
Not Just a Bystander: The Emerging Role of Astrocytes and Research Tools in Studying Cognitive Dysfunctions in Schizophrenia


{kind=link}
{kind=link}
Abstract
:1. Schizophrenia and Unmet Needs in the Treatment of Schizophrenia
2. The Emerging Role of Astrocytes in Schizophrenia
2.1. Astrocytic Gene and Astrocyte-Related Gene Expression in Schizophrenia
2.2. The Role of Astrocytes in Cognitive Deficits in Schizophrenia
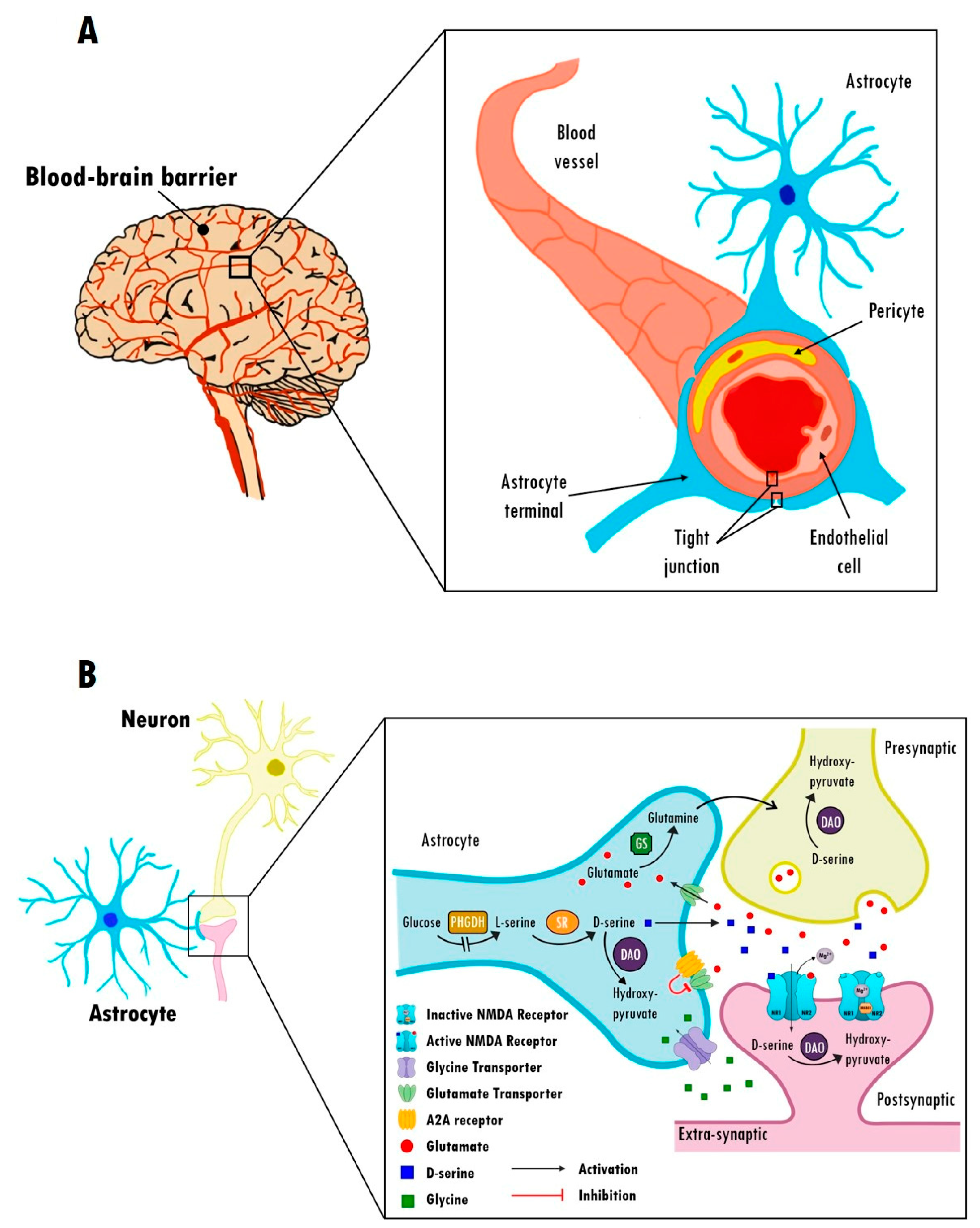
2.3. Astrocytic Modulation of the BBB in Schizophrenia
2.4. Astrocytic Regulation of Glutamate Transmission in Schizophrenia
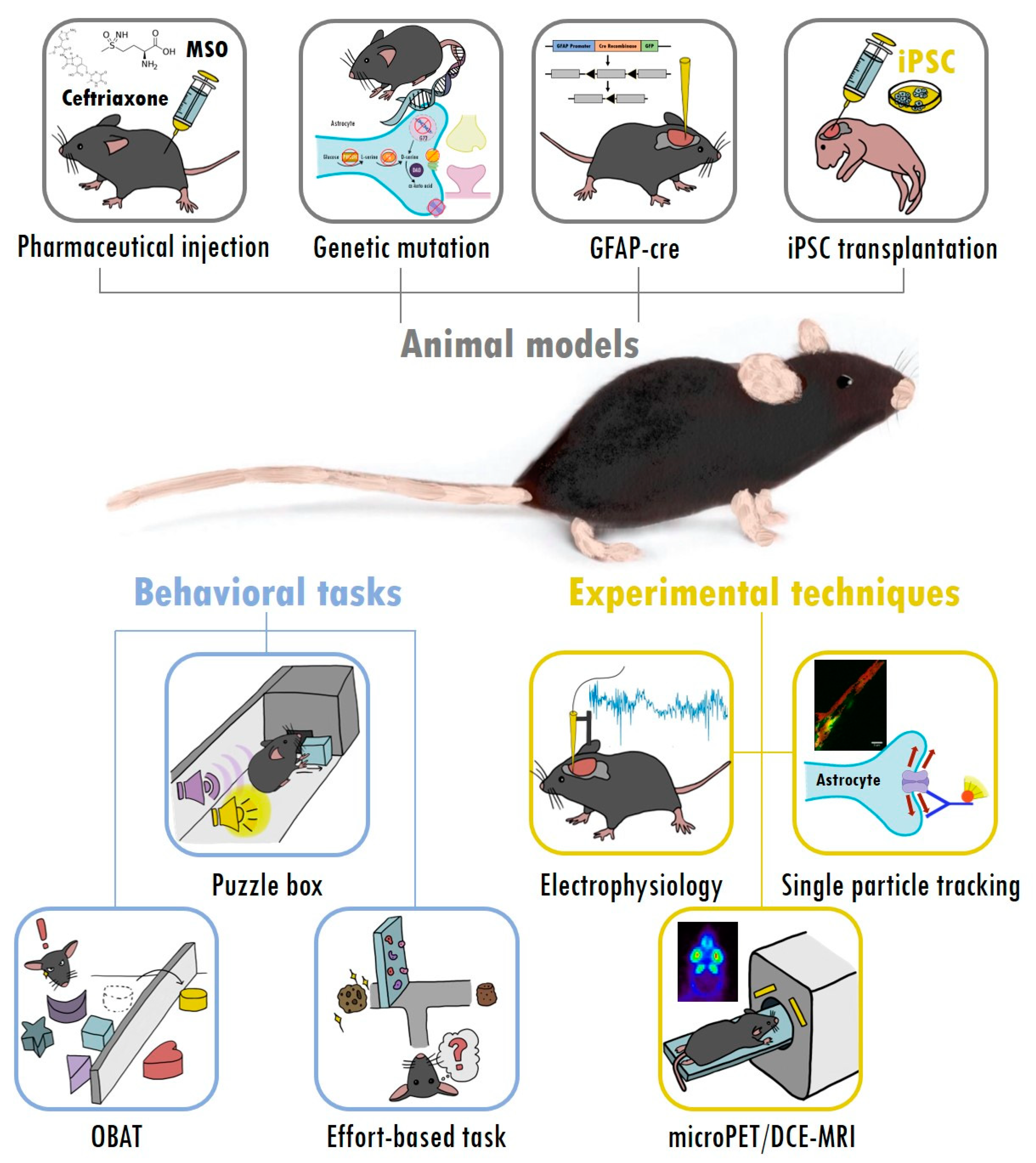
3. Taking Advantage of Mouse Models and Experimental Tools to Study Astrocytes and Cognitive Deficits in Schizophrenia
3.1. Using Animal Models to Investigate Astrocytic Regulation of Glutamate Transmission in Schizophrenia
3.2. A Selection of Behavioral Tasks for Assessing Cognitive Deficits and Negative Symptoms of Schizophrenia in Animal Models
3.3. Experimental Tools to Evaluate the Function of Astrocytes
4. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, A. Schizophrenia: The drug deadlock. Nature 2010, 468, 158–159. [Google Scholar] [CrossRef] [Green Version]
