
Impaired Leptin Signalling in Obesity: Is Leptin a New Thermolipokine?

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Genetics of Leptin
3. Leptin and the Regulation of Thermogenesis
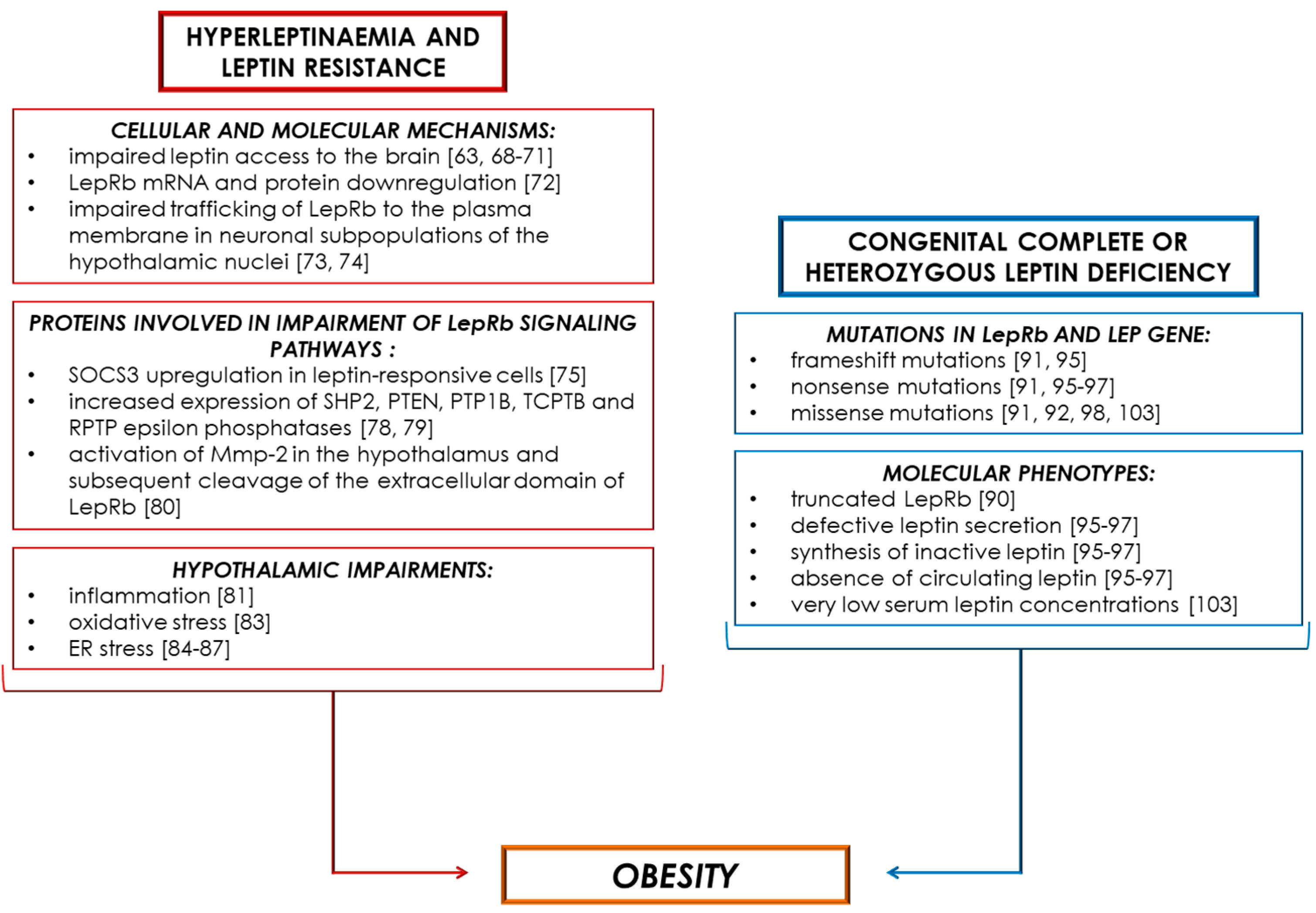
4. Obesity-Associated Hyperleptinaemia and Leptin Resistance
5. Obesity-Associated Leptin Deficiency

6. Pharmacotherapy of Leptin Signalling
7. Pharmacological Treatment of Leptin Deficiency

{kind=link}
{kind=link}
| Drugs | Species | In Vitro | In Vivo/Ex Vivo | Effects |
|---|---|---|---|---|
| For Hyperleptinaemia | ||||
| hLep3 antibodies | Mouse | - | ↑ UCP-1 and PGC-1α mRNA in WAT and BAT [111] ↓ SOCS3, TNF-α and IL-1β mRNA in the hypothalamus [111] | ↑ Glucose tolerance [111] ↑ EE [111] ↓ Body weight [111] ↓ Hypothalamus inflammation [111] ↑ Leptin sensitivity [111] |
| Telmisartan | Mouse | ↑ PPAR-δ signalling [119] ↓ Adipogenesis [119] | ↑ UCP-1 protein in BAT [119] | ↑ Leptin sensitivity [115] ↓ Hypothalamus inflammation [117] ↑ Leptin transport through the BBB [118] ↓ Body weight [118] ↓ Insulin resistance [120] ↑ Sympathetic nervous system [120] |
| Human | - | - | ↑ Cardiac function [121] ↓ Plasma leptin levels [122] | |
| PAI-1 inhibitor M5441 | Mouse | - | ↑ UCP-1, DIO-2, CIDEA and PRDM-16 mRNA in BAT [126] | ↑ Leptin sensitivity [126] ↓ Plasma leptin levels [126] ↓ Body weight [126] ↑ Lipolysis [130] |
| Panax notoginseng saponins | Mouse | - | ↑ UCP-1, PGC-1α and DIO-2 mRNA/protein in BAT [7] ↑ UCP-1, PRDM-16 and PGC-1α mRNA/protein in WAT [7] ↑ AMPK-α/STAT3 signalling in WAT [7] | ↑ EE [7] ↓ Body weight [7] |
| For Leptin Deficiency | ||||
| Metreleptin | Human | - | - | ↓ Triglycerides [101] ↑ HDL [101] ↑ T-cell responsiveness [101] ↓ Body weight [135] ↑ Physical activity [135] ↓ Hunger [135] ↑ Fat oxidation [136] ↓ Fall in EE during caloric restriction [136] |
| Metreleptin/pramlintide | Mouse | - | - | ↓ Body weight [143] |
| Human | - | - | ↓ Body weight [144] | |
| Melatonin | Mouse | - | ↓ TNF-α, resistin and visfatin proteins in adipose tissue [147] ↑ UCP-1 and PGC-1α proteins in WAT [148] | ↓ Body weight [147] ↓ Adipose tissue inflammation [147] |
| Melanocortin receptor agonists | Mouse | - | - | ↓ Body weight [149] ↓ Food intake [149] |
| Human | - | - | ↓ Body weight [150] |
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Porro, S.; Genchi, V.A.; Cignarelli, A.; Natalicchio, A.; Laviola, L.; Giorgino, F.; Perrini, S. Dysmetabolic adipose tissue in obesity: Morphological and functional characteristics of adipose stem cells and mature adipocytes in healthy and unhealthy obese subjects. J. Endocrinol. Investig. 2020. [Google Scholar] [CrossRef]
- Cignarelli, A.; Genchi, V.A.; Perrini, S.; Natalicchio, A.; Laviola, L.; Giorgino, F. Insulin and insulin receptors in adipose tissue development. Int. J. Mol. Sci. 2019, 20, 759. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- Perrini, S. Leptin: A marker of renal injury. Intern. Emerg. Med. 2019, 14, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Bjørbæk, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, N.; Tan, H.Y.; Li, S.; Zhang, C.; Zhang, Z.; Feng, Y. Panax notoginseng saponins modulate the gut microbiota to promote thermogenesis and beige adipocyte reconstruction via leptin-mediated AMPKα/STAT3 signaling in diet-induced obesity. Theranostics 2020, 10, 11302–11323. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor γ (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Ding, X.; Cao, Y.; Wang, H.; Zeng, W. Dense Intra-adipose Sympathetic Arborizations Are Essential for Cold-Induced Beiging of Mouse White Adipose Tissue. Cell Metab. 2017, 26, 686–692.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakir, I.; Diaz-Martinez, M.; Lining Pan, P.; Welch, E.B.; Patel, S.; Ghamari-Langroudi, M. Leptin receptor signaling in sim1-expressing neurons regulates body temperature and adaptive thermogenesis. Endocrinology 2019, 160, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, S.R.; Abu-Asab, M.; Glasow, A.; Päth, G.; Hauner, H.; Tsokos, M.; Chrousos, G.P.; Scherbaum, W.A. Immunohistochemical and ultrastructural localization of leptin and leptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes 2000, 49, 532–538. [Google Scholar] [CrossRef] [Green Version]
- Wagoner, B.; Hausman, D.B.; Harris, R.B.S. Direct and indirect effects of leptin on preadipocyte proliferation and differentiation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290. [Google Scholar] [CrossRef] [Green Version]
