
Genetic and Epigenetic Variations of HPV52 in Cervical Precancer


 ,
,
Abstract
:1. Introduction
2. Results
2.1. CpG Methylation Determined by Pyrosequencing
2.2. CpG Methylation Determined by Next-Generation Sequencing (NGS)
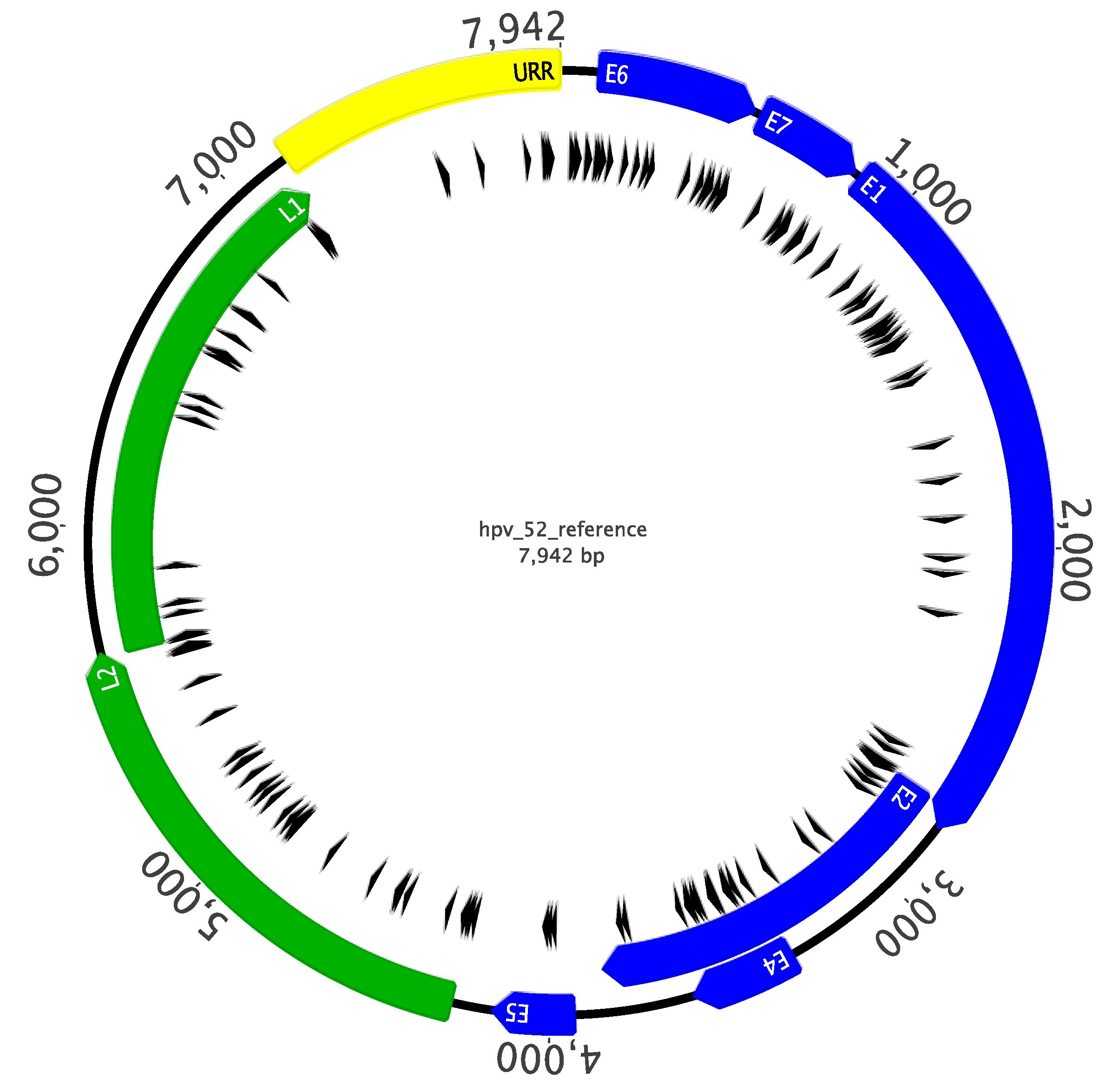
2.3. Sanger Sequencing Analysis of Complete HPV52 Genomes
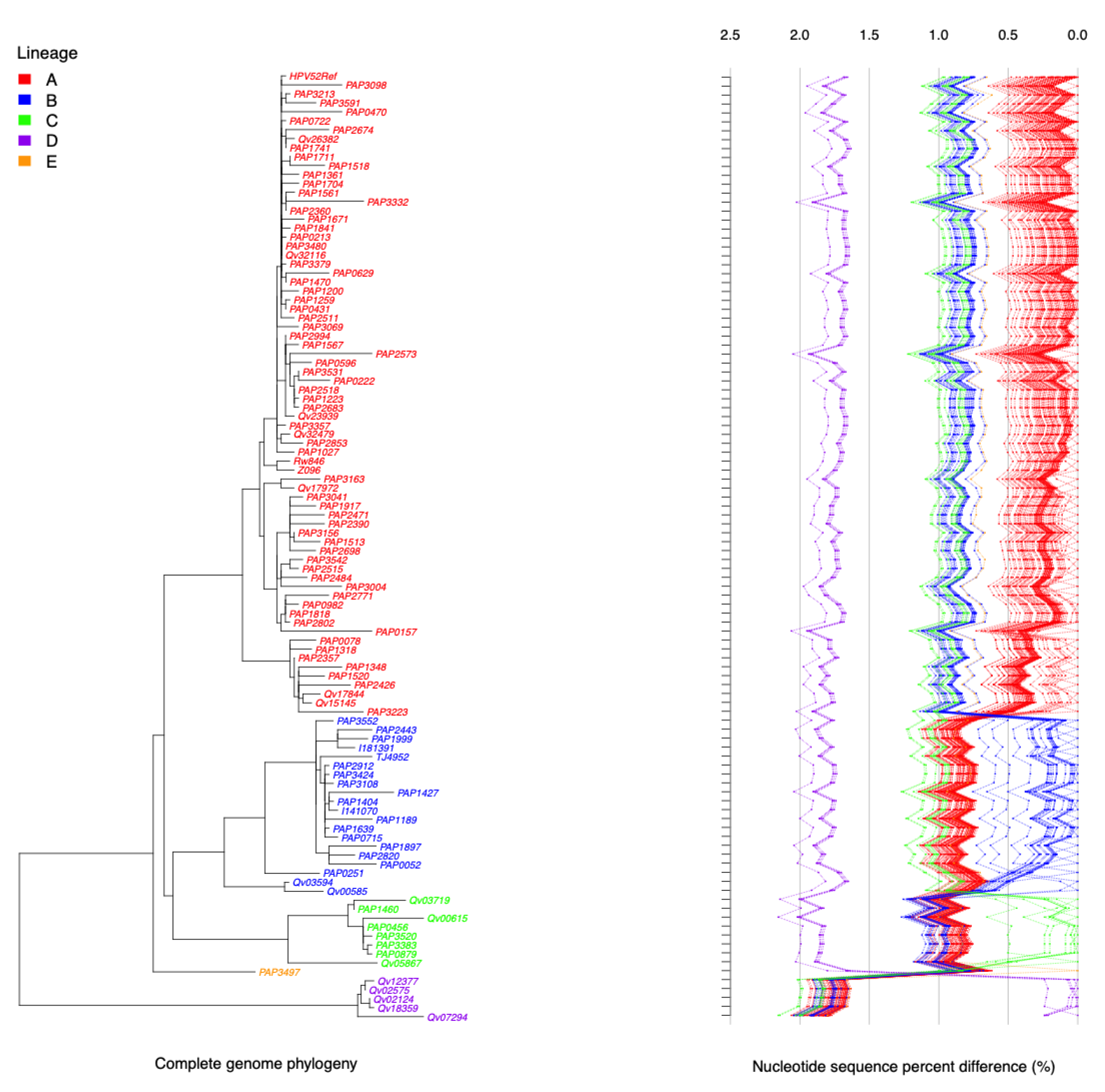
2.4. HPV52 Phylogenetic Analysis of 106 Isolates
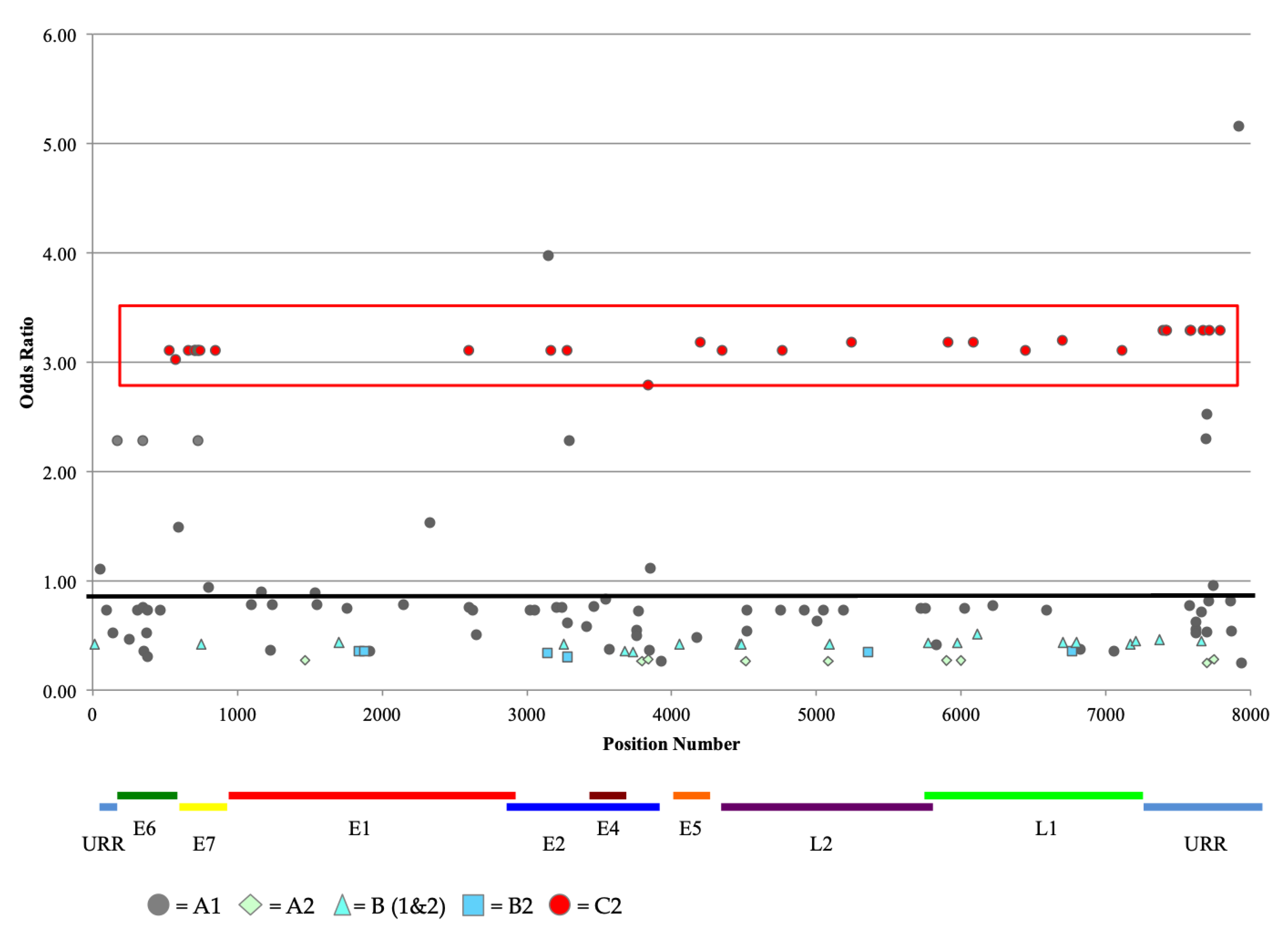
2.5. Association of Variant Lineage and Disease Status
2.6. Analysis of Nucleotide Variations at CpG Sites
3. Discussion
4. Materials and Methods
4.1. Study Design and Population
4.2. Sodium Bisulfite Treatment
4.3. Pyrosequencing
4.4. Next-Generation Sequenicng (NGS)
4.5. Bioinformatic Analysis of NGS Reads
4.6. Sequencing of HPV52 Complete Genomes
4.7. Construction of Phylogentic Tree
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
Abbreviations
| HPV | Human papillomavirus |
| HR-HPV | High-risk human papillomavirus |
| Alpha9 | Alphapapillomavirus species 9 (HPV16-related) |
| Alpha7 | Alphapapillomavirus species 7 (HPV18-related) |
| CIN3 | Cervical intraepithelial neoplasia grade 3 |
| CpG site | CG dinucleotide |
| AIS | Adenocarcinoma in situ |
| SCC | Squamous cell cancer |
| URR | Upstream regulatory region |
| OR | Odds ratio |
| CI | Confidence interval |
| AUC | Area under the curve |
| SNP | Single nucleotide polymorphism |
| NGS | Next-generation sequencing |
| NCR1 | Noncoding region 1 |
| VWAS | Viral genome wide association study |
| PaP cohort | HPV Persistence and Progression |
| KPNC | Kaiser Permanente Northern California |
| NCI | National Cancer Institute |
| HC2 | Hybrid capture 2 |
| NILM | Negative for intraepithelial lesions or malignancy |
| bp | Base pair |
| ML | Maximum likelihood |
| MCMC | Markov chain Monte Carlo |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.; Clifford, G.M. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, A.T.; Fregnani, J.H.; Villa, L.L.; Sichero, L.; Nunes, E.M.; Longatto-Filho, A. Diversity of human papillomavirus typing among women population living in rural and remote areas of Brazilian territory. Papillomavirus Res. 2019, 8, 100186. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, T.; Li, L.; Wang, M.Z.; Wu, Z.; Cui, J.; Liu, B.; Zhang, X.; Qiao, Y.; Chen, W. Clustering patterns of type-type combination in multiple genotypes infections of human papillomavirus in cervical adenocarcinoma. J. Med. Virol. 2019, 91, 2001–2008. [Google Scholar] [CrossRef]
