawa, T. Fructose and uric acid in diabetic nephropathy. Diabetologia 2015, 58, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef]
- Aldaba-Muruato, L.R.; Moreno Gil, M.; Shibayama, M.; Tsutsumi, V.; Muriel, P. Protective effects of allopurinol against acute liver damage and cirrhosis induced by carbon tetrachloride: Modulation of NF-κB, cytokine production and oxidative stress. Biochim. Biophys. Acta 2012, 1820, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Mottaran, G.; Occhino, E.; Reale, M.V. Review article: Role of oxidative stress in the progression of non-alcoholic steatosis. Aliment. Pharmacol. Ther. 2005, 22, 71–73. [Google Scholar] [CrossRef]
- Zhou, Y.; Fang, L.; Jiang, L.; Wen, P.; Cao, H.; He, W.; Dai, C.; Yang, J. Uric Acid Induces Renal Inflammation via Activating Tubular NF-κB Signaling Pathway. PLoS ONE 2012, 7, e39738. [Google Scholar] [CrossRef] [Green Version]
- Spiga, R.; Marini, M.A.; Mancuso, E.; Di Fatta, C.; Fuoco, A.; Perticone, F.; Andreozzi, F.; Mannino, G.C.; Sesti, G. Uric Acid Is Associated with Inflammatory Biomarkers and Induces Inflammation Via Activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1241–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, P.; Clare, K.; George, J.; Dillon, J.F. Determining the role for uric acid in non-alcoholic steatohepatitis development and the utility of urate metabolites in diagnosis: An opinion review. World J. Gastroenterol. 2020, 26, 1683–1690. [Google Scholar] [CrossRef]
- Oral; Sahin; Turker; Kocak Relationship Between Serum Uric Acid Levels and Nonalcoholic Fatty Liver Disease in Non-Obese Patients. Medicina 2019, 55, 600. [CrossRef] [Green Version]
- Yang, Y.; Zhou, Y.; Cheng, S.; Sun, J.L.; Yao, H.; Ma, L. Effect of uric acid on mitochondrial function and oxidative stress in hepatocytes. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Jhang, J.-J.; Cheng, Y.-T.; Ho, C.-Y.; Yen, G.-C. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell. Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarfeshani, A.; Ngo, S.; Sheppard, A. MicroRNA Expression Relating to Dietary-Induced Liver Steatosis and NASH. J. Clin. Med. 2015, 4, 1938–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanousková, B.; Neprašová, B.; Skálová, L.; Maletínská, L.; Zemanová, K.; Ambrož, M.; Matoušková, P. High-fructose drinks affect microRNAs expression differently in lean and obese mice. J. Nutr. Biochem. 2019, 68, 42–50. [Google Scholar] [CrossRef]
- Dong, X.; Liu, H.; Chen, F.; Li, D.; Zhao, Y. MiR-214 Promotes the Alcohol-Induced Oxidative Stress via Down-Regulation of Glutathione Reductase and Cytochrome P450 Oxidoreductase in Liver Cells. Alcohol. Clin. Exp. Res. 2014, 38, 68–77. [Google Scholar] [CrossRef]
- Dai, B.-H.; Geng, L.; Wang, Y.; Sui, C.-J.; Xie, F.; Shen, R.-X.; Shen, W.-F.; Yang, J.-M. microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis. 2013, 4, e604. [Google Scholar] [CrossRef]
- Jimenez Calvente, C.; Del Pilar, H.; Tameda, M.; Johnson, C.D.; Feldstein, A.E. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol. Ther. 2020, 28, 653–663. [Google Scholar] [CrossRef]
- Pan, J.H.; Cha, H.; Tang, J.; Lee, S.; Lee, S.H.; Le, B.; Redding, M.C.; Kim, S.; Batish, M.; Kong, B.C.; et al. The role of microRNA-33 as a key regulator in hepatic lipogenesis signaling and a potential serological biomarker for NAFLD with excessive dietary fructose consumption in C57BL/6N mice. Food Funct. 2021, 12, 656–667. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lanaspa, M.A.; Millan, I.S.; Fini, M.; Rivard, C.J.; Sanchez-Lozada, L.G.; Andres-Hernando, A.; Tolan, D.R.; Johnson, R.J. Fructose contributes to the Warburg effect for cancer growth. Cancer Metab. 2020, 8, 16. [Google Scholar] [CrossRef]
- Healy, M.E.; Chow, J.D.Y.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, M.E.; Lahiri, S.; Hargett, S.R.; Chow, J.D.Y.; Byrne, F.L.; Breen, D.S.; Kenwood, B.M.; Taddeo, E.P.; Lackner, C.; Caldwell, S.H.; et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci. Rep. 2016, 6, 22292. [Google Scholar] [CrossRef]
- Kim, J.; Kang, J.; Kang, Y.-L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.-W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef]
- Krause, N.; Wegner, A. Fructose Metabolism in Cancer. Cells 2020, 9, 2635. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Yamada, S.; Tanabe, H.; Takami, H.; Inokawa, Y.; Sonohara, F.; Shimizu, D.; Hattori, N.; Kanda, M.; Tanaka, C.; et al. High Serum Uric Acid Levels Could Be a Risk Factor of Hepatocellular Carcinoma Recurrences. Nutr. Cancer 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Kitade, M.; Nishimura, N.; Kaji, K.; Asada, K.; Namisaki, T.; Moriya, K.; Kawaratani, H.; Okura, Y.; Takaya, H.; et al. Oral administration of fructose exacerbates liver fibrosis and hepatocarcinogenesis via increased intestinal permeability in a rat steatohepatitis model. Oncotarget 2018, 9, 28638–28651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Systemic effects of overconsumption of fructose. Elevated fructose intake is implicated in increased oxidative stress, inflammation, higher uric acid levels, hypertriglyceridemia, higher systolic blood pressure, and insulin resistance, which are associated with the development or worsening of liver diseases.