- Machinal-Quélin, F.; Dieudonné, M.N.; Leneveu, M.C.; Pecquery, R.; Giudicelli, Y. Proadipogenic effect of leptin on rat preadipocytes in vitro: Activation of MAPK and STAT3 signaling pathways. Am. J. Physiol. Cell Physiol. 2002, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isse, N.; Ogawa, Y.; Tamura, N.; Masuzaki, H.; Mori, K.; Okazaki, T.; Satoh, N.; Shigemoto, M.; Yoshimasa, Y.; Nishi, S.; et al. Structural organization and chromosomal assignment of the human obese gene. J. Biol. Chem. 1995, 270, 27728–27733. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less is more. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Huvenne, H.; Le Beyec, J.; Pépin, D.; Alili, R.; Kherchiche, P.P.; Jeannic, E.; Frelut, M.L.; Lacorte, J.M.; Nicolino, M.; Viard, A.; et al. Seven novel deleterious LEPR mutations found in early-onset obesity: A Δexon6-8 shared by subjects from Reunion Island, France, suggests a founder effect. J. Clin. Endocrinol. Metab. 2015, 100, E757–E766. [Google Scholar] [CrossRef] [PubMed]
- Foucan, L.; Larifla, L.; Durand, E.; Rambhojan, C.; Armand, C.; Michel, C.T.; Billy, R.; Dhennin, V.; Graeve, D.F.; Rabearivelo, I.; et al. High prevalence of rare monogenic forms of obesity in obese guadeloupean afro-caribbean children. J. Clin. Endocrinol. Metab. 2018, 103, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of Leptin Deficiency, Inefficiency, and Leptin Receptors in Obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, F. Adipose tissue function and dysfunction: Organ cross talk and metabolic risk. Am. J. Physiol. Endocrinol. Metab. 2009, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghalandari, H.; Hosseini-Esfahani, F.; Mirmiran, P. The association of polymorphisms in leptin/leptin receptor genes and ghrelin/ghrelin receptor genes with overweight/obesity and the related metabolic disturbances: A review. Int. J. Endocrinol. Metab. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Hoffsted, J.; Eriksson, P.; Mottagui-Tabar, S.; Arner, P. A polymorphism in the leptin promoter region (-2548 G/A) influences gene expression and adipose tissue secretion of leptin. Horm. Metab. Res. 2002, 34, 355–359. [Google Scholar] [CrossRef]
- Şahin, S.; Rüstemoǧlu, A.; Tekcan, A.; Taşliyurt, T.; Güven, H.; Yiǧit, S. Investigation of associations between obesity and LEP G2548A and LEPR 668A/G polymorphisms in a Turkish population. Dis. Markers 2013, 35, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.H.; Say, Y.H. Leptin and leptin receptor gene polymorphisms and their association with plasma leptin levels and obesity in a multi-ethnic Malaysian suburban population. J. Physiol. Anthropol. 2014, 33. [Google Scholar] [CrossRef] [Green Version]
- Constantin, A.; Costache, G.; Sima, V.A.; Glavce, C.S.; Vladica, M.; Popov, D.L. Leptin G-2548A and leptin receptor Q223R gene polymorphisms are not associated with obesity in Romanian subjects. Biochem. Biophys. Res. Commun. 2010, 391, 282–286. [Google Scholar] [CrossRef]
- Shahid, A.; Rana, S.; Mahmood, S.; Saeed, S. Role of leptin G-2548A polymorphism in age- and gender-specific development of obesity. J. Biosci. 2015, 40, 521–530. [Google Scholar] [CrossRef]
- Hinuy, H.M.; Hirata, M.H.; Forti, N.; Diament, J.; Sampaio, M.F.; Armaganijan, D.; Salazar, L.A.; Hirata, R.D.C. Leptin G-2548A promoter polymorphism is associated with increased plasma leptin and BMI in Brazilian women. Arq. Bras. Endocrinol. Metabol. 2008, 52, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.M.; Jhang, J.Y.; Wu, S.; Teng, M.S.; Hsu, L.A.; Ko, Y.L. Modification effect of sex and obesity on the correlation of LEP polymorphisms with leptin levels in Taiwanese obese women. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef]
- Sahin, D.S.; Tumer, C.; Demir, C.; Celik, M.M.; Celik, M.; Ucar, E.; Gunesacar, R. Association with Leptin Gene c.-2548 GA Polymorphism, Serum Leptin Levels, and Body Mass Index in Turkish Obese Patients. Cell Biochem. Biophys. 2013, 65, 243–247. [Google Scholar] [CrossRef]
- Mammès, O.; Betoulle, D.; Aubert, R.; Herbeth, B.; Siest, G.; Fumeron, F. Association of the G-2548A polymorphism in the 5′ region of the LEP gene with overweight. Ann. Hum. Genet. 2000, 64, 391–394. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, L.H.; Xiao, Y.; Lu, M.Y.; Zhang, L.J.; Wang, Y. Association of leptin gene -2548 G/A polymorphism with obesity: A meta-analysis. Ann. Nutr. Metab. 2014, 64, 127–136. [Google Scholar] [CrossRef]