- Toh, Z.Q.; Kosasih, J.; Russell, F.M.; Garland, S.M.; Mulholland, E.K.; Licciardi, P.V. Recombinant human papillomavirus nonavalent vaccine in the prevention of cancers caused by human papillomavirus. Infect. Drug Resist. 2019, 12, 1951–1967. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Sun, X.; Li, M.; Liu, T.; Hu, G.; He, Y.; Mao, L.; Yan, L.; Xie, L.; Zou, H.; et al. Cervical human papillomavirus among women in Guangdong, China 2008-2017: Implication for screening and vaccination. J. Med. Virol. 2019, 91, 1856–1865. [Google Scholar] [CrossRef]
- Pity, I.S.; Abdo, H.M.; Goreal, A.A. Human Papillomavirus Genotyping among Different Cervical Smears in Duhok/Iraq. Asian Pac. J. Cancer Prev. 2019, 20, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Vyankandondera, J.; Wambua, S.; Irungu, E.; Mandaliya, K.; Temmerman, M.; Ryan, C.; Mohamed, Y.; Vanden Broeck, D.; Verhelst, R.; Chersich, M.F.; et al. Type-Specific Human Papillomavirus Prevalence, Incident Cases, Persistence, and Associated Pregnancy Outcomes Among HIV-Infected Women in Kenya. Sex Transm. Dis. 2019, 46, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, D.; Wang, K.; Wang, J.; Zhang, Z.; Chen, Y.; Zhang, X.; Ma, C. HPV genotype prevalence and distribution during 2009–2018 in Xinjiang, China: Baseline surveys prior to mass HPV vaccination. BMC Womens Health 2019, 19, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Liu, Y.; Zuo, T.; Cui, X.; Li, M.; Zhang, J.; Yu, H.; Piao, H. The prevalence, trends, and geographical distribution of human papillomavirus infection in China: The pooled analysis of 1.7 million women. Cancer Med. 2019, 8, 5373–5385. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ye, M.; Chen, Y.; Gong, Q.; Mei, B. Genetic variation of E6 and E7 genes of human papillomavirus 52 from Central China. J. Med. Virol. 2021, 93, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- McHome, B.L.; Kjaer, S.K.; Manongi, R.; Swai, P.; Waldstroem, M.; Iftner, T.; Wu, C.; Mwaiselage, J.; Rasch, V. HPV types, cervical high-grade lesions and risk factors for oncogenic human papillomavirus infection among 3416 Tanzanian women. Sex Transm. Infect. 2021, 97, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Sweet, K.; Bosire, C.; Sanusi, B.; Sherrod, C.J.; Kwatampora, J.; Waweru, W.; Mugo, N.; Kimani, J.; Ting, J.; Clark, J.; et al. Prevalence, incidence, and distribution of human papillomavirus types in female sex workers in Kenya. Int. J. STD AIDS 2020, 31, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Tounkara, F.K.; Teguete, I.; Guedou, F.A.; Goma-Matsetse, E.; Kone, A.; Behanzin, L.; Traore, S.; Aza-Gnandji, M.; Keita, B.; Guenoun, J.; et al. Human papillomavirus genotype distribution and factors associated among female sex workers in West Africa. PLoS ONE 2020, 15, e0242711. [Google Scholar] [CrossRef]
