
Proteomic Profiling of BRAFV600E Mutant Colon Cancer Cells Reveals the Involvement of Nucleophosmin/c-Myc Axis in Modulating the Response and Resistance to BRAF Inhibition by Vemurafenib

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Proteomic Profiling of Colon Cancer Cell Lines Differing in BRAF Mutation Status
2.2. Bioinformatics Analysis of Proteomic Data
2.2.1. Functional and Pathway Enrichment Analysis
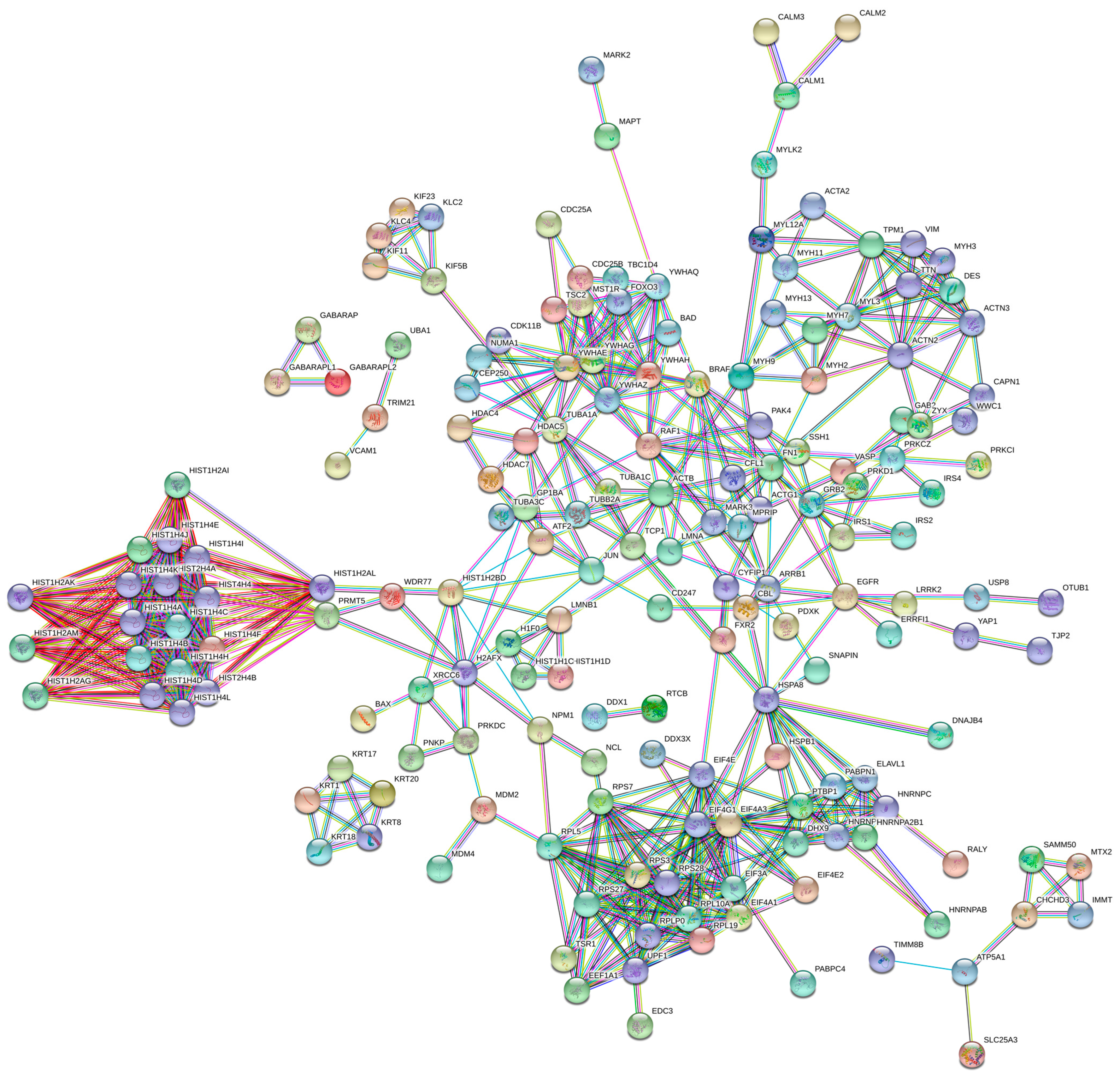
2.2.2. PPI Network Construction and Module Analysis
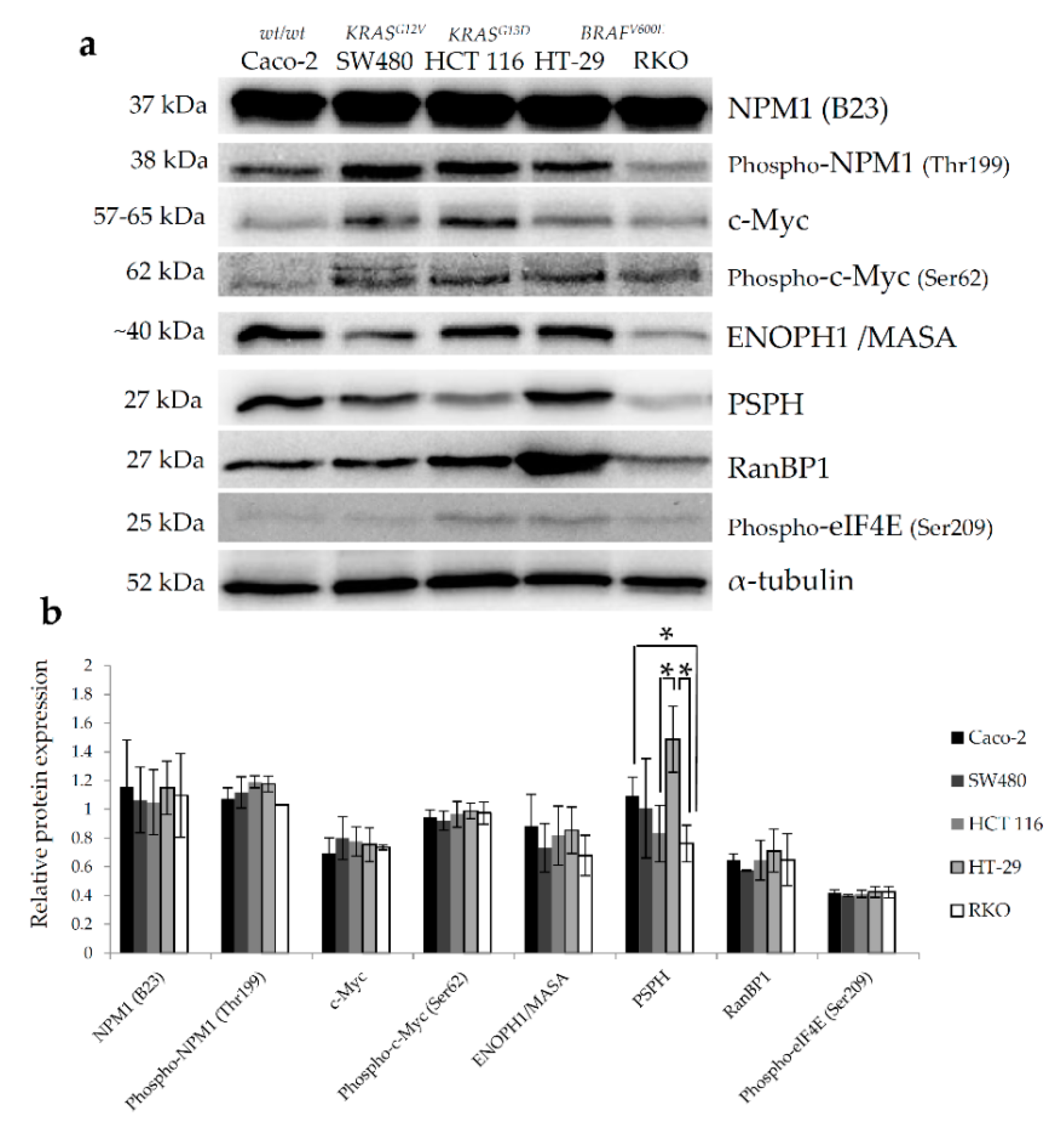
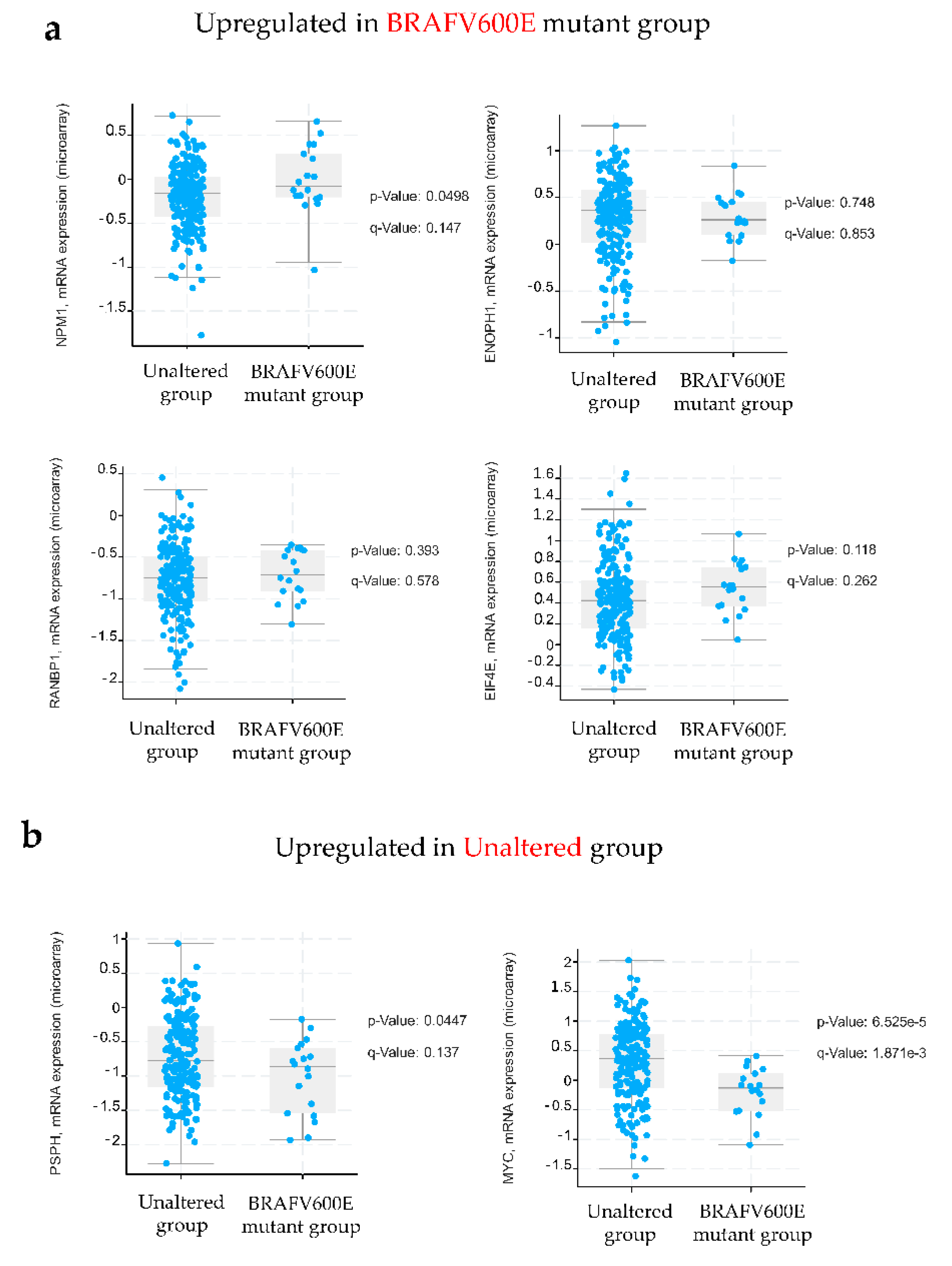
2.3. In Vitro and In Silico Validation of Proteomic Data Reveals Increased Abundance of Nucleophosmin in BRAFV600E-Mutated Colon Cancer
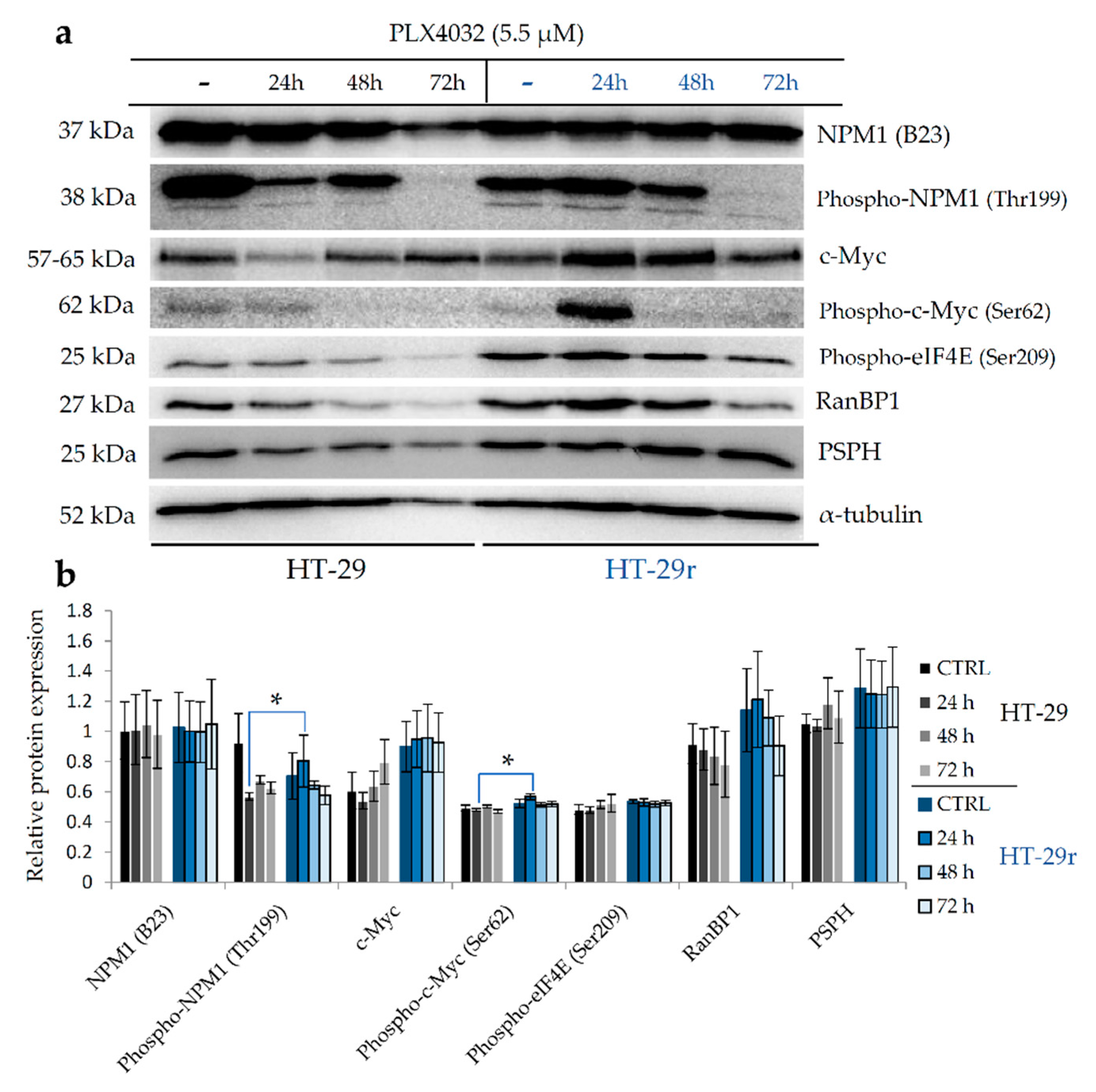
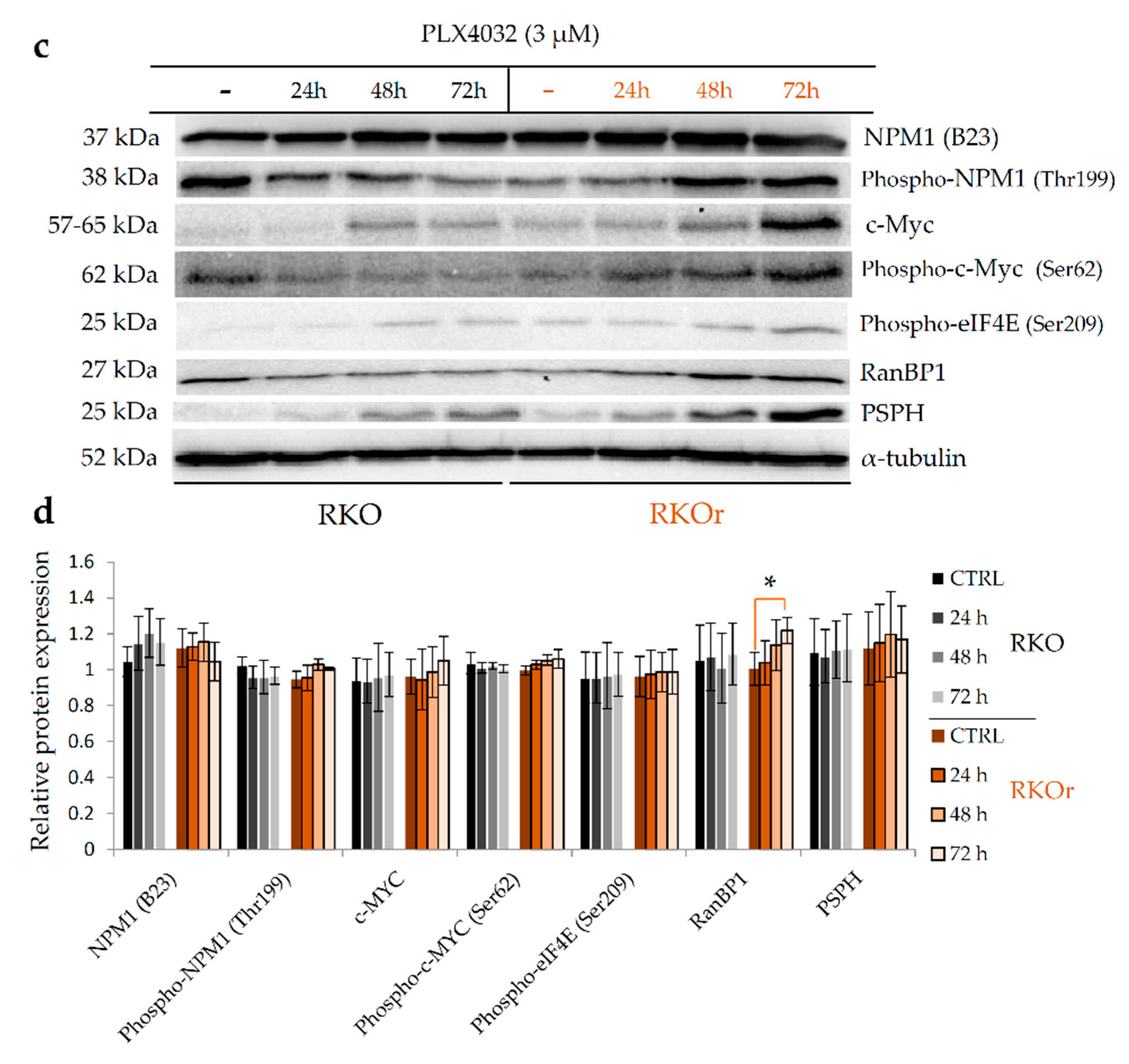
2.4. Nucleophosmin and c-Myc Mediate Resistance to BRAFV600E Inhibition by PLX4032 in BRAFV600E Mutant Colon Cancer Cell Lines
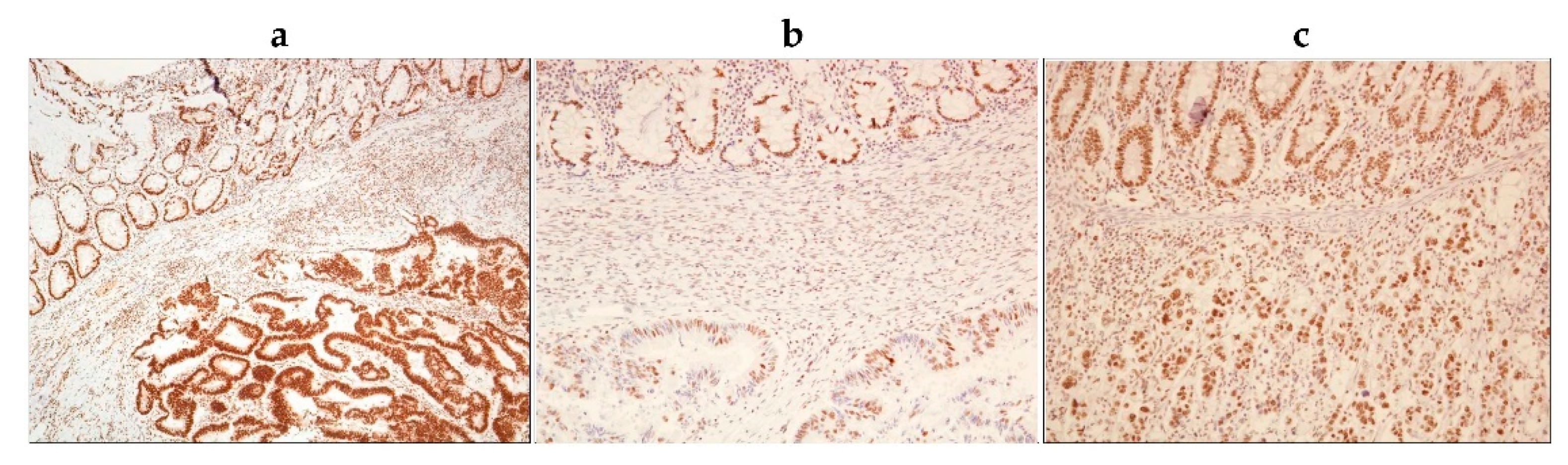
2.5. Tumour Tissues from BRAF-Mutated Colon Adenocarcinoma Patients Have Significantly Increased Abundance of Cytoplasmic p-NPM1 (Thr199)
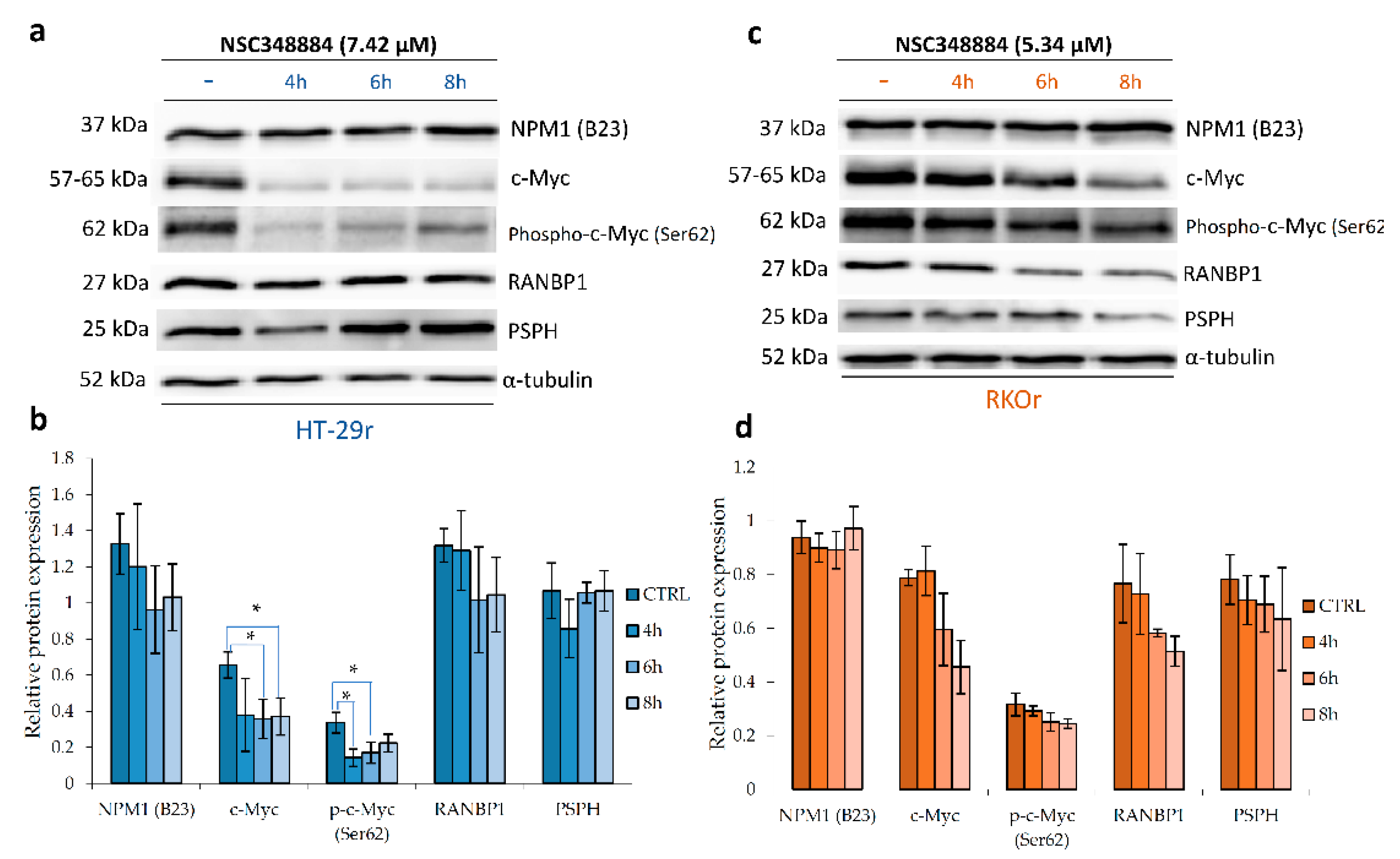
2.6. Pharmacological Inhibition of Oncogenic Nucleophosmin/MYC Axis Restores Sensitivity of BRAF Mutant Colon Cancer Cells to PLX4032
3. Discussion
4. Materials and Methods
4.1. Cell Culturing and Development of Vemurafenib-Resistant Colon Cancer Cell Lines
4.2. Cell Viability Assay
4.3. Two-Dimensional Gel Electrophoresis and Image Analysis
4.4. MALDI-TOF/TOF Mass Spectrometry Analysis
4.5. Bioinformatic Analyses
4.6. Human Tissue Samples
4.7. Immunohistochemistry Analysis
4.8. Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Morris, V.K.; Bekaii-Saab, T. Improvements in Clinical Outcomes for BRAFV600E -Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2020, 26, 4435–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, E.; Pretta, A.; Impera, V.; Mariani, S.; Giampieri, R.; Casula, L.; Pusceddu, V.; Coni, P.; Fanni, D.; Puzzoni, M.; et al. BRAF-Mutant Colorectal Cancer, a Different Breed Evolving. Expert Rev. Mol. Diagn. 2018, 18, 499–512. [Google Scholar] [CrossRef]
- Barras, D.; Missiaglia, E.; Wirapati, P.; Sieber, O.M.; Jorissen, R.N.; Love, C.; Molloy, P.L.; Jones, I.T.; McLaughlin, S.; Gibbs, P.; et al. BRAF V600E Mutant Colorectal Cancer Subtypes Based on Gene Expression. Clin Cancer Res 2017, 23, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Simon, I.; Moreno, V.; Roepman, P.; Tabernero, J.; Snel, M.; van’t Veer, L.; Salazar, R.; Bernards, R.; Capella, G. A