
Protease–Antiprotease Imbalance in Bronchiectasis

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Search Strategy and Selection Criteria
3. Airway Inflammation in Bronchiectasis
4. An Overview on Proteases and Antiproteases in the Airways
4.1. Serine Proteases
Neutrophil Elastase
4.2. Matrix Metalloproteinases
4.3. Cysteine Proteases
4.4. Antiproteases
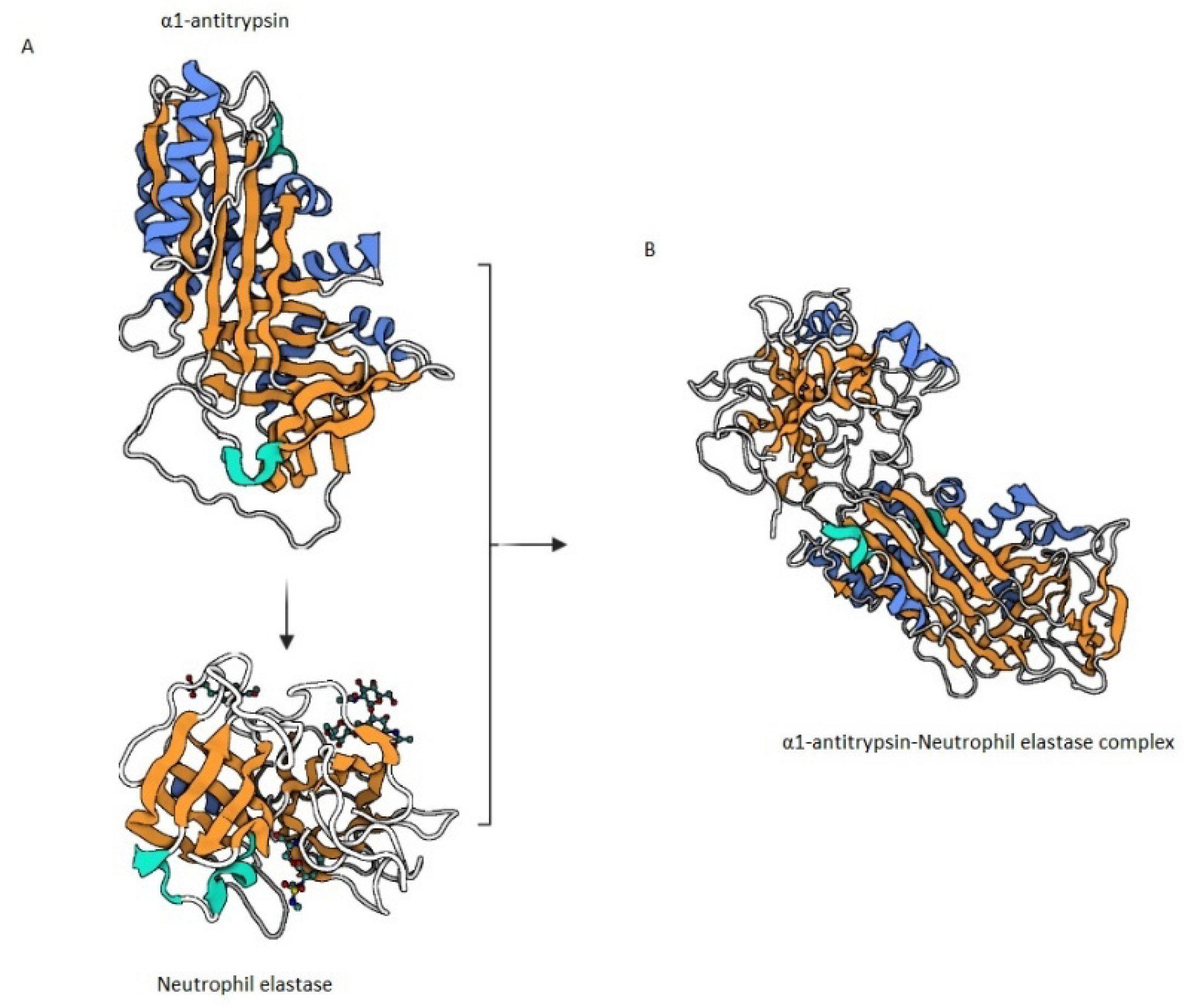
4.4.1. α1 Antitrypsin (AAT)
4.4.2. Antileukoprotease (ALP) Superfamily
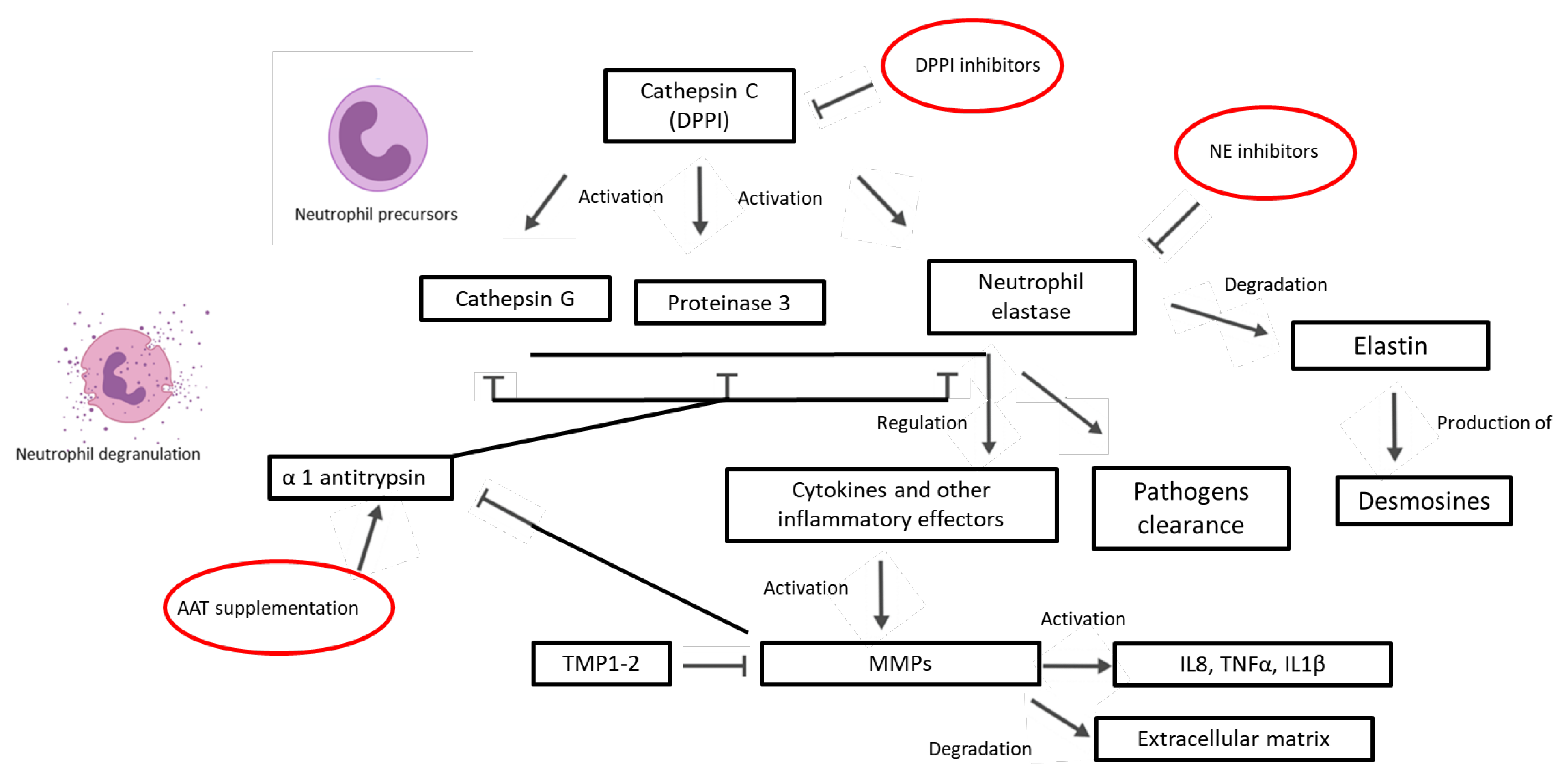
5. The Role of Proteases and Antiproteases in Bronchiectasis
5.1. Neutrophil Elastase in Bronchiectasis
5.2. Matrix Metalloproteinases in Bronchiectasis
5.3. Airway Proteases and Microbial Community in Bronchiectasis
5.4. Antiproteases in Bronchiectasis
α1 Antitrypsin Deficiency (AATD) in Bronchiectasis
6. Treatments of Protease–Antiprotease Imbalance in Bronchiectasis
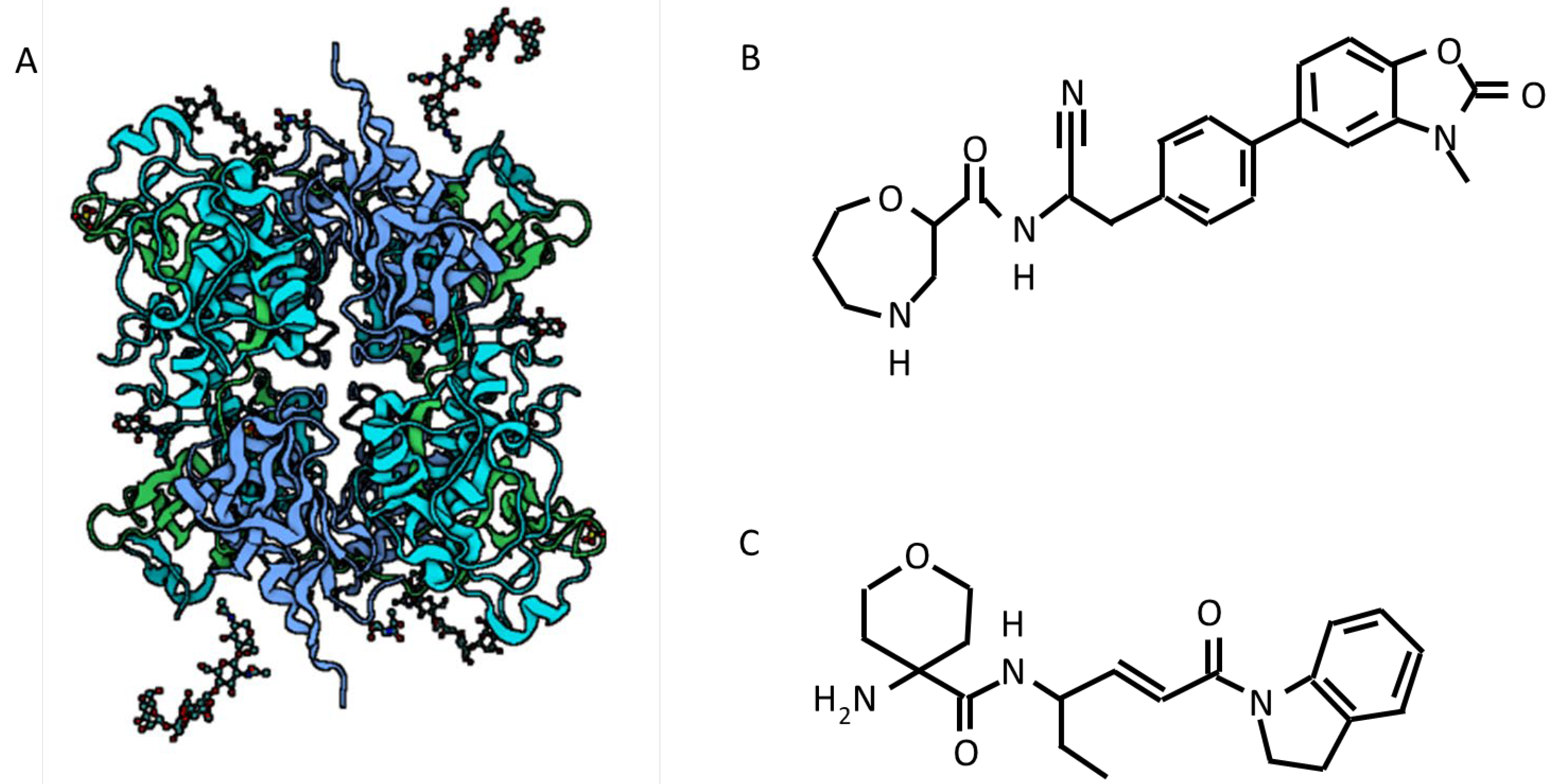
6.1. Active Neutrophil Elastase Inhibitors
6.2. DPP1 Inhibitors
6.3. AAT Supplementation
6.4. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Polverino, E.; Goeminne, P.C.; McDonnell, M.J.; Aliberti, S.; Marshall, S.E.; Loebinger, M.R.; Murris, M.; Cantón, R.; Torres, A.; Dimakou, K.; et al. European Respiratory Society guidelines for the management of adult bronchiectasis. Eur. Respir. J. 2017, 50, 1700629. [Google Scholar] [CrossRef]
- Aliberti, S.; Sotgiu, G.; Lapi, F.; Gramegna, A.; Cricelli, C.; Blasi, F. Prevalence and incidence of bronchiectasis in Italy. BMC Pulm. Med. 2020, 20, 15. [Google Scholar] [CrossRef]
- Quint, J.K.; Millett, E.R.C.; Joshi, M.; Navaratnam, V.; Thomas, S.L.; Hurst, J.R.; Smeeth, L.; Brown, J.S. Changes in the incidence, prevalence and mortality of bronchiectasis in the UK from 2004 to 2013: A population-based cohort study. Eur. Respir. J. 2016, 47, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, M.; Rodríguez-Blanco, T.; Barrecheguren, M.; Simonet, P.; Miravitlles, M. Prevalence and incidence of bronchiectasis in Catalonia, Spain: A population-based study. Respir. Med. 2016, 121, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonni, S.; Chalmers, J.D.; Goeminne, P.C.; McDonnell, M.J.; Dimakou, K.; De Soyza, A.; Polverino, E.; Van de Kerkhove, C.; Rutherford, R.; Davison, J.; et al. Etiology of Non-Cystic Fibrosis Bronchiectasis in Adults and Its Correlation to Disease Severity. Ann. Am. Thorac. Soc. 2015, 12, 1764–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Chalmers, J.D.; Olivier, K.N. Advances in bronchiectasis: Endotyping, genetics, microbiome, and disease heterogeneity. Lancet 2018, 392, 880–890. [Google Scholar] [CrossRef] [Green Version]
- Cole, P.J. Inflammation: A two-edged sword—The model of bronchiectasis. Eur. J. Respir. Dis. Suppl. 1986, 147, 6–15. [Google Scholar]
- Amati, F.; Simonetta, E.; Gramegna, A.; Tarsia, P.; Contarini, M.; Blasi, F.; Aliberti, S. The biology of pulmonary exacerbations in bronchiectasis. Eur. Respir. Rev. 2019, 28, 190055. [Google Scholar] [CrossRef] [Green Version]