- Wu, W.; Song, L.; Yang, Y.; Wang, J.; Liu, H.; Zhang, L. Exploring the dynamics and interplay of human papillomavirus and cervical tumorigenesis by integrating biological data into a mathematical model. BMC Bioinform. 2020, 21, 152. [Google Scholar] [CrossRef]
- Murao, K.; Tetsutani, M.; Ishigami, T.; Kubo, Y.; Arase, S. Bowen disease of the palm associated with human papillomavirus 52. Clin. Exp. Dermatol. 2013, 38, 489–491. [Google Scholar] [CrossRef]
- Martinez-Bailon, C.; Mantilla-Morales, A.; Mendez-Matias, G.; Alvarado-Cabrero, I.; Maldonado-Rodriguez, R.; Quintero-Becerra, J.; Arias-Flores, R.; Pina-Sanchez, P. Human papillomavirus genotypes and P16INK4A expression in squamous penile carcinoma in Mexican patients. BMC Infect. Dis. 2019, 19, 1068. [Google Scholar] [CrossRef]
- Schiffman, M.; Herrero, R.; Desalle, R.; Hildesheim, A.; Wacholder, S.; Rodriguez, A.C.; Bratti, M.C.; Sherman, M.E.; Morales, J.; Guillen, D.; et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 2005, 337, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.D.; Harari, A.; Chen, Z. Human papillomavirus genome variants. Virology 2013, 445, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, G.Y.; Bierman, R.; Beardsley, L.; Chang, C.J.; Burk, R.D. Natural history of cervicovaginal papillomavirus infection in young women. N. Engl. J. Med. 1998, 338, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Wentzensen, N. Human papillomavirus infection and the multistage carcinogenesis of cervical cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCredie, M.R.; Sharples, K.J.; Paul, C.; Baranyai, J.; Medley, G.; Jones, R.W.; Skegg, D.C. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: A retrospective cohort study. Lancet Oncol. 2008, 9, 425–434. [Google Scholar] [CrossRef]
- Fontham, E.T.H.; Wolf, A.M.D.; Church, T.R.; Etzioni, R.; Flowers, C.R.; Herzig, A.; Guerra, C.E.; Oeffinger, K.C.; Shih, Y.T.; Walter, L.C.; et al. Cervical cancer screening for individuals at average risk: 2020 guideline update from the American Cancer Society. CA Cancer J. Clin. 2020, 70, 321–346. [Google Scholar] [CrossRef]
- Wentzensen, N.; Schiffman, M.; Palmer, T.; Arbyn, M. Triage of HPV positive women in cervical cancer screening. J. Clin. Virol. 2016, 76 (Suppl. 1), S49–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.; Wudayagiri, R.; Valluru, L. Cervical cancer: Biomarkers for diagnosis and treatment. Clin. Chim. Acta 2015, 445, 7–11. [Google Scholar] [CrossRef]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimer, M.; Sun, C.; McAndrew, T.; Smith, B.; Harari, A.; Chen, Z.; Mirabello, L.; Wentzensen, N.; Goldberg, G.L.; Rodriguez, A.C.; et al. HPV16 CpG methyl-haplotypes are associated with cervix precancer and cancer in the Guanacaste natural history study. Gynecol. Oncol. 