Combined Oncogenic Pathway Signature of BRAF, KRAS and PI3KCA Mutation Improves Colorectal Cancer Classification and Cetuximab Treatment Prediction. Gut 2013, 62, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici, V.; Budinska, E.; Tejpar, S.; Weinrich, S.; Estrella, H.; Hodgson, G.; Van Cutsem, E.; Xie, T.; Bosman, F.T.; Roth, A.D.; et al. Identification of a Poor-Prognosis BRAF-Mutant-like Population of Patients with Colon Cancer. J. Clin. Oncol. 2012, 30, 1288–1295. [Google Scholar] [CrossRef]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.-J.; Kang, H.C.; Jang, S.-G.; Kim, K.; Ahn, S.-A.; Yoon, H.-J.; Yoon, S.N.; Park, J.-G. Oligonucleotide Microarray Analysis of Distinct Gene Expression Patterns in Colorectal Cancer Tissues Harboring BRAF and K- Ras Mutations. Carcinogenesis 2006, 27, 392–404. [Google Scholar] [CrossRef]
- San Lucas, F.A.; Fowler, J.; Chang, K.; Kopetz, S.; Vilar, E.; Scheet, P. Cancer in silico Drug Discovery: A Systems Biology Tool for Identifying Candidate Drugs to Target Specific Molecular Tumor Subtypes. Mol. Cancer 2014, 13, 3230–3240. [Google Scholar] [CrossRef] [Green Version]
- Mossmann, D.; Park, S.; Hall, M.N. MTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting MTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- Fritsche-Guenther, R.; Zasada, C.; Mastrobuoni, G.; Royla, N.; Rainer, R.; Roßner, F.; Pietzke, M.; Klipp, E.; Sers, C.; Kempa, S. Alterations of MTOR Signaling Impact Metabolic Stress Resistance in Colorectal Carcinomas with BRAF and KRAS Mutations. Sci. Rep. 2018, 8, 9204. [Google Scholar] [CrossRef]
- Coffee, E.M.; Faber, A.C.; Roper, J.; Sinnamon, M.J.; Goel, G.; Keung, L.; Wang, W.V.; Vecchione, L.; de Vriendt, V.; Weinstein, B.J.; et al. Concomitant BRAF and PI3K/MTOR Blockade Is Required for Effective Treatment of BRAFV600E Colorectal Cancer. Clin. Cancer Res. 2013, 19, 2688–2698. [Google Scholar] [CrossRef] [Green Version]
- Zeller, K.I.; Haggerty, T.J.; Barrett, J.F.; Guo, Q.; Wonsey, D.R.; Dang, C.V. Characterization of Nucleophosmin (B23) as a Myc Target by Scanning Chromatin Immunoprecipitation. J. Biol. Chem. 2001, 276, 48285–48291. [Google Scholar] [CrossRef] [Green Version]