- Faverio, P.; Stainer, A.; Bonaiti, G.; Zucchetti, S.C.; Simonetta, E.; Lapadula, G.; Marruchella, A.; Gori, A.; Blasi, F.; Codecasa, L.; et al. Characterizing Non-Tuberculous Mycobacteria Infection in Bronchiectasis. Int. J. Mol. Sci. 2016, 17, 1913. [Google Scholar] [CrossRef] [Green Version]
- Bonaiti, G.; Pesci, A.; Marruchella, A.; Lapadula, G.; Gori, A.; Aliberti, S. Nontuberculous Mycobacteria in Noncystic Fibrosis Bronchiectasis. Biomed. Res. Int. 2015, 2015, 197950. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Zariwala, M.; Leigh, M. Primary Ciliary Dyskinesia. Clin. Chest Med. 2016, 37, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Contarini, M.; Shoemark, A.; Rademacher, J.; Finch, S.; Gramegna, A.; Gaffuri, M.; Roncoroni, L.; Seia, M.; Ringshausen, F.C.; Welte, T.; et al. Why, when and how to investigate primary ciliary dyskinesia in adult patients with bronchiectasis. Multidiscip. Respir. Med. 2018, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Balázs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54, S5–S12. [Google Scholar] [CrossRef] [Green Version]
- Ramzi, N.; Jamee, M.; Bakhtiyari, M.; Rafiemanesh, H.; Zainaldain, H.; Tavakol, M.; Rezaei, A.; Kalvandi, M.; Zian, Z.; Mohammadi, H.; et al. Bronchiectasis in common variable immunodeficiency: A systematic review and meta-analysis. Pediatr. Pulmonol. 2020, 55, 292–299. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, J.; Lau, J.; Wang, S.; Taneja, V.; Matteson, E.L.; Vassallo, R. Mechanisms of lung disease development in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 581–596. [Google Scholar] [CrossRef]
- Shoemark, A.; Cant, E.; Carreto, L.; Smith, A.; Oriano, M.; Keir, H.R.; Perea, L.; Canto, E.; Terranova, L.; Vidal, S.; et al. A point-of-care neutrophil elastase activity assay identifies bronchiectasis severity, airway infection and risk of exacerbation. Eur. Respir. J. 2019, 53, 1900303. [Google Scholar] [CrossRef] [Green Version]
- Oriano, M.; Gramegna, A.; Terranova, L.; Sotgiu, G.; Sulaiman, I.; Ruggiero, L.; Saderi, L.; Wu, B.; Chalmers, J.D.; Segal, L.N.; et al. Sputum Neutrophil Elastase associates with microbiota and P. aeruginosa in bronchiectasis. Eur. Respir. J. 2020, 56, 2000769. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, A.; Aliberti, S.; Sibila, O.; Di Francesco, C.; Sotgiu, G.; Perea, L.; Terranova, L.; Oriano, M.; Pilocane, T.; Saderi, L.; et al. Sputum neutrophil elastase in bronchiectasis: A Southern European cohort study. Eur. Respir. J. 2020, 56, 2001702. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Attucci, S.; Juliano, M.A.; Kalupov, T.; Jourdan, M.L.; Juliano, L.; Gauthier, F. Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat. Protoc. 2008, 3, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Elkington, P.T.G.; Friedland, J.S. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006, 61, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, J.D.; Moffitt, K.L.; Suarez-Cuartin, G.; Sibila, O.; Finch, S.; Furrie, E.; Dicker, A.; Wrobel, K.; Elborn, J.S.; Walker, B.; et al. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am. J. Respir. Crit. Care Med. 2017, 195, 1384–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, H.R.; Shoemark, A.; Dicker, A.J.; Perea, L.; Pollock, J.; Giam, Y.H.; Suarez-Cuartin, G.; Crichton, M.L.; Lonergan, M.; Oriano, M.; et al. Neutrophil Extracellular Traps are Increased in Severe Bronchiectasis and Reduced by Long-Term Azithromycin Treatment. Lancet 2020. [Google Scholar] [CrossRef]