- Nesrine, Z.; Haithem, H.; Imen, B.; Fadoua, N.; Asma, O.; Fadhel, N.M.; Ali, B. Leptin and Leptin receptor polymorphisms, plasma Leptin levels and obesity in Tunisian volunteers. Int. J. Exp. Pathol. 2018, 99, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, U.; Kidd, E.; Malagamuwa, T.; Walley, A. Genetics of Severe Obesity. Curr. Diab. Rep. 2018, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, S.C.; Chung, W.K.; Wu-Peng, X.S.; Zhang, Y.; Liu, S.M.; Tartaglia, L.; Leibel, R.L. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 1996, 271, 994–996. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Volders, K.; Stanhope, R.; Heuschkel, R.; White, A.; Lank, E.; Keogh, J.; O’Rahilly, S.; Creemers, J.W.M. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. In Proceedings of the Journal of Clinical Endocrinology and Metabolism; Endocrine Society (USA): Washington, DC, USA, 2007; Volume 92, pp. 3369–3373. [Google Scholar]
- Matsuoka, N.; Ogawa, Y.; Hosoda, K.; Matsuda, J.; Masuzaki, H.; Miyawaki, T.; Azuma, N.; Natsui, K.; Nishimura, H.; Yoshimasa, Y.; et al. Human leptin receptor gene in obese Japanese subjects: Evidence against either obesity-causing mutations or association of sequence variants with obesity. Diabetologia 1997, 40, 1204–1210. [Google Scholar] [CrossRef]
- Foucan, L.; Bassien-Capsa, V.; Rambhojan, C.; Lacorte, J.M.; Larifla, L. Influence of K656N Polymorphism of the Leptin Receptor Gene on Obesity-Related Traits in Nondiabetic Afro-Caribbean Individuals. Metab. Syndr. Relat. Disord. 2019, 17, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.A.; Huang, S.; Walet, A.C.E.; Zhang, Z.C.; Leow, M.K.S.; Liu, M.; Sun, L. Adipocyte long-Noncoding RNA transcriptome analysis of obese mice identified lnc-Leptin, which regulates leptin. Diabetes 2018, 67, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Marsh, A.J.; Fontes, M.A.P.; Killinger, S.; Pawlak, D.B.; Polson, J.W.; Dampney, R.A.L. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 2003, 42, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minokoshi, Y.; Haque, M.S.; Shimazu, T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 1999, 48, 287–291. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Yu, S.; Jiang, Y.; Laque, A.; Schwartzenburg, C.; Morrison, C.D.; Derbenev, V.A.; Zsombok, A.; Münzberg, H. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol. Metab. 2014, 3, 681–693. [Google Scholar] [CrossRef]
- Harlan, S.M.; Morgan, D.A.; Agassandian, K.; Guo, D.F.; Cassell, M.D.; Sigmund, C.D.; Mark, A.L.; Rahmouni, K. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res. 2011, 108, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Gamber, K.; Greeley, S.; Silva, J.; Huntoon, N.; Leng, X.H.; Bjørbæk, C. Leptin-Dependent Control of Glucose Balance and Locomotor Activity by POMC Neurons. Cell Metab. 2009, 9, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.; Lemko, H.M.D.; Castorena, C.M.; Fujikawa, T.; Lee, S.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Holland, W.L.; Liu, C.; et al. POMC neurons expressing leptin receptors coordinate metabolic responses to fasting via suppression of leptin levels. eLife 2018, 7. [Google Scholar] [CrossRef]
- Balthasar, N.; Coppari, R.; McMinn, J.; Liu, S.M.; Lee, C.E.; Tang, V.; Kenny, C.D.; McGovern, R.A.; Chua, S.C.; Elmquist, J.K.; et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004, 42, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Sorrell, J.E.; Clegg, D.J.; Woods, S.C.; Seeley, R.J. The roles of leptin receptors on POMC neurons in the regulation of sex-specific energy homeostasis. Physiol. Behav. 2010, 100, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berglund, E.D.; Vianna, C.R.; Donato, J.; Kim, M.H.; Chuang, J.C.; Lee, C.E.; Lauzon, D.A.; Lin, P.; Brule, L.J.; Scott, M.M.; et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J. Clin. Investig. 2012, 122, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Carmo, J.M.; Da Silva, A.A.; Cai, Z.; Lin, S.; Dubinion, J.H.; Hall, J.E. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension 2011, 57, 918–926. [Google Scholar] [CrossRef]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin–BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.W.; Cannon, B.; Nedergaard, J. Leptin: Is it thermogenic? Endocr. Rev. 2019, 41, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.W.; Hoefig, C.S.; Abreu-Vieira, G.; de Jong, J.M.A.; Petrovic, N.; Mittag, J.; Cannon, B.; Nedergaard, J. Leptin Raises Defended Body Temperature without Activating Thermogenesis. Cell Rep. 2016, 14, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plum, L.; Rother, E.; Münzberg, H.; Wunderlich, F.T.; Morgan, D.A.; Hampel, B.; Shanabrough, M.; Janoschek, R.; Könner, A.C.; Alber, J.; et al. Enhanced Leptin-Stimulated Pi3k Activation in the CNS Promotes White Adipose Tissue Transdifferentiation. Cell Metab. 2007, 6, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Ruan, B.H.; Dietrich, M.O.; Liu, Z.W.; Zimmer, M.R.; Li, M.D.; Singh, J.P.; Zhang, K.; Yin, R.; Wu, J.; Horvath, T.L.; et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 2014, 159, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ge, J.; Cao, H.; Zhang, X.; Guo, Y.; Li, X.; Xia, B.; Yang, G.; Shi, X. Leptin Promotes White Adipocyte Browning by Inhibiting the Hh Signaling Pathway. Cells 2019, 8, 372. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Bellisari, A. Evolutionary origins of obesity. Obes. Rev. 2008, 9, 165–180. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Martínez-Sánchez, N.; Milbank, E.; Contreras, C. Incendiary leptin. Nutrients 2020, 12, 472. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Myers, M.G., Jr.; Heymsfield, S.B.; Haft, C.; Kahn, B.B.; Laughlin, M.; Leibel, R.L.; Tschöp, M.H.; Yanovski, J.A. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012, 15, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, A.L. Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R566. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Murphy, E.M.; Heymsfield, S.B.; Matthews, D.E.; Leibel, R.L. Low Dose Leptin Administration Reverses Effects of Sustained Weight-Reduction on Energy Expenditure and Circulating Concentrations of Thyroid Hormones. J. Clin. Endocrinol. Metab. 2002, 87, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Van Heek, M.; Compton, D.S.; France, C.F.; Tedesco, R.P.; Fawzi, A.B.; Graziano, M.P.; Sybertz, E.J.; Strader, C.D.; Davis, H.R. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Investig. 1997, 99, 385–390. [Google Scholar] [CrossRef]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Montez, J.M.; Soukas, A.; Asilmaz, E.; Fayzikhodjaeva, G.; Fantuzzi, G.; Friedman, J.M. Acute leptin deficiency, leptin resistance, and the physiologic response to leptin withdrawal. Proc. Natl. Acad. Sci. USA 2005, 102, 2537–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpace, P.J.; Matheny, M.; Tümer, N.; Cheng, K.Y.; Zhang, Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia 2005, 48, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Kastin, A.J.; Huang, W.; Jaspan, J.B.; Maness, L.M. Leptin enters the brain by a saturable system independent of insulin. Peptides 1996, 17, 305–311. [Google Scholar] [CrossRef]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.R.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Halaas, J.L.; Boozer, C.; Blair-West, J.; Fidahusein, N.; Denton, D.A.; Friedman, J.M. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8878–8883. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.W.; Peskind, E.; Raskind, M.; Boyko, E.J.; Porte, D. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nat. Med. 1996, 2, 589–593. [Google Scholar] [CrossRef]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, V.R. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Martin, R.L.; Perez, E.; He, Y.J.; Dawson, R.; Millard, W.J. Leptin resistance is associated with hypothalamic leptin receptor mRNA and protein downregulation. Metabolism. 2000, 49, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Guo, D.F.; Bugge, K.; Morgan, D.A.; Rahmouni, K.; Sheffield, V.C. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum. Mol. Genet. 2009, 18, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.F.; Cui, H.; Zhang, Q.; Morgan, D.A.; Thedens, D.R.; Nishimura, D.; Grobe, J.L.; Sheffield, V.C.; Rahmouni, K. The BBSome Controls Energy Homeostasis by Mediating the Transport of the Leptin Receptor to the Plasma Membrane. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef]
- Bjørbæk, C.; El-Haschimi, K.; Frantz, J.D.; Flier, J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999, 274, 30059–30065. [Google Scholar] [CrossRef] [Green Version]