- Marinkovic, D.; Marinkovic, T.; Kokai, E.; Barth, T.; Möller, P.; Wirth, T. Identification of Novel Myc Target Genes with a Potential Role in Lymphomagenesis. Nucleic Acids Res. 2004, 32, 5368–5378. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.V. The Role of C- Myc in Regulation of Translation Initiation. Oncogene 2004, 23, 3217–3221. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. CMyc-Mediated Activation of Serine Biosynthesis Pathway Is Critical for Cancer Progression under Nutrient Deprivation Conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, N.; Nagano, H.; Tanaka, T. The Role of Tumor Suppressor P53 in Metabolism and Energy Regulation, and Its Implication in Cancer and Lifestyle-Related Diseases. Endocr. J. 2019, 66, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Pascal, L.E.; True, L.D.; Campbell, D.S.; Deutsch, E.W.; Risk, M.; Coleman, I.M.; Eichner, L.J.; Nelson, P.S.; Liu, A.Y. Correlation of MRNA and Protein Levels: Cell Type-Specific Gene Expression of Cluster Designation Antigens in the Prostate. BMC Genom. 2008, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, Y.; Horn, H.F.; Kawamura, K.; Tarapore, P.; Fukasawa, K. Specific Phosphorylation of Nucleophosmin on Thr199 by Cyclin- Dependent Kinase 2-Cyclin E and Its Role in Centrosome Duplication. J. Biol. Chem. 2001, 276, 21529–21537. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Budhu, A.; Forgues, M.; Wang, X.W. Temporal and Spatial Control of Nucleophosmin by the Ran–Crm1 Complex in Centrosome Duplication. Nat. Cell Biol. 2005, 7, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Shakalya, K.; Stejskal, A.; Goldman, A.; Beeck, S.; Cooke, L.; Mahadevan, D. NSC348884, a Nucleophosmin Inhibitor Disrupts Oligomer Formation and Induces Apoptosis in Human Cancer Cells. Oncogene 2008, 27, 4210–4220. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.-H.; Wang, Y.-Q.; Yu, Z.-Y.; Hu, L.-N.; Ou, T.-M.; Chen, S.-B.; Huang, Z.-S.; Tan, J.-H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Rochlitz, C.F.; Heide, I.; Thiede, C.; Herrmann, R.; de Kant, E. Evidence for a Mutual Regulation of P53 and C-Myc Expression in Human Colorectal Cancer Metastases. Ann. Oncol. 1995, 6, 981–986. [Google Scholar] [CrossRef]
- Frazier, M.W.; He, X.; Wang, J.; Gu, Z.; Cleveland, J.L.; Zambetti, G.P. Activation of C-Myc Gene Expression by Tumor-Derived P53 Mutants Requires a Discrete C-Terminal Domain. Mol. Cell Biol. 1998, 18, 3735–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, T.; Oikonomou, E.; Kosmidou, V.; Makrodouli, E.; Bantounas, I.; Avlonitis, S.; Zografos, G.; Pintzas, A. A Molecular Signature for Oncogenic BRAF in Human Colon Cancer Cells Is Revealed by Microarray Analysis. Curr. Cancer Drug Targets 2012, 12, 873–898. [Google Scholar] [CrossRef]