- Potempa, J.A.N.; Banbula, A.; Travis, J.I.M. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol. 2000 2000, 24, 153–192. [Google Scholar] [CrossRef] [PubMed]
- Sandri, A.; Ortombina, A.; Boschi, F.; Cremonini, E.; Boaretti, M.; Sorio, C.; Melotti, P.; Bergamini, G.; Lleo, M. Inhibition of Pseudomonas aeruginosa secreted virulence factors reduces lung inflammation in CF mice. Virulence 2018, 9, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dente, F.L.; Bilotta, M.; Bartoli, M.L.; Bacci, E.; Cianchetti, S.; Latorre, M.; Malagrinò, L.; Nieri, D.; Roggi, M.A.; Vagaggini, B.; et al. Neutrophilic Bronchial Inflammation Correlates with Clinical and Functional Findings in Patients with Noncystic Fibrosis Bronchiectasis. Mediat. Inflamm. 2015, 2015, 642503. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.J. Tissue destruction by neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [CrossRef]
- Craig, A.; Mai, J.; Cai, S.; Jeyaseelan, S. Neutrophil Recruitment to the Lungs during Bacterial Pneumonia. Infect. Immun. 2009, 77, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Weissmann, G.; Smolen, J.E.; Korchak, H.M. Release of Inflammatory Mediators from Stimulated Neutrophils. N. Engl. J. Med. 1980, 303, 27–34. [Google Scholar] [CrossRef]
- Rossaint, J.; Zarbock, A. Tissue-Specific Neutrophil Recruitment into the Lung, Liver, and Kidney. J. Innate Immun. 2013, 5, 348–357. [Google Scholar] [CrossRef]
- Aliberti, S.; Lonni, S.; Dore, S.; Mcdonnell, M.J.; Goeminne, P.C.; Dimakou, K.; Fardon, T.C.; Rutherford, R.; Pesci, A.; Restrepo, M.I.; et al. Clinical phenotypes in adult patients with bronchiectasis. Eur. Respir. J. 2016, 47, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, J.D.; Smith, M.P.; McHugh, B.J.; Doherty, C.; Govan, J.R.; Hill, A.T. Short- and Long-Term Antibiotic Treatment Reduces Airway and Systemic Inflammation in Non–Cystic Fibrosis Bronchiectasis. Am. J. Respir. Crit. Care Med. 2012, 186, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Keir, H.R.; Shoemark, A.; Dicker, A.J.; Perea, L.; Pollock, J.; Giam, Y.H.; Suarez-Cuartin, G.; Crichton, M.L.; Lonergan, M.; Oriano, M.; et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: An international, observational, multicohort study. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Sibila, O.; Perea, L.; Cantó, E.; Shoemark, A.; Cassidy, D.; Smith, A.H.; Suarez-Cuartin, G.; Rodrigo-Troyano, A.; Keir, H.R.; Oriano, M.; et al. Antimicrobial peptides, disease severity and exacerbations in bronchiectasis. Thorax 2019, thoraxjnl-2018-212895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuschillo, S.; De Felice, A.; Balzano, G. Mucosal inflammation in idiopathic bronchiectasis: Cellular and molecular mechanisms. Eur. Respir. J. 2008, 31, 396–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angrill, J.; Agustí, C.; De Celis, R.; Filella, X.; Rañó, A.; Elena, M.; De La Bellacasa, J.P.; Xaubet, A.; Torres, A. Bronchial inflammation and colonization in patients with clinically stable bronchiectasis. Am. J. Respir. Crit. Care Med. 2001, 164, 1628–1632. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.A.; Posadas, T.; Sotgiu, G.; Blasi, F.; Saderi, L.; Aliberti, S. Role of inhaled corticosteroids in reducing exacerbations in bronchiectasis patients with blood eosinophilia pooled post-hoc analysis of 2 randomized clinical trials. Respir. Med. 2020, 172, 106127. [Google Scholar] [CrossRef]
- Perea, L.; Cantó, E.; Suarez-Cuartin, G.; Aliberti, S.; Chalmers, J.D.; Sibila, O.; Vidal, S. A Cluster Analysis of Bronchiectasis Patients Based on the Airway Immune Profile. Chest 2020, 159, 1758–1767. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.A.; Posadas, T.; Sotgiu, G.; Blasi, F.; Saderi, L.; Aliberti, S. Repeteability of Circulating Eosinophil Measures and Inhaled Corticosteroids Effect in Bronchiectasis: A Post Hoc Analysis of a Randomized Clinical Trial. Arch. Bronconeumol. 2020, 56, 681–683. [Google Scholar] [CrossRef]
- Aliberti, S.; Sotgiu, G.; Martinez Garcia, M.-A. Blood eosinophils do not predict inhaled budesonide response in bronchiectasis. Eur. Respir. J. 2020, 56, 2002210. [Google Scholar] [CrossRef]
- Rademacher, J.; Konwert, S.; Fuge, J.; Dettmer, S.; Welte, T.; Ringshausen, F.C. Anti-IL5 and anti-IL5Rα therapy for clinically significant bronchiectasis with eosinophilic endotype: A case series. Eur. Respir. J. 2020, 55, 1901333. [Google Scholar] [CrossRef]
- López-Boado, Y.S.; Espinola, M.; Bahr, S.; Belaaouaj, A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 2004, 172, 509–515. [Google Scholar] [CrossRef]
- Small, D.M.; Brown, R.R.; Doherty, D.F.; Abladey, A.; Zhou-Suckow, Z.; Delaney, R.J.; Kerrigan, L.; Dougan, C.M.; Borensztajn, K.S.; Holsinger, L.; et al. Targeting of Cathepsin S Reduces Cystic Fibrosis-like Lung Disease. Eur. Respir. J. 2019, 1801523. [Google Scholar] [CrossRef]
- Segal, A.W. Europe PMC Funders Group How Neutrophils Kill Microbes. Annu. Rev. Immunol. 2007, 2. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedor, J.; Hogue, L.; Akers, K.; Boslaugh, S.; Schreiber, J.; Ferkol, T. Cathepsin-G interferes with clearance of Pseudomonas aeruginosa from mouse lungs. Pediatr. Res. 2007, 61, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Crisford, H.; Sapey, E.; Stockley, R.A. Proteinase 3; a potential target in chronic obstructive pulmonary disease and other chronic inflammatory diseases. Respir. Res. 2018, 19, 180. [Google Scholar] [CrossRef]