Figure 1.
Systemic effects of overconsumption of fructose. Elevated fructose intake is implicated in increased oxidative stress, inflammation, higher uric acid levels, hypertriglyceridemia, higher systolic blood pressure, and insulin resistance, which are associated with the development or worsening of liver diseases.

Figure 2.
Fructose’s effects on the gut. Excessive fructose intake induces lipogenesis, oxidative stress, uric acid production, inflammation, and dysbiosis on the gut, which trigger necrosis and fibrosis in nonalcoholic steatohepatitis (NASH).
Figure 2.
Fructose’s effects on the gut. Excessive fructose intake induces lipogenesis, oxidative stress, uric acid production, inflammation, and dysbiosis on the gut, which trigger necrosis and fibrosis in nonalcoholic steatohepatitis (NASH).

Figure 3.
Metabolism of fructose in the liver. Fructose enters hepatocytes and is phosphorylated to fructose 1-phosphate by ketohexokinase C (KHK-C). Fructose 1-phosphate is converted to glyceraldehyde-3 phosphate and is a substrate for the synthesis of triacylglycerols and very-low-density lipoproteins (VLDL). Fructose intake can upregulate the ATP citrate lyase (ACLY) enzyme, promoting citrate breakdown to acetyl-CoA. Acetyl-CoA is converted to malonyl-coenzyme A (malonyl-CoA) by acetyl-CoA carboxylase (ACC-1). Elevated levels of malonyl-CoA inhibit β-oxidation through limiting carnitine palmitoyl transferase 1A, which promotes the accumulation of diacylglycerols and ceramides, causing hepatic dyslipidemia. Moreover, increased KHK-C activity exhausts adenosine triphosphate (ATP), generating adenosine diphosphate (ADP), which is converted to adenosine monophosphate (AMP). In turn, AMP is transformed to inosine monophosphate (IMP), increasing purine production. Xanthine oxidoreductase (XO) produces oxygen reactive species (ROS), hydrogen peroxide (H2O2), 4-hydroxynonenal (4-HNE), and xanthine. Then, xanthine is metabolized, resulting in the overproduction of uric acid and ROS, which induce oxidative stress. Uric acid activates nuclear factor-κB (NF-κB), triggering inflammation.
Figure 3.
Metabolism of fructose in the liver. Fructose enters hepatocytes and is phosphorylated to fructose 1-phosphate by ketohexokinase C (KHK-C). Fructose 1-phosphate is converted to glyceraldehyde-3 phosphate and is a substrate for the synthesis of triacylglycerols and very-low-density lipoproteins (VLDL). Fructose intake can upregulate the ATP citrate lyase (ACLY) enzyme, promoting citrate breakdown to acetyl-CoA. Acetyl-CoA is converted to malonyl-coenzyme A (malonyl-CoA) by acetyl-CoA carboxylase (ACC-1). Elevated levels of malonyl-CoA inhibit β-oxidation through limiting carnitine palmitoyl transferase 1A, which promotes the accumulation of diacylglycerols and ceramides, causing hepatic dyslipidemia. Moreover, increased KHK-C activity exhausts adenosine triphosphate (ATP), generating adenosine diphosphate (ADP), which is converted to adenosine monophosphate (AMP). In turn, AMP is transformed to inosine monophosphate (IMP), increasing purine production. Xanthine oxidoreductase (XO) produces oxygen reactive species (ROS), hydrogen peroxide (H2O2), 4-hydroxynonenal (4-HNE), and xanthine. Then, xanthine is metabolized, resulting in the overproduction of uric acid and ROS, which induce oxidative stress. Uric acid activates nuclear factor-κB (NF-κB), triggering inflammation.