- Klein, R.M.; Spofford, L.S.; Abel, E.V.; Ortiz, A.; Aplin, A.E. B-RAF Regulation of Rnd3 Participates in Actin Cytoskeletal and Focal Adhesion Organization. Mol. Biol. Cell 2008, 19, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.-H.; Lee, J.-K.; Jung, E.; Kim, J. Actin Remodeling Confers BRAF Inhibitor Resistance to Melanoma Cells through YAP/TAZ Activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.C.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol. Cell Proteom. 2016, 15, 2924–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.Y.; Kim, H.M.; Jung, W.-H.; Koo, J.S. Expression of Serine/Glycine Metabolism-Related Proteins Is Different According to the Thyroid Cancer Subtype. J. Transl. Med. 2016, 14, 168. [Google Scholar] [CrossRef] [Green Version]
- Ross, K.C.; Andrews, A.J.; Marion, C.D.; Yen, T.J.; Bhattacharjee, V. Identification of the Serine Biosynthesis Pathway as a Critical Component of BRAF Inhibitor Resistance of Melanoma, Pancreatic, and Non-Small Cell Lung Cancer Cells. Mol. Cancer 2017, 16, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, F.; Zhang, X.-F.; Qi, L.-S.; Yang, L.; Guo, H.; Zhang, N. Expression of Nucleophosmin/NPM1 Correlates with Migration and Invasiveness of Colon Cancer Cells. J. Biomed. Sci. 2012, 19, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesmann, N.; Gieringer, R.; Grus, F.; Brieger, J. Phosphoproteome Profiling Reveals Multifunctional Protein NPM1 as Part of the Irradiation Response of Tumor Cells. Transl. Oncol. 2019, 12, 308–319. [Google Scholar] [CrossRef]
- Chartier, N.T.; Oddou, C.I.; Lainé, M.G.; Ducarouge, B.; Marie, C.A.; Block, M.R.; Jacquier-Sarlin, M.R. Cyclin-Dependent Kinase 2/Cyclin E Complex Is Involved in P120 Catenin (P120ctn)-Dependent Cell Growth Control: A New Role for P120ctn in Cancer. Cancer Res. 2007, 67, 9781–9790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, L.E.; Bleiler, M.; Giardina, C. A Look into Centrosome Abnormalities in Colon Cancer Cells, How They Arise and How They Might Be Targeted Therapeutically. Biochem. Pharm. 2018, 147, 1–8. [Google Scholar] [CrossRef]
- Zhang, M.S.; Arnaoutov, A.; Dasso, M. RanBP1 Governs Spindle Assembly by Defining Mitotic Ran-GTP Production. Dev. Cell 2014, 31, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, A.; Ciciarello, M.; Mangiacasale, R.; Roscioli, E.; Rensen, W.M.; Lavia, P. RANBP1 Localizes a Subset of Mitotic Regulatory Factors on Spindle Microtubules and Regulates Chromosome Segregation in Human Cells. J. Cell Sci. 2007, 120, 3748–3761. [Google Scholar] [CrossRef] [Green Version]