2015, 138, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Frimer, M.; Harari, A.; McAndrew, T.; Smith, B.; Chen, Z.; Wentzensen, N.; Wacholder, S.; Castle, P.E.; Raine-Bennett, T.; et al. HPV16 methyl-haplotypes determined by a novel next-generation sequencing method are associated with cervical precancer. Int. J. Cancer 2015, 136, E146–E153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorincz, A.T.; Brentnall, A.R.; Scibior-Bentkowska, D.; Reuter, C.; Banwait, R.; Cadman, L.; Austin, J.; Cuzick, J.; Vasiljevic, N. Validation of a DNA methylation HPV triage classifier in a screening sample. Int. J. Cancer 2016, 138, 2745–2751. [Google Scholar] [CrossRef] [Green Version]
- Wentzensen, N.; Sun, C.; Ghosh, A.; Kinney, W.; Mirabello, L.; Wacholder, S.; Shaber, R.; LaMere, B.; Clarke, M.; Lorincz, A.T.; et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J. Natl. Cancer Inst. 2012, 104, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Anastos, K.; Segondy, M.; Sahasrabuddhe, V.V.; Gravitt, P.E.; Hsing, A.W.; Burk, R.D. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS ONE 2011, 6, e20183. [Google Scholar] [CrossRef]
- Clarke, M.A.; Gradissimo, A.; Schiffman, M.; Lam, J.; Sollecito, C.C.; Fetterman, B.; Lorey, T.; Poitras, N.; Raine-Bennett, T.R.; Castle, P.E.; et al. Human Papillomavirus DNA Methylation as a Biomarker for Cervical Precancer: Consistency across 12 Genotypes and Potential Impact on Management of HPV-Positive Women. Clin. Cancer Res. 2018, 24, 2194–2202. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.; Chen, Z.; Reimers, L.; van Doorslaer, K.; Schiffman, M.; Desalle, R.; Herrero, R.; Yu, K.; Wacholder, S.; Wang, T.; et al. Sequence imputation of HPV16 genomes for genetic association studies. PLoS ONE 2011, 6, e21375. [Google Scholar] [CrossRef] [Green Version]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174 e1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, M.; Gage, J.C.; Clifford, G.M.; Demarco, M.; Cheung, L.C.; Chen, Z.; Yeager, M.; Cullen, M.; Boland, J.F.; Chen, X.; et al. Association of HPV35 with cervical carcinogenesis among women of African ancestry: Evidence of viral-host interaction with implications for disease intervention. Int. J. Cancer 2020, 147, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, A.R.; Vasiljevic, N.; Scibior-Bentkowska, D.; Cadman, L.; Austin, J.; Cuzick, J.; Lorincz, A.T. HPV33 DNA methylation measurement improves cervical pre-cancer risk estimation of an HPV16, HPV18, HPV31 and EPB41L3 methylation classifier. Cancer Biomark. 2015, 15, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Louvanto, K.; Franco, E.L.; Ramanakumar, A.V.; Vasiljevic, N.; Scibior-Bentkowska, D.; Koushik, A.; Cuzick, J.; Coutlee, F.; Lorincz, A.T.; Biomarkers of Cervical Cancer Risk Study Team. Methylation of viral and host genes and severity of cervical lesions associated with human papillomavirus type 16. Int. J. Cancer 2015, 136, E638–E645. [Google Scholar] [CrossRef]