- Benabid, R.; Wartelle, J.; Malleret, L.; Guyot, N.; Gangloff, S.; Lebargy, F.; Belaaouaj, A. Neutrophil elastase modulates cytokine expression: Contribution to host defense against pseudomonas aeruginosa-induced pneumonia. J. Biol. Chem. 2012, 287, 34883–34894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosi, M.F.; Zakem, H.; Berger, M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J. Clin. Investig. 1990, 86, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, F.; Cooley, J.; Accurso, F.J.; Remold-O’Donnell, E. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax 2004, 59, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramegna, A.; Amati, F.; Terranova, L.; Sotgiu, G.; Tarsia, P.; Miglietta, D.; Calderazzo, M.A.; Aliberti, S.; Blasi, F. Neutrophil elastase in bronchiectasis. Respir. Res. 2017, 18, 1–13. [Google Scholar] [CrossRef]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaître, V.; D’Armiento, J. Matrix metalloproteinases in development and disease. Birth Defects Res. Part C Embryo Today Rev. 2006, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, X.; Shapiro, S.D.; Shipley, J.M.; Twining, S.S.; Diaz, L.A.; Senior, R.M.; Werb, Z. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell 2000, 102, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Wolters, P.J.; Laig-Webster, M.; Caughey, G.H. Dipeptidyl peptidase I cleaves matrix-associated proteins and is expressed mainly by mast cells in normal dog airways. Am. J. Respir. Cell Mol. Biol. 2000, 22, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Chapman, H.A. Importance of lysosomal cysteine proteases in lung disease. Respir. Res. 2000, 1, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, J.D.; Haworth, C.S.; Metersky, M.L.; Loebinger, M.R.; Blasi, F.; Sibila, O.; O’Donnell, A.E.; Sullivan, E.J.; Mange, K.C.; Fernandez, C.; et al. Phase 2 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis. N. Engl. J. Med. 2020, 383, 2127–2137. [Google Scholar] [CrossRef]
- Fagerhol, M.K.; Laurell, C.-B. The polymorphism of “prealbumins” and α1-antitrypsin in human sera. Clin. Chim. Acta 1967, 16, 199–203. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A review of α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Veith, M.; Tüffers, J.; Peychev, E.; Klemmer, A.; Kotke, V.; Janciauskiene, S.; Wilhelm, S.; Bals, R.; Koczulla, A.R.; Vogelmeier, C.F.; et al. The Distribution of Alpha-1 Antitrypsin Genotypes Between Patients with COPD/Emphysema, Asthma and Bronchiectasis. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 2827–2836. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Thun, G.A.; Zorzetto, M.; Ottaviani, S.; Imboden, M.; Schindler, C.; Von Eckardstein, A.; Rohrer, L.; Rochat, T.; Russi, E.W.; et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax 2012, 67, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Alpha-1 antitrypsin deficiency. Respir. Med. CME 2011, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sallenave, J.M. The role of secretory leukocyte proteinase inhibitor and elafin (elastase-specific inhibitor/skin-derived antileukoprotease) as alarm antiproteinases in inflammatory lung disease. Respir. Res. 2000, 1, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Abe, S.; Shibata, Y.; Yuki, H.; Suzuki, H.; Saito, H.; Sata, M.; Kato, S.; Tomoike, H. Elevated