- Pedroso, J.A.B.; Silveira, M.A.; Lima, L.B.; Furigo, I.C.; Zampieri, T.T.; Ramos-Lobo, A.M.; Buonfiglio, D.C.; Teixeira, P.D.S.; Frazão, R.; Donato, J. Changes in leptin signaling by SOCS3 modulate fasting-induced hyperphagia and weight regain in Mice. Endocrinology 2016, 157, 3901–3914. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Tremblay, M.L. Modulation of leptin resistance by protein tyrosine phosphatases. Cell Metab. 2012, 15, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Sharir, Y.; Kuperman, Y.; Apelblat, D.; Hertog, J.D.; Spiegel, I.; Knobler, H.; Elson, A. Protein tyrosine phosphatase alpha inhibits hypothalamic leptin receptor signaling and regulates body weight in vivo. FASEB J. 2019, 33, 5101–5111. [Google Scholar] [CrossRef]
- Mazor, R.; Friedmann-Morvinski, D.; Alsaigh, T.; Kleifeld, O.; Kistler, E.B.; Rousso-Noori, L.; Huang, C.; Li, J.B.; Verma, I.M.; Schmid-Schönbein, G.W. Cleavage of the leptin receptor by matrix metalloproteinase-2 promotes leptin resistance and obesity in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- De Git, K.C.G.; Adan, R.A.H. Leptin resistance in diet-induced obesity: The role of hypothalamic inflammation. Obes. Rev. 2015, 16, 207–224. [Google Scholar] [CrossRef]
- Lian, Y.; Zhao, F.; Wang, W. Central leptin resistance and hypothalamic inflammation are involved in letrozole-induced polycystic ovary syndrome rats. Biochem. Biophys. Res. Commun. 2016, 476, 306–312. [Google Scholar] [CrossRef]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Liu, G.; Guo, J.; Su, Z. Hypothalamic endoplasmic reticulum stress as a key mediator of obesity-induced leptin resistance. Obes. Rev. 2018, 19, 770–785. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G.; Ozcan, U. Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Hosoi, T.; Sasaki, M.; Miyahara, T.; Hashimoto, C.; Matsuo, S.; Yoshii, M.; Ozawa, K. Endoplasmic reticulum stress induces leptin resistance. Mol. Pharmacol. 2008, 74, 1610–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Farr, O.M.; Gavrieli, A.; Mantzoros, C.S. Leptin applications in 2015: What have we learned about leptin and obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, A.M.; Mantzoros, C.S. Drug Insight: The role of leptin in human physiology and pathophysiology—Emerging clinical applications. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Wangensteen, T.; Collins, S.; Kimber, W.; Matarese, G.; Keogh, J.M.; Lank, E.; Bottomley, B.; Lopez-Fernandez, J.; Ferraz-Amaro, I.; et al. Clinical and Molecular Genetic Spectrum of Congenital Deficiency of the Leptin Receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazen, I.; El-Gammal, M.; Abdel-Hamid, M.; Farooqi, I.S.; Amr, K. Homozygosity for a novel missense mutation in the leptin receptor gene (P316T) in two Egyptian cousins with severe early onset obesity. Mol. Genet. Metab. 2011, 102, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Hannema, S.E.; Wit, J.M.; Houdijk, M.E.C.A.M.; van Haeringen, A.; Bik, E.C.; Verkerk, A.J.M.H.; Uitterlinden, A.G.; Kant, S.G.; Oostdijk, W.; Bakker, E.; et al. Novel Leptin Receptor Mutations Identified in Two Girls with Severe Obesity Are Associated with Increased Bone Mineral Density. Horm. Res. Paediatr. 2016, 85, 412–420. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Manzoor, J.; Philippe, J.; Durand, E.; Arshad, M.; Sand, O.; Butt, T.A.; Falchi, M.; Arslan, M.; et al. Novel LEPR mutations in obese Pakistani children identified by PCR-based enrichment and next generation sequencing. Obesity 2014, 22, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Wabitsch, M.; Funcke, J.B.; Von Schnurbein, J.; Denzer, F.; Lahr, G.; Mazen, I.; El-Gammal, M.; Denzer, C.; Moss, A.; Debatin, K.M.; et al. Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J. Clin. Endocrinol. Metab. 2015, 100, 3227–3230. [Google Scholar] [CrossRef] [Green Version]
- Saeed, S.; Bonnefond, A.; Manzoor, J.; Shabir, F.; Ayesha, H.; Philippe, J.; Durand, E.; Crouch, H.; Sand, O.; Ali, M.; et al. Genetic variants in LEP, LEPR, and MC4R explain 30% of severe obesity in children from a consanguineous population. Obesity 2015, 23, 1687–1695. [Google Scholar] [CrossRef]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 214–215. [Google Scholar] [CrossRef]
- Saeed, S.; Butt, T.A.; Anwer, M.; Arslan, M.; Froguel, P. High prevalence of leptin and melanocortin-4 receptor gene mutations in children with severe obesity from Pakistani consanguineous families. Mol. Genet. Metab. 2012, 106, 121–126. [Google Scholar] [CrossRef]