- Ramirez, A.T.; Sanchez, G.I.; Nedjai, B.; Agudelo, M.C.; Brentnall, A.R.; Cuschieri, K.; Castaneda, K.M.; Cuzick, J.; Lorincz, A.T.; Group, A.-U.-C.T. Effective methylation triage of HPV positive women with abnormal cytology in a middle-income country. Int. J. Cancer 2021, 148, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Gillio-Tos, A.; Fiano, V.; Grasso, C.; Trevisan, M.; Gori, S.; Mongia, A.; De Marco, L.; Ronco, G.; New Technologies for Cervical Cancer Screening Working Group. Assessment of viral methylation levels for high risk HPV types by newly designed consensus primers PCR and pyrosequencing. PLoS ONE 2018, 13, e0194619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Knebel Doeberitz, M.; Prigge, E.S. Role of DNA methylation in HPV associated lesions. Papillomavirus Res. 2019, 7, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Murakami, I.; Fujii, T.; Dan, K.; Saito, M.; Ohno, A.; Iwata, T.; Aoki, D. Methylation of human papillomavirus-52 and -58 is a candidate biomarker in cervical neoplasia. J. Clin. Virol. 2013, 58, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.W.; Huang, R.L.; Su, P.H.; Chen, Y.C.; Wang, H.C.; Liao, C.C.; Lai, H.C. Genotype-specific methylation of HPV in cervical intraepithelial neoplasia. J. Gynecol. Oncol. 2017, 28, e56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradissimo, A.; Lam, J.; Attonito, J.D.; Palefsky, J.; Massad, L.S.; Xie, X.; Eltoum, I.E.; Rahangdale, L.; Fischl, M.A.; Anastos, K.; et al. Methylation of High-Risk Human Papillomavirus Genomes Are Associated with Cervical Precancer in HIV-Positive Women. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Aho, J.; Hankins, C.; Tremblay, C.; Lang, F.; Forest, P.; Pourreaux, K.; Rouah, F.; Coutlee, F.; The Canadian Women’s HIV Study Group. Molecular analysis of human papillomavirus type 52 isolates detected in the genital tract of human immunodeficiency virus-seropositive and -seronegative women. J. Infect. Dis. 2003, 188, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Van Doorslaer, K.; Burk, R.D. Evolution of human papillomavirus carcinogenicity. Adv. Virus Res. 2010, 77, 41–62. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Lu, Z.; Liu, J.; Wang, G.; Zhou, W.; Yang, L.; Liu, C.; Wang, B.; Ruan, Q. Genetic variations of E6 and long control region of human papillomavirus type 16 from patients with cervical lesion in Liaoning, China. BMC Cancer 2013, 13, 459. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Ki, E.Y.; Zhang, C.; Ho, W.C.; Lee, S.J.; Jeong, M.J.; Chan, P.K.; Park, J.S. Analysis of Sequence Variation and Risk Association of Human Papillomavirus 52 Variants Circulating in Korea. PLoS ONE 2016, 11, e0168178. [Google Scholar] [CrossRef]