levels of cytokeratin 19 in the bronchoalveolar lavage fluid of patients with chronic airway inflammatory diseases—A specific marker for bronchial epithelial injury. Am. J. Respir. Crit. Care Med. 1997, 155, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Ip, M.; Shum, D.; Lauder, I.; Lam, W.K.; So, S.Y. Effect of antibiotics on sputum inflammatory contents in acute exacerbations of bronchiectasis. Respir. Med. 1993, 87, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Sibila, O.; Laserna, E.; Shoemark, A.; Keir, H.R.; Finch, S.; Rodrigo-Troyano, A.; Perea, L.; Lonergan, M.; Goeminne, P.C.; Chalmers, J.D. Airway Bacterial Load and Inhaled Antibiotic Response in Bronchiectasis. Am. J. Respir. Crit. Care Med. 2019, 200, 33–41. [Google Scholar] [CrossRef]
- Oriano, M.; Terranova, L.; Sotgiu, G.; Saderi, L.; Bellofiore, A.; Retucci, M.; Marotta, C.; Gramegna, A.; Miglietta, D.; Carnini, C.; et al. Evaluation of active neutrophil elastase in sputum of bronchiectasis and cystic fibrosis patients: A comparison among different techniques. Pulm. Pharmacol. Ther. 2019, 59, 1–6. [Google Scholar] [CrossRef]
- Huang, J.T.-J.; Kuzmanova, E.; Dicker, A.; Keir, H.; Finch, S.; Aliberti, S.; Fardon, T.; Chalmers, J. Desmosine is a predictor of long-term cardiovascular mortality in bronchiectasis. Eur. Respir. J. 2020, 56, 4718. [Google Scholar] [CrossRef]
- Guan, W.-J.; Gao, Y.-H.; Xu, G.; Lin, Z.-Y.; Tang, Y.; Gu, Y.-Y.; Liu, G.-H.; Li, H.-M.; Chen, R.-C.; Zhong, N.-S. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: Clinical correlates and prognostic implications. Respirology 2015, 20, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Pifferi, M.; Bush, A.; Caramella, D.; Metelli, M.R.; Di Cicco, M.; Piras, M.; Gherarducci, G.; Capristo, C.; Maggi, F.; Peroni, D.; et al. Matrix metalloproteinases and airway remodeling and function in primary ciliary dyskinesia. Respir. Med. 2017, 124, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac Aogáin, M.; Narayana, J.K.; Tiew, P.Y.; Ali, N.A.B.M.; Yong, V.F.L.; Jaggi, T.K.; Lim, A.Y.H.; Keir, H.R.; Dicker, A.J.; Thng, K.X.; et al. Integrative microbiomics in bronchiectasis exacerbations. Nat. Med. 2021, 27, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.D.; Chotirmall, S.H. Bronchiectasis: New therapies and new perspectives. Lancet Respir. Med. 2018, 6, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Carreto, L.; Morrison, M.; Donovan, J.; Finch, S.; Tan, G.L.; Fardon, T.; Wilson, R.; Furrie, E.; Loebinger, M.; Chalmers, J.D. Utility of routine screening for alpha-1 antitrypsin deficiency in patients with bronchiectasis. Thorax 2020, 75, 592–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvelier, A.; Muir, J.F.; Hellot, M.F.; Benhamou, D.; Martin, J.P.; Bénichou, J.; Sesboüé, R. Distribution of alpha(1)-antitrypsin alleles in patients with bronchiectasis. Chest 2000, 117, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.; De Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Watz, H.; Nagelschmitz, J.; Kirsten, A.; Pedersen, F.; van der Mey, D.; Schwers, S.; Bandel, T.