- Mazen, I.; El-Gammal, M.; Abdel-Hamid, M.; Amr, K. A novel homozygous missense mutation of the leptin gene (N103K) in an obese Egyptian patient. Mol. Genet. Metab. 2009, 97, 305–308. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Heni, M.; Moss, A.; Von Schnurbein, J.; Fritsche, A.; Häring, H.U.; Farooqi, S.; Preissl, H.; Wabitsch, M. Leptin therapy in a congenital leptin-deficient patient leads to acute and long-term changes in homeostatic, reward, and food-related brain areas. J. Clin. Endocrinol. Metab. 2011, 96. [Google Scholar] [CrossRef] [Green Version]
- Yupanqui-Lozno, H.; Bastarrachea, R.A.; Yupanqui-Velazco, M.E.; Alvarez-Jaramillo, M.; Medina-Méndez, E.; Giraldo-Peña, A.P.; Arias-Serrano, A.; Torres-Forero, C.; Garcia-Ordoñez, A.M.; Mastronardi, C.A.; et al. Congenital leptin deficiency and leptin gene missense mutation found in two colombian sisters with severe obesity. Genes 2019, 10, 342. [Google Scholar] [CrossRef] [Green Version]
- Blüher, S.; Mantzoros, C.S. Leptin in humans: Lessons from translational research. Am. J. Clin. Nutr. 2009, 89, 991S–997S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.F.; Bargut, T.C.L.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Thermogenesis, fatty acid synthesis with oxidation, and inflammation in the brown adipose tissue of ob/ob (−/−) mice. Ann. Anat. 2017, 210, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, A.A.; Ravussin, Y.; Leibel, R.L.; LeDuc, C.A. Energy homeostasis in leptin deficient Lepob/ob mice. PLoS ONE 2017, 12, e0189784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozata, M.; Ozdemir, I.C.; Licinio, J. Human leptin deficiency caused by a missense mutation: Multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 1999, 84, 3686–3695. [Google Scholar] [CrossRef] [PubMed]
- Sadaf Farooqi, I.; O’Rahilly, S. Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef]
- Von Schnurbein, J.; Heni, M.; Moss, A.; Nagel, S.A.; MacHann, J.; Muehleder, H.; Debatin, K.M.; Farooqi, S.; Wabitsch, M. Rapid improvement of hepatic steatosis after initiation of leptin substitution in a leptin-deficient girl. Horm. Res. Paediatr. 2013, 79, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Betz, M.J.; Enerbäck, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab. 2019, 30, 706–719.e6. [Google Scholar] [CrossRef]
- Kotzbeck, P.; Giordano, A.; Mondini, E.; Murano, I.; Severi, I.; Venema, W.; Cecchini, M.P.; Kershaw, E.E.; Barbatelli, G.; Haemmerle, G.; et al. Brown adipose tissue whitening leads to brown adipocyte death and adipose tissue inflammation. J. Lipid Res. 2018, 59, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Banks, W. The Blood-Brain Barrier as a Cause of Obesity. Curr. Pharm. Des. 2008, 14, 1606–1614. [Google Scholar] [CrossRef]
- Kintscher, U.; Bramlage, P.; Paar, W.D.; Thoenes, M.; Unger, T. Irbesartan for the treatment of hypertension in patients with the metabolic syndrome: A sub analysis of the Treat to Target post authorization survey. Prospective observational, two armed study in 14,200 patients. Cardiovasc. Diabetol. 2007, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Fielitz, H.; Lau, M.; Geißler, C.; Werner, L.; Winkler, M.; Raasch, W. Preventing leptin resistance by blocking angiotensin II at1 receptors in diet-induced obese rats. Br. J. Pharmacol. 2015, 172, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza-Mello, V.; Gregório, B.M.; Cardoso-De-Lemos, F.S.; De Carvalho, L.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. Comparative effects of telmisartan, sitagliptin and metformin alone or in combination on obesity, insulin resistance, and liver and pancreas remodelling in C57BL/6 mice fed on a very high-fat diet. Clin. Sci. 2010, 119, 239–250. [Google Scholar] [CrossRef]
- Rawish, E.; Prevents Development of Obesity; Normalizes Hypothalamic Lipid Droplets; Nickel, L.; Schuster, F.; Stölting, I.; Frydrychowicz, A.; Saar, K.; Hübner, N.; Othman, A.; et al. Telmisartan prevents development of obesity and normalizes hypothalamic lipid droplets. J. Endocrinol. 2020, 244, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Schuster, F.; Huber, G.; Stölting, I.; Wing, E.E.; Saar, K.; Hübner, N.; Banks, W.A.; Raasch, W. Telmisartan prevents diet-induced obesity and preserves leptin transport across the blood-brain barrier in high-fat diet-fed mice. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1673–1689. [Google Scholar] [CrossRef]