- Chang, Y.J.; Chen, H.C.; Lee, B.H.; You, S.L.; Lin, C.Y.; Pan, M.H.; Chou, Y.C.; Hsieh, C.Y.; Chen, Y.M.; Cheng, Y.J.; et al. Unique variants of human papillomavirus genotypes 52 and 58 and risk of cervical neoplasia. Int. J. Cancer 2011, 129, 965–973. [Google Scholar] [CrossRef]
- Sun, Z.; Lu, Z.; Liu, J.; Wang, G.; Zhou, W.; Yang, L.; Liu, C.; Ruan, Q. Genomic polymorphism of human papillomavirus type 52 in women from Northeast China. Int. J. Mol. Sci. 2012, 13, 14962–14972. [Google Scholar] [CrossRef] [PubMed]
- Formentin, A.; Archambault, J.; Koushik, A.; Richardson, H.; Brassard, P.; Franco, E.L.; Coutlee, F. Human papillomavirus type 52 polymorphism and high-grade lesions of the uterine cervix. Int. J. Cancer 2013, 132, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, A.; Matsushita, K.; Hoang, H.T.; Agdamag, D.M.; Nguyen, C.H.; Tran, V.T.; Sasagawa, T.; Saikawa, K.; Lihana, R.; Pham, H.V.; et al. E6 and E7 variants of human papillomavirus-16 and -52 in Japan, the Philippines, and Vietnam. J. Med. Virol. 2013, 85, 1069–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Liu, Z.; Su, J.; Yan, D.; Sun, W.; Zeng, Z. Human papillomavirus type-specific prevalence in women referred for colposcopic examination in Beijing. J. Med. Virol. 2014, 86, 1937–1943. [Google Scholar] [CrossRef]
- Zhang, C.; Park, J.S.; Grce, M.; Hibbitts, S.; Palefsky, J.M.; Konno, R.; Smith-McCune, K.K.; Giovannelli, L.; Chu, T.Y.; Picconi, M.A.; et al. Geographical distribution and risk association of human papillomavirus genotype 52-variant lineages. J. Infect. Dis. 2014, 210, 1600–1604. [Google Scholar] [CrossRef] [Green Version]
- Tenjimbayashi, Y.; Onuki, M.; Hirose, Y.; Mori, S.; Ishii, Y.; Takeuchi, T.; Tasaka, N.; Satoh, T.; Morisada, T.; Iwata, T.; et al. Whole-genome analysis of human papillomavirus genotypes 52 and 58 isolated from Japanese women with cervical intraepithelial neoplasia and invasive cervical cancer. Infect. Agent Cancer 2017, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frati, E.R.; Bianchi, S.; Amendola, A.; Colzani, D.; Petrelli, F.; Zehender, G.; Tanzi, E. Genetic characterization of variants of HPV16, HPV18 and HPV52 circulating in Italy among general and highrisk populations. Mol. Med. Rep. 2020, 21, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Raiol, T.; Wyant, P.S.; de Amorim, R.M.; Cerqueira, D.M.; Milanezi, N.; Brigido Mde, M.; Sichero, L.; Martins, C.R. Genetic variability and phylogeny of the high-risk HPV-31, -33, -35, -52, and -58 in central Brazil. J. Med. Virol. 