-J.; Rabe, K.F. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85-8501 for the treatment of non-cystic fibrosis bronchiectasis: A randomized controlled trial. Pulm. Pharmacol. Ther. 2019, 56, 86–93. [Google Scholar] [CrossRef]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Sellier Kessler, O.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2020, 19, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.E.; Mayer, R.J.; Goyal, N.; Bal, J.; Dallow, N.; Boyce, M.; Carpenter, D.; Churchill, A.; Heslop, T.; Lazaar, A.L. Epithelial desquamation observed in a phase I study of an oral cathepsin C inhibitor (GSK2793660). Br. J. Clin. Pharmacol. 2017, 83, 2813–2820. [Google Scholar] [CrossRef]
- Campos, M.A.; Kueppers, F.; Stocks, J.M.; Strange, C.; Chen, J.; Griffin, R.; Wang-Smith, L.; Brantly, M.L. Safety and Pharmacokinetics of 120 mg/kg versus 60 mg/kg Weekly Intravenous Infusions of Alpha-1 Proteinase Inhibitor in Alpha-1 Antitrypsin Deficiency: A Multicenter, Randomized, Double-Blind, Crossover Study (SPARK). COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 687–695. [Google Scholar] [CrossRef]
- Sandhaus, R.A.; Stocks, J.; Rouhani, F.N.; Brantly, M.; Strauss, P. Biochemical Efficacy and Safety of a New, Ready-to-Use, Liquid Alpha-1-Proteinase Inhibitor, GLASSIA (Alpha1-Proteinase Inhibitor (Human), Intravenous). COPD J. Chronic Obstr. Pulm. Dis. 2014, 11, 17–25. [Google Scholar] [CrossRef]
- Seersholm, N.; Sandhaus, R.; Chapman, K.R.; Burdon, J.; Piitulainen, E.; Stocks, J.; Tortorici, M.; Rosenberg, T.; Vit, O.; Bexon, M.; et al. Safety of bi-weekly infusion of A1-PI augmentation therapy in RAPID. Eur. Respir. J. 2015, 46, PA999. [Google Scholar] [CrossRef]
- Barker, A.F.; Campos, M.A.; Brantly, M.L.; Stocks, J.M.; Sandhaus, R.A.; Lee, D.; Steinmann, K.; Lin, J.; Sorrells, S. Bioequivalence of a Liquid Formulation of Alpha(1)-Proteinase Inhibitor Compared with Prolastin®-C (Lyophilized Alpha(1)-PI) in Alpha(1)-Antitrypsin Deficiency. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 590–596. [Google Scholar] [CrossRef]
- Gunawardena, K.A.; Gullstrand, H.; Perrett, J. Pharmacokinetics and safety of AZD9668, an oral neutrophil elastase inhibitor, in healthy volunteers and patients with COPD. Int. J. Clin. Pharmacol. Ther. 2013, 51, 288–304. [Google Scholar] [CrossRef]
- Nagelschmitz, J.; Kaufel, D.; Stephan, S.; von Nussbaum, F.; Li, V.; Delbeck, M.; Lustig, K.; Bandel, T.; Watz, H. The novel elastase inhibitor BAY 85-8501 provides a new approach in the treatment of pulmonary diseases. Eur. Respir. J. 2014, 44, 3416. [Google Scholar]
- Carnini, C.; Brogin, G.; Patacchini, R.; Miglietta, D.; Stefani, M.; Finch, H.; Fitzgerald, M.; Fox, C.; Puccini, P.; Villetti, G.; et al. CHF6333: Pharmacological and pharmacokinetic characterization of a novel potent inhaled inhibitor of neutrophil elastase. In B80-A: Mechanisms and Models of Acute Lung Injury; American Thoracic Society International Conference Abstracts; American Thoracic Society: New York, NY, USA, 2017; p. A4420. [Google Scholar]