- He, H.; Yang, D.; Ma, L.; Luo, Z.; Ma, S.; Feng, X.; Cao, T.; Yan, Z.; Liu, D.; Tepel, M.; et al. Telmisartan prevents weight gain and obesity through activation of peroxisome proliferator-activated receptor-δ-dependent pathways. Hypertension 2010, 55, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Penna-de-Carvalho, A.; Graus-Nunes, F.; Rabelo-Andrade, J.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Enhanced pan-peroxisome proliferator-activated receptor gene and protein expression in adipose tissue of diet-induced obese mice treated with telmisartan. Exp. Physiol. 2014, 99, 1663–1678. [Google Scholar] [CrossRef] [Green Version]
- Bochar, O.M.; Sklyarova, H.Y.; Faynyk, A.F.; Bochar, V.T.; Kuzminov, Y.B. The effect of therapy with olmesartan or telmisartan in patients with arterial hypertension combined with obesity. Wiad Lek 2020, 73, 321–324. [Google Scholar] [CrossRef]
- Nedogoda, V.S.; Ledyaeva, A.A.; Chumachok, V.E.; Tsoma, V.V.; Mazina, G.; Salasyuk, A.S.; Barykina, I.N. Randomized trial of perindopril, enalapril, losartan and telmisartan in overweight or obese patients with hypertension. Clin. Drug Investig. 2013, 33, 553–561. [Google Scholar] [CrossRef]
- Sudi, K.M.; Gallistl, S.; Weinhandl, G.; Muntean, W.; Borkenstein, M.H. Relationship between plasminogen activator inhibitor-1 antigen, leptin, and fat mass in obese children and adolescents. Metabolism. 2000, 49, 890–895. [Google Scholar] [CrossRef]
- Gur-Wahnon, D.; Mizrachi, T.; Maaravi-Pinto, F.Y.; Lourbopoulos, A.; Grigoriadis, N.; Higazi, A.A.R.; Brenner, T. The plasminogen activator system: Involvement in central nervous system inflammation and a potential site for therapeutic intervention. J. Neuroinflammation 2013, 10, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, M.K.; Mahalle, N.; Dutta, M.K. Adipokines (adiponectin and plasminogen activator inhhibitor-1) in metabolic syndrome. Indian J. Endocrinol. Metab. 2012, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, S.; Yamada, T.; Takahashi, K.; Dan, T.; Kaneko, K.; Kodama, S.; Asai, Y.; Munakata, Y.; Endo, A.; Sugawara, H.; et al. Inhibition of Plasminogen Activator Inhibitor-1 Activation Suppresses High Fat Diet-Induced Weight Gain via Alleviation of Hypothalamic Leptin Resistance. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Taeye, D.B.; Smith, L.H.; Vaughan, D.E. Plasminogen activator inhibitor-1: A common denominator in obesity, diabetes and cardiovascular disease. Curr. Opin. Pharmacol. 2005, 5, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, L.; Liu, Z.; Liu, Y.; Luo, M.; Chen, N.; Deng, X.; Luo, Y.; He, J.; Zhang, L.; et al. PAI-1 Exacerbates White Adipose Tissue Dysfunction and Metabolic Dysregulation in High Fat Diet-Induced Obesity. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Mackie, A.; Beussink-Nelson, L.; Kamide, C.E.; Henkel, A.S.; Place, A.T.; Eren, M.; Lloyd-Jones, D.; Shah, S.J.; Miyata, T.; et al. Abstract 179: Targeted Inhibition of Plasminogen Activator Inhibitor-1 Attenuates Weight Gain and Prevents Vascular Dysfunction Following a High Fat Diet|Circulation Research. Circ. Res. 2015, 117, A179. [Google Scholar]
- Levine, J.A.; Olivares, S.; Miyata, T.; Vaughan, D.E.; Henkel, A.S. Inhibition of PAI-1 Promotes Lipolysis and Enhances Weight Loss in Obese Mice. Obesity 2021, oby.23112. [Google Scholar] [CrossRef]
- Schéle, E.; Grahnemo, L.; Anesten, F.; Halleń, A.; Bäckhed, F.; Jansson, J.O. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 2013, 154, 3643–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Da Silva, T.F.; Casarotti, S.N.; de Oliveira, G.L.V.; Penna, A.L.B. The impact of probiotics, prebiotics, and synbiotics on the biochemical, clinical, and immunological markers, as well as on the gut microbiota of obese hosts. Crit. Rev. Food Sci. Nutr. 2020, 61, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, Z.; Feng, F.; Wu, T.; Luo, D.; Hu, C.; Sun, C. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. Int. J. Obes. 2018, 42, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Licinio, J.; Caglayan, S.; Ozata, M.; Yildiz, B.O.; De Miranda, P.B.; O’Kirwan, F.; Whitby, R.; Liang, L.; Cohen, P.; Bhasin, S.; et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl. Acad. Sci. USA 2004, 101, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Galgani, J.E.; Greenway, F.L.; Caglayan, S.; Wong, M.L.; Licinio, J.; Ravussin, E. Leptin replacement prevents weight loss-induced metabolic adaptation in congenital leptin-deficient patients. J. Clin. Endocrinol. Metab. 2010, 95, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Jebb, S.A.; Langmack, G.; Lawrence, E.; Cheetham, C.H.; Prentice, A.M.; Hughes, I.A.; McCamish, M.A.; O’Rahilly, S. Effects of Recombinant Leptin Therapy in a Child with Congenital Leptin Deficiency. N. Engl. J. Med. 1999, 341, 879–884. [Google Scholar] [CrossRef]