2009, 81, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Rosl, F.; Arab, A.; Klevenz, B.; zur Hausen, H. The effect of DNA methylation on gene regulation of human papillomaviruses. J. Gen. Virol. 1993, 74, 791–801. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef] [Green Version]
- Castle, P.E.; Shaber, R.; LaMere, B.J.; Kinney, W.; Fetterma, B.; Poitras, N.; Lorey, T.; Schiffman, M.; Dunne, A.; Ostolaza, J.M.; et al. Human Papillomavirus (HPV) Genotypes in Women with Cervical Precancer and Cancer at Kaiser Permanente Northern California. Cancer Epidemiol. Biomark. Prev. 2011, 20, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Castle, P.E.; Schiffman, M.; Gravitt, P.E.; Kendall, H.; Fishman, S.; Dong, H.; Hildesheim, A.; Herrero, R.; Bratti, M.C.; Sherman, M.E.; et al. Comparisons of HPV DNA detection by MY09/11 PCR methods. J. Med. Virol. 2002, 68, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamady, M.; Walker, J.J.; Harris, J.K.; Gold, N.J.; Knight, R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 2008, 5, 235–237. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Median | Man-Whitney (Wilcoxon Rank Sum) | ROC Curve | 95% CI Interval | Univariate Regression High HPV Methylation vs. Low/Medium Methylation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Region Name | Genome Position | Control (n = 39) | Case (n = 50) | Difference | p Value | AUC | OR | 95% CI | p Value | |
| E7 | c_754 c_776 c_779 c_787 | 1.40 13.20 1.20 5.90 | 1.80 13.30 1.20 6.40 | 0.40 0.10 0.00 0.50 | 0.010910 0.542481 0.771323 0.141489 | 0.6583 0.5379 0.5181 0.5916 | (0.54, 0.78) (0.42, 0.66) (0.39, 0.65) (0.47, 0.71) | 4.64 2.36 0.50 1.99 | (1.88, 11.47) (0.95, 5.89) (0.18, 1.39) (0.82, 4.81) | 0.000886 0.064880 0.182842 0.125934 |
| E1 | c_1122 c_1141 | 1.85 6.20 | 1.55 7.55 | −0.30 1.35 | 0.319005 0.057360 | 0.4405 0.6187 | (0.32, 0.56) (0.50, 0.74) | 0.61 1.85 | (0.23, 1.59) (0.76, 4.46) | 0.313096 0.173033 |
| L2 | c_4274 c_4283 c_4292 c_4919 c_4926 c_4931 c_4949 | 8.00 18.50 10.90 2.40 4.30 4.40 32.60 | 13.50 17.20 19.90 3.00 5.25 4.70 39.50 | 5.50 −1.30 9.00 0.60 0.95 0.30 6.90 | 0.001171 0.003156 0.001699 0.085078 0.251942 0.295395 0.371808 | 0.7013 0.6831 0.6946 0.6062 0.5710 0.5649 0.5554 | (0.59, 0.82) (0.57, 0.80) (0.58, 0.81) (0.49, 0.72) (0.45, 0.69) (0.45, 0.68) (0.43, 0.68) | 2.44 2.25 2.44 2.25 1.50 1.56 1.38 | (1.01, 5.86) (0.94, 5.41) (1.01, 5.86) (0.94, 5.41) (0.62, 3.63) (0.63, 3.84) (0.57, 3.35) | 0.046613 0.070045 0.046613 0.070045 0.368990 0.333509 0.478098 |