- López, D.J.; Rodríguez, J.A.; Bañuelos, S. Nucleophosmin, a Multifunctional Nucleolar Organizer with a Role in DNA Repair. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140532. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Boone, D.; Hann, S.R. Nucleophosmin Interacts Directly with C-Myc and Controls c-Myc-Induced Hyperproliferation and Transformation. Proc. Natl. Acad. Sci. USA 2008, 105, 18794–18799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hann, S.R. Nucleophosmin Is Essential for C-Myc Nucleolar Localization and c-Myc-Mediated RDNA Transcription. Oncogene 2013, 32, 1988–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, K.R.; Crawford, L.; Tsui, E.; Manchester, H.E.; Maertens, O.; Liu, X.; Liberti, M.V.; Magpusao, A.N.; Stein, E.M.; Tingley, J.P.; et al. Melanoma Therapeutic Strategies That Select against Resistance by Exploiting MYC-Driven Evolutionary Convergence. Cell Rep. 2017, 21, 2796–2812. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Suman, S.; Ranjan, R. Network Analysis Reveals Potential Markers for Pediatric Adrenocortical Carcinoma. OTT 2016, 9, 4569–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot Number | Accession ID | Protein Name p-Value (HT-29 vs. Caco-2; HT-29 vs. SW480) | Molecular Weight (kDa) | Peptide Matches | Sequence Coverage (%) | SCORE |
|---|---|---|---|---|---|---|
| 160 | MIC60_HUMAN | MICOS complex subunit MIC60 (0.002; 0.044) | 83.60 | 4 | 10.00 | 307.41 |
| 517 | K1C20_HUMAN | Keratin, type I cytoskeletal 20 (0.001; 0.001) | 48.50 | 4 | 9.90 | 145.21 |
| 678 | ROAA_HUMAN | Heterogeneous nuclear ribonucleoprotein A/B (0.010; 0.011) | 36.20 | 2 | 3.90 | 60.25 |
| 731 | NPM_HUMAN | Nucleophosmin (0.028; 0.016) | 32.60 | 4 | 16.30 | 191.18 |
| 717 | K2C1_HUMAN | Keratin, type II cytoskeletal 1 (0.009; 0.003) | 66.00 | 1 | 2.50 | 44.11 |
| 925 | TPM1_HUMAN | Tropomyosin alpha-1 chain (0.030; 0.001) | 32.70 | 2 | 8.80 | 79.33 |
| 983 | RANG_HUMAN | Ran-specific GTPase-activating protein (0.001; 0.0003) | 23.47 | 16 | 47.00 | 76.00 |
| 996 | UCHL3_HUMAN | Ubiquitin carboxyl-terminal hydrolase isozyme L3 (0.039; 0.023) | 26.34 | 18 | 70.00 | 178.00 |
| 1191 | ARI5B_HUMAN | AT-rich interactive domain-containing protein 5B (0.006; 0.002) | 133.43 | 14 | 17.00 | 37.00 |
| 985 | MIXTURE 1 SERB_HUMAN IF4E_HUMAN | Phosphoserine phosphatase Eukaryotic translation initiation factor 4E (0.040; 0.030) | 25.16 25.31 | 16 14 | 62.00 48.00 | 100.00 85.00 |
| 961 | YWHAZ _HUMAN | 14-3-3 protein theta (0.043;0.061) | 28.032 | 13 | 40.00 | 53.00 |