- Gibson, W.T.; Farooqi, I.S.; Moreau, M.; DePaoli, A.M.; Lawrence, E.; O’Rahilly, S.; Trussell, R.A. Congenital leptin deficiency due to homozygosity for the Δ133G mutation: Report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab. 2004, 89, 4821–4826. [Google Scholar] [CrossRef] [Green Version]
- Berman, S.M.; Paz-Filho, G.; Wong, M.L.; Kohno, M.; Licinio, J.; London, E.D. Effects of leptin deficiency and replacement on cerebellar response to food-related cues. Cerebellum 2013, 12, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Santos, C.; Zhang, Z.; Morgan, D.A.; Guo, D.F.; Russo, A.F.; Rahmouni, K. Amylin acts in the central nervous system to increase sympathetic nerve activity. Endocrinology 2013, 154, 2481–2488. [Google Scholar] [CrossRef] [Green Version]
- Lutz, T.A.; Coester, B.; Whiting, L.; Dunn-Meynell, A.A.; Boyle, C.N.; Bouret, S.G.; Levin, B.E.; Le Foll, C. Amylin selectively signals onto POMC neurons in the arcuate nucleus of the hypothalamus. Diabetes 2018, 67, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Turek, V.F.; Trevaskis, J.L.; Levin, B.E.; Dunn-Meynell, A.A.; Irani, B.; Gu, G.; Wittmer, C.; Griffin, P.S.; Vu, C.; Parkes, D.G.; et al. Mechanisms of Amylin/Leptin Synergy in Rodent Models. Endocrinology 2010, 151, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, J.L.; Coffey, T.; Cole, R.; Lei, C.; Wittmer, C.; Walsh, B.; Weyer, C.; Koda, J.; Baron, A.D.; Parkes, D.G.; et al. Amylin-Mediated Restoration of Leptin Responsiveness in Diet-Induced Obesity: Magnitude and Mechanisms. Endocrinology 2008, 149, 5679–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced Weight Loss With Pramlintide/Metreleptin: An Integrated Neurohormonal Approach to Obesity Pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonfiglio, D.; Tchio, C.; Furigo, I.; Donato, J.; Baba, K.; Cipolla-Neto, J.; Tosini, G. Removing melatonin receptor type 1 signaling leads to selective leptin resistance in the arcuate nucleus. J. Pineal Res. 2019, 67. [Google Scholar] [CrossRef]
- Favero, G.; Stacchiotti, A.; Castrezzati, S.; Bonomini, F.; Albanese, M.; Rezzani, R.; Rodella, L.F. Melatonin reduces obesity and restores adipokine patterns and metabolism in obese (ob/ob) mice. Nutr. Res. 2015, 35, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Aranda, A.; Fernández-Vázquez, G.; Campos, D.; Tassi, M.; Velasco-Perez, L.; Tan, D.-X.; Reiter, R.J.; Agil, A. Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J. Pineal Res. 2013, 55, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Blüher, S.; Ziotopoulou, M.; Bullen, J.W.; Moschos, S.J.; Ungsunan, L.; Kokkotou, E.; Maratos-Flier, E.; Mantzoros, C.S. Responsiveness to Peripherally Administered Melanocortins in Lean and Obese Mice. Diabetes 2004, 53, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Clément, K.; van den Akker, E.; Argente, J.; Bahm, A.; Chung, W.K.; Connors, H.; De Waele, K.; Farooqi, I.S.; Gonneau-Lejeune, J.; Gordon, G.; et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: Single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020, 8, 960–970. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genchi, V.A.; D’Oria, R.; Palma, G.; Caccioppoli, C.; Cignarelli, A.; Natalicchio, A.; Laviola, L.; Giorgino, F.; Perrini, S. Impaired Leptin Signalling in Obesity: Is Leptin a New Thermolipokine? Int. J. Mol. Sci. 2021, 22, 6445. https://doi.org/10.3390/ijms22126445
Genchi VA, D’Oria R, Palma G, Caccioppoli C, Cignarelli A, Natalicchio A, Laviola L, Giorgino F, Perrini S. Impaired Leptin Signalling in Obesity: Is Leptin a New Thermolipokine? International Journal of Molecular Sciences. 2021; 22(12):6445. https://doi.org/10.3390/ijms22126445
Chicago/Turabian StyleGenchi, Valentina Annamaria, Rossella D’Oria, Giuseppe Palma, Cristina Caccioppoli, Angelo Cignarelli, Annalisa Natalicchio, Luigi Laviola, Francesco Giorgino, and Sebastio Perrini. 2021. "Impaired Leptin Signalling in Obesity: Is Leptin a New Thermolipokine?" International Journal of Molecular Sciences 22, no. 12: 6445. https://doi.org/10.3390/ijms22126445