| L1 | c_5606 c_5609 c_5615 c_5621 c_5648 c_5655 c_7098 c_7111 c_7119 | 18.00 15.00 30.20 5.00 26.80 35.90 3.10 5.90 3.40 | 33.20 25.90 48.10 8.05 32.15 51.20 3.90 10.30 5.70 | 15.20 10.90 17.90 3.05 5.35 15.30 0.80 4.40 2.30 | 0.001393 0.007199 0.000257 0.001651 0.034620 0.001799 0.024831 0.016845 0.032968 | 0.6982 0.6667 0.7267 0.6951 0.631 0.6936 0.6397 0.6489 0.6327 | (0.58, 0.81) (0.55, 0.79) (0.62, 0.83) (0.58, 0.81) (0.51, 0.75) (0.58, 0.80) (0.52, 0.76) (0.53, 0.77) (0.51, 0.76) | 3.38 2.64 4.78 2.86 2.08 3.67 1.99 2.76 3.55 | (1.39, 8.17) (1.10, 6.36) (1.94, 11.80) (1.19, 6.90) (0.86, 5.00) (1.51, 8.92) (0.82, 4.81) (1.14, 6.68) (1.46, 8.65) | 0.007037 0.030232 0.000684 0.019101 0.102620 0.004099 0.125934 0.024124 0.005258 |
| URR | c_7557 c_7563 | 2.50 2.80 | 2.50 3.05 | 0.00 0.25 | 0.885024 0.423051 | 0.5096 0.553 | (0.37, 0.64) (0.42, 0.68) | 0.81 1.71 | (0.30, 2.19) (0.68, 2.01) | 0.680344 0.579475 |
| Median | Man-Whitney (Wilcoxon Rank Sum) | ROC Curve | 95% CI Interval | Univariate Regression High HPV Methylation vs. Low/Medium Methylation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Region Name | Genome Position | Control (n = 38) | Case (n = 50) | Difference | p Value | AUC | OR | 95% CI | p Value | |
| L1 | c_5606 c_5609 c_5615 c_5621 c_5648 c_5655 | 27.36 18.84 40.90 5.34 32.21 48.22 | 30.45 22.11 46.76 6.26 37.07 52.92 | 3.09 3.27 5.86 0.92 4.86 4.70 | 0.005258 0.003606 0.003330 0.052489 0.003512 0.005895 | 0.6731 0.6805 0.6821 0.6203 0.681 0.6708 | (0.56, 0.79) (0.56, 0.80) (0.57, 0.80) (0.50, 0.74) (0.57, 0.80) (0.55, 0.79) | 3.38 1.92 3.67 1.92 2.86 2.86 | (1.39, 8.17) (0.80, 4.61) (1.51, 8.92) (0.80, 4.61) (1.19, 6.90) (1.19, 6.90) | 0.007037 0.146627 0.004099 0.146627 0.019101 0.019101 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bee, K.J.; Gradissimo, A.; Chen, Z.; Harari, A.; Schiffman, M.; Raine-Bennett, T.; Castle, P.E.; Clarke, M.; Wentzensen, N.; Burk, R.D. Genetic and Epigenetic Variations of HPV52 in Cervical Precancer. Int. J. Mol. Sci. 2021, 22, 6463. https://doi.org/10.3390/ijms22126463
Bee KJ, Gradissimo A, Chen Z, Harari A, Schiffman M, Raine-Bennett T, Castle PE, Clarke M, Wentzensen N, Burk RD. Genetic and Epigenetic Variations of HPV52 in Cervical Precancer. International Journal of Molecular Sciences. 2021; 22(12):6463. https://doi.org/10.3390/ijms22126463
Chicago/Turabian StyleBee, Katharine J., Ana Gradissimo, Zigui Chen, Ariana Harari, Mark Schiffman, Tina Raine-Bennett, Philip E. Castle, Megan Clarke, Nicolas Wentzensen, and Robert D. Burk. 2021. "Genetic and Epigenetic Variations of HPV52 in Cervical Precancer" International Journal of Molecular Sciences 22, no. 12: 6463. https://doi.org/10.3390/ijms22126463