| 1162 | NMES1_HUMAN | Normal mucosa of oesophagus-specific gene 1 protein (0.005; 0.004) | 9.611 | 4 | 55.00 | 37.00 |
| 855 | ENOPH_HUMAN | Enolase-phosphatase E1 (0.027; 0.0978) | 28.90 | 6 | 34.10 | 378.18 |
| 971 | YWHAQ_HUMAN | 14-3-3 protein zeta/delta (0.073; 0.043) | 27.899 | 21 | 38.00 | 114 |
| 655 | CAPG_HUMAN | Macrophage-capping protein (0.0003; 0.077) | 38.50 | 4 | 10.30 | 223.83 |
| Spot Number | Accession ID | Protein Name p-Value (HT-29 vs. Caco-2; HT-29 vs. SW480) | Molecular Weight (kDa) | Peptide Matches | Sequence Coverage (%) | SCORE |
|---|---|---|---|---|---|---|
| 183 | MOES_HUMAN | Moesin (0.001; 0.005) | 67.80 | 3 | 3.50 | 119.80 |
| 631 | K1C19_HUMAN | Keratin, type I cytoskeletal 19 (<0.0001; <0.0001) | 44.10 | 2 | 7.80 | 142.88 |
| 643 | K1C19_HUMAN | Keratin, type I cytoskeletal 19 (<0.0001; <0.0001) | 44.10 | 9 | 27.30 | 527.99 |
| 632 | K1C19_HUMAN | Keratin, type I cytoskeletal 19 (<0.0001; <0.0001) | 44.10 | 11 | 36.00 | 856.99 |
| 840 | ANXA3_HUMAN | Annexin A3 (0.0001; 0.0002) | 36.40 | 2 | 13.90 | 149.55 |
| 1048 | PCNA_HUMAN | Proliferating cell nuclear antigen (0.016; 0.002) | 29.09 | 13 | 59.00 | 97.00 |
| 1171 | MIXTURE 1 DBNL_HUMAN TBA1B_HUMAN | Drebrin-like protein Tubulin alpha-1B chain (0.034; 0.001) | 48.46 50.80 | 17 16 | 34.00 39.00 | 88.00 86.00 |
| 1209 | T191A_HUMAN | Transmembrane protein 191A (0.001; 0.014) | 18.06 | 4 | 33.00 | 30.00 |
| 1147 | EPHA6_HUMAN | Ephrin type-A receptor 6 (0.0772; <0.0001) | 117.901 | 8 | 13.00 | 40 |
| 794 | HSP7C_HUMAN | Heat shock cognate 71 kDa protein (0.026; 0.131) | 70.90 | 3 | 7.60 | 214.36 |
| Pathway Identifier | Pathway Name | #Entities Found | #Entities Total | Entities p-Value | Entities FDR | Submitted Entities Found |
|---|---|---|---|---|---|---|
| R-HSA-9614399 | Regulation of localization of FOXO transcription factors | 2 | 12 | 1.44 × 10−4 | 0.009 | YWHAQ; YWHAZ |
| R-HSA-75035 | Chk1/Chk2(Cds1) mediated inactivation of Cyclin B: Cdk1 complex | 2 | 13 | 1.69 × 10−4 | 0.009 | YWHAQ; YWHAZ |
| R-HSA-111447 | Activation of BAD and translocation to mitochondria | 2 | 15 | 2.25 × 10−4 | 0.009 | YWHAQ; YWHAZ |
| R-HSA-114452 | Activation of BH3-only proteins | 2 | 30 | 8.88 × 10−4 | 0.026 | YWHAQ; YWHAZ |
| R-HSA-390522 | Striated Muscle Contraction | 2 | 36 | 0.001 | 0.028 | TPM1 |
| R-HSA-445355 | Smooth Muscle Contraction | 2 | 39 | 0.001 | 0.028 | TPM1 |
| R-HSA-109606 | Intrinsic Pathway for Apoptosis | 2 | 55 | 0.003 | 0.041 | YWHAQ; YWHAZ |
| R-HSA-5625740 | RHO GTPases activate PKNs | 2 | 63 | 0.004 | 0.046 | YWHAQ; YWHAZ |