Figure 4.
Molecular mechanisms by which fructose induces nonalcoholic steatohepatitis. Increased intestinal permeability (“leaky gut”) and dysbiosis produced by high fructose intake promote lipopolysaccharide (LPS) translocation from the intestine to the portal blood to reach the liver. Then, LPS activates the Toll-like receptor (TLR)-4/MyD88 signaling pathway, inducing tumor necrosis factor-alpha (TNF-α) through the nuclear translocation of transcription nuclear factor kappa B (NF-κB), which reinforces the inflammatory process through NLRP3 inflammasome activation and the subsequent maturation of interleukin (IL)-1 beta (β), caspase 1, and IL-18. Additionally, TNF-α and caspase 1 promote sterol-responsive element-binding protein 1 c (SREBP1c) activation and nuclear factor E2-related factor 2 (Nrf2) inhibition, while IL-6 drives hepatic stellate cell (HSC) activation, an orchestrated interaction of various molecular factors, leading to oxidative stress, inflammation, steatosis, and fibrogenesis, which pave the way to nonalcoholic steatohepatitis (NASH) development.
Figure 4.
Molecular mechanisms by which fructose induces nonalcoholic steatohepatitis. Increased intestinal permeability (“leaky gut”) and dysbiosis produced by high fructose intake promote lipopolysaccharide (LPS) translocation from the intestine to the portal blood to reach the liver. Then, LPS activates the Toll-like receptor (TLR)-4/MyD88 signaling pathway, inducing tumor necrosis factor-alpha (TNF-α) through the nuclear translocation of transcription nuclear factor kappa B (NF-κB), which reinforces the inflammatory process through NLRP3 inflammasome activation and the subsequent maturation of interleukin (IL)-1 beta (β), caspase 1, and IL-18. Additionally, TNF-α and caspase 1 promote sterol-responsive element-binding protein 1 c (SREBP1c) activation and nuclear factor E2-related factor 2 (Nrf2) inhibition, while IL-6 drives hepatic stellate cell (HSC) activation, an orchestrated interaction of various molecular factors, leading to oxidative stress, inflammation, steatosis, and fibrogenesis, which pave the way to nonalcoholic steatohepatitis (NASH) development.

Figure 5.
The fructose–pyroptosis axis. High fructose intake induces uric acid production in the intestine and liver, increasing reactive oxygen species (ROS). The resulting oxidative stress promotes the intracellular translocation of the thioredoxin-interacting protein (TXNIP) in the mitochondria; then, the interaction between TXNIP and NOD-like receptor family pyrin domain containing 3 (NLRP3) leads to NLRP3 inflammasome activation. The assembly of the inflammasome machinery enhances the pyroptosis of hepatocytes by the gasdermin D pathway and leads to the activation of Kupffer cells and transforming growth factor-beta (TGF-β) secretion, which results in HSC activation, triggering fibrogenesis in nonalcoholic steatohepatitis (NASH).
Figure 5.
The fructose–pyroptosis axis. High fructose intake induces uric acid production in the intestine and liver, increasing reactive oxygen species (ROS). The resulting oxidative stress promotes the intracellular translocation of the thioredoxin-interacting protein (TXNIP) in the mitochondria; then, the interaction between TXNIP and NOD-like receptor family pyrin domain containing 3 (NLRP3) leads to NLRP3 inflammasome activation. The assembly of the inflammasome machinery enhances the pyroptosis of hepatocytes by the gasdermin D pathway and leads to the activation of Kupffer cells and transforming growth factor-beta (TGF-β) secretion, which results in HSC activation, triggering fibrogenesis in nonalcoholic steatohepatitis (NASH).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Muriel, P.; López-Sánchez, P.; Ramos-Tovar, E. Fructose and the Liver. Int. J. Mol. Sci. 2021, 22, 6969. https://doi.org/10.3390/ijms22136969
AMA Style
Muriel P, López-Sánchez P, Ramos-Tovar E. Fructose and the Liver. International Journal of Molecular Sciences. 2021; 22(13):6969. https://doi.org/10.3390/ijms22136969
Chicago/Turabian StyleMuriel, Pablo, Pedro López-Sánchez, and Erika Ramos-Tovar. 2021. "Fructose and the Liver" International Journal of Molecular Sciences 22, no. 13: 6969. https://doi.org/10.3390/ijms22136969
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.