- Barros-Tizón, J.C.; Torres, M.L.; Blanco, I.; Martínez, M.T. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Ther. Adv. Respir. Dis. 2012, 6, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantly, M.L.; Lascano, J.E.; Shahmohammadi, A. Intravenous Alpha-1 Antitrypsin Therapy for Alpha-1 Antitrypsin Deficiency: The Current State of the Evidence. Chronic Obstr. Pulm. Dis. 2018, 6, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Molecule | Administration | Update | Main Evidence | Safety/Adverse Effects | References or Trial Registration |
|---|---|---|---|---|---|
| Neutrophil elastase inhibitors | |||||
| AZD9668 | Oral | Phase 2 completed | Increased pulmonary function and decreased inflammatory biomarkers (IL-6-IL-8) in patients treated vs. non treated | Safe and tolerable | [76] |
| BAY 85–8501 | Oral | Phase 2a completed | No evidence in increasing pulmonary function and quality of life after 4 weeks’ treatment | Safe and tolerable | [77] |
| CHF6333 | Inhaled powder | Phase 1 completed | Paper not published to date | Safe and tolerable | NCT04010799 |
| POL6014 | Inhaled | Phase 1 completed | No data available on bronchiectasis | Safe and tolerable | [78] |
| Cathepsin C/DPP1 inhibitors | |||||
| GSK2793660 | Oral | Phase 1 | - | Terminated because of adverse events | [79] |
| Brensocatib | Oral | Phase 2 completed | Effective in reducing time to the first exacerbation and rate of severe exacerbations | Safe and tolerable | [57] |
| AATD therapy | |||||
| Prolastin C | Intravenous | Post-marketing | Effective in increasing AAT serum levels. It reduces the decline in lung density. | Safe and tolerable | [80] |
| API-GLASSIA | Intravenous | Post-marketing | Bioequivalent to Prolastin-C | Safe and tolerable | [81] |
| Zemaira | Intravenous | Post-marketing | Dose confirmed, higher dose may be associated with greater effect. Prevents emphysema in AATD patients | Safe and tolerable | [82] |
| Liquid Alpha1-PI | Intravenous | Post-marketing | Bioequivalent to Prolastin-C | Safe and tolerable | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oriano, M.; Amati, F.; Gramegna, A.; De Soyza, A.; Mantero, M.; Sibila, O.; Chotirmall, S.H.; Voza, A.; Marchisio, P.; Blasi, F.; et al. Protease–Antiprotease Imbalance in Bronchiectasis. Int. J. Mol. Sci. 2021, 22, 5996. https://doi.org/10.3390/ijms22115996
Oriano M, Amati F, Gramegna A, De Soyza A, Mantero M, Sibila O, Chotirmall SH, Voza A, Marchisio P, Blasi F, et al. Protease–Antiprotease Imbalance in Bronchiectasis. International Journal of Molecular Sciences. 2021; 22(11):5996. https://doi.org/10.3390/ijms22115996
Chicago/Turabian StyleOriano, Martina, Francesco Amati, Andrea Gramegna, Anthony De Soyza, Marco Mantero, Oriol Sibila, Sanjay H. Chotirmall, Antonio Voza, Paola Marchisio, Francesco Blasi, and et al. 2021. "Protease–Antiprotease Imbalance in Bronchiectasis" International Journal of Molecular Sciences 22, no. 11: 5996. https://doi.org/10.3390/ijms22115996