| R-HSA-9614085 | FOXO-mediated transcription | 2 | 66 | 0.004 | 0.046 | YWHAQ; YWHAZ |
| R-HSA-1445148 | Translocation of SLC2A4 (GLUT4) to the plasma membrane | 2 | 72 | 0.005 | 0.049 | YWHAQ; YWHAZ |
| R-HSA-69473 | G2/M DNA damage checkpoint | 2 | 78 | 0.006 | 0.059 | YWHAQ; YWHAZ |
| R-HSA-5628897 | TP53 Regulates Metabolic Genes | 2 | 88 | 0.007 | 0.059 | YWHAQ; YWHAZ |
| R-HSA-8869496 | TFAP2A acts as a transcriptional repressor during retinoic acid induced cell differentiation | 1 | 5 | 0.007 | 0.059 | NPM1 |
| R-HSA-1237112 | Methionine salvage pathway | 1 | 6 | 0.009 | 0.061 | ENOPH1 |
| R-HSA-977347 | Serine biosynthesis | 1 | 9 | 0.013 | 0.078 | PSPH |
| R-HSA-1640170 | Cell Cycle | 4 | 670 | 0.015 | 0.078 | NPM1; YWHAQ; CAPG; YWHAZ |
| R-HSA-3700989 | Transcriptional Regulation by TP53 | 3 | 367 | 0.016 | 0.078 | NPM1; YWHAQ; YWHAZ |
| R-HSA-2514853 | Condensation of Prometaphase Chromosomes | 1 | 11 | 0.016 | 0.078 | CAPG |
| R-HSA-9013700 | NOTCH4 Activation and Transmission of Signal to the Nucleus | 1 | 11 | 0.016 | 0.078 | YWHAZ |
| R-HSA-69481 | G2/M Checkpoints | 2 | 151 | 0.020 | 0.078 | YWHAQ; YWHAZ |
| R-HSA-392517 | Rap1 signaling | 1 | 16 | 0.023 | 0.078 | YWHAZ |
| R-HSA-450604 | KSRP (KHSRP) binds and destabilizes mRNA | 1 | 17 | 0.025 | 0.078 | YWHAZ |
| R-HSA-109581 | Apoptosis | 2 | 182 | 0.029 | 0.078 | YWHAQ; YWHAZ |
| R-HSA-6804115 | TP53 regulates transcription of additional cell cycle genes whose exact role in the p53 pathway remain uncertain | 1 | 21 | 0.030 | 0.078 | NPM1 |
| R-HSA-397014 | Muscle contraction | 2 | 196 | 0.033 | 0.078 | TPM1 |
| R-HSA-166208 | mTORC1-mediated signaling | 1 | 24 | 0.035 | 0.078 | EIF4E |
| R-HSA-429947 | Deadenylation of mRNA | 1 | 25 | 0.036 | 0.078 | EIF4E |
| R-HSA-3214842 | HDMs demethylate histones | 1 | 26 | 0.037 | 0.078 | ARID5B |
| R-HSA-5357801 | Programmed Cell Death | 2 | 217 | 0.040 | 0.078 | YWHAQ; YWHAZ |
| R-HSA-1614635 | Sulfur amino acid metabolism | 1 | 28 | 0.040 | 0.078 | ENOPH1 |
| R-HSA-8866652 | Synthesis of active ubiquitin: roles of E1 and E2 enzymes | 1 | 30 | 0.043 | 0.078 | UCHL3 |
| R-HSA-8949613 | Cristae formation | 1 | 31 | 0.045 | 0.078 | IMMT |
| R-HSA-9013424 | RHOV GTPase cycle | 1 | 33 | 0.047 | 0.078 | TPM1 |
| BRAF mut | BRAF wt | ||
|---|---|---|---|
| N = 7 (33%) | N = 14 (67%) | ||
| Staining intensity | mean | mean | p-value |
| Nuclear | 2 | 1.43 | 0.009 |
| Cytoplasmic | 1.86 | 0.86 | 0.005 |
| RKOr | NSC348884 | ||||||
|---|---|---|---|---|---|---|---|
| 2 h | 4 h | ||||||
| - | IC50 (2.67 µM) | 2 × IC50 (5.34 µM) | - | IC50 (2.67 µM) | 2 × IC50 (5.34 µM) | ||
| PLX4032 | IC50 (µM) | 31.45 ± 1.17 | 19.75 ± 1.76 | 11.88 ± 1.34 | 33.17 ± 0.95 | 18.55 ± 1.98 | <0.01 |
| LC50 (µM) | 92.15 ± 13.07 | 73.63 ± 5.32 | 73.38 ± 5.96 | 84.94 ± 0.61 | 72.98 ± 2.56 | 59.69 ± 9.97 | |
| HT-29r | NSC348884 | ||||||
| 2 h | 4 h | ||||||
| - | IC50 (3.71 µM) | 2 × IC50 (7.42 µM) | - | IC50 (3.71 µM) | 2 × IC50 (7.42 µM) | ||
| PLX4032 | IC50 (µM) | 41.10 ± 2.40 | 32.95 ± 4.15 | 20.18 ± 1.42 | 35.19 ± 3.12 | 25.16 ± 1.17 | <0.01 |
| LC50 (µM) | >100 | 93.98 ± 5.31 | 84.57 ± 3.28 | 85.61 ± 8.86 | 79.14 ± 6.96 | 46.56 ± 16.13 | |
| RKOr | IZCZ-3 | ||||||
|---|---|---|---|---|---|---|---|
| 6 h | 8 h | ||||||
| - | IC50 (0.58 µM) | 2 × IC50 (1.16 µM) | - | IC50 (0.58 µM) | 2 × IC50 (1.16 µM) | ||
| PLX4032 | IC50 (µM) | 30.22 ± 0.34 | 8.73 ± 0.01 | 4.57 ± 0.63 | 29.71 ± 1.03 | 7.74 ± 0.66 | 2.36 ± 1.30 |
| LC50 (µM) | 82.89 ± 6.48 | 71.09 ± 2.92 | 66.42 ± 1.49 | 85.88 ± 1.94 | 77.06 ± 1.47 | 61.80 ± 12.90 | |
| HT-29r | IZCZ-3 | ||||||
| 6 h | 8 h | ||||||
| - | IC50 (0.35 µM) | 2 × IC50 (0.70 µM) | - | IC50 (0.35 µM) | 2 × IC50 (0.70 µM) | ||
| PLX4032 | IC50 (µM) | 37.81 ± 4.17 | 31.76 ± 3.29 | 26.43 ± 4.51 | 39.99 ± 1.49 | 32.03 ± 3.13 | 17.90 ± 3.23 |
| LC50 (µM) | 90.13 ± 1.02 | 75.49 ± 1.76 | 77.49 ± 0.52 | 94.27 ± 9.43 | 80.13 ± 1.28 | 73.30 ± 1.00 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grbčić, P.; Fučkar Čupić, D.; Gamberi, T.; Kraljević Pavelić, S.; Sedić, M. Proteomic Profiling of BRAFV600E Mutant Colon Cancer Cells Reveals the Involvement of Nucleophosmin/c-Myc Axis in Modulating the Response and Resistance to BRAF Inhibition by Vemurafenib. Int. J. Mol. Sci. 2021, 22, 6174. https://doi.org/10.3390/ijms22126174
Grbčić P, Fučkar Čupić D, Gamberi T, Kraljević Pavelić S, Sedić M. Proteomic Profiling of BRAFV600E Mutant Colon Cancer Cells Reveals the Involvement of Nucleophosmin/c-Myc Axis in Modulating the Response and Resistance to BRAF Inhibition by Vemurafenib. International Journal of Molecular Sciences. 2021; 22(12):6174. https://doi.org/10.3390/ijms22126174
Chicago/Turabian StyleGrbčić, Petra, Dora Fučkar Čupić, Tania Gamberi, Sandra Kraljević Pavelić, and Mirela Sedić. 2021. "Proteomic Profiling of BRAFV600E Mutant Colon Cancer Cells Reveals the Involvement of Nucleophosmin/c-Myc Axis in Modulating the Response and Resistance to BRAF Inhibition by Vemurafenib" International Journal of Molecular Sciences 22, no. 12: 6174. https://doi.org/10.3390/ijms22126174