- Green, M.F.; Kern, R.S.; Heaton, R.K. Longitudinal studies of cognition and functional outcome in schizophrenia: Implications for MATRICS. Schizophr. Res. 2004, 72, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.J.; Tsuang, H.C.; Chiang, A.; Liu, C.M.; Hsieh, M.H.; Hwang, T.J.; Liu, S.K.; Hsiao, P.C.; Faraone, S.V.; Tsuang, M.T.; et al. The multidimensionality of schizotypy in nonpsychotic relatives of patients with schizophrenia and its applications in ordered subsets linkage analysis of schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Elert, E. Aetiology: Searching for schizophrenia’s roots. Nature 2014, 508, S2–S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citrome, L.; Stensbol, T.B.; Maeda, K. The preclinical profile of brexpiprazole: What is its clinical relevance for the treatment of psychiatric disorders? Expert Rev. Neurother. 2015, 15, 1219–1229. [Google Scholar] [CrossRef]
- Koychev, I.; Joyce, D.; Barkus, E.; Ettinger, U.; Schmechtig, A.; Dourish, C.T.; Dawson, G.R.; Craig, K.J.; Deakin, J.F. Cognitive and oculomotor performance in subjects with low and high schizotypy: Implications for translational drug development studies. Transl. Psychiatry 2016, 6, C. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, M.T.; D’Amato, L.C. Astrocyte-Physiology and Pathology. 2018. Available online: https://www.intechopen.com/books/astrocyte-physiology-and-pathology (accessed on 18 May 2021).
- Roussos, P.; Giakoumaki, S.G.; Adamaki, E.; Georgakopoulos, A.; Robakis, N.K.; Bitsios, P. The association of schizophrenia risk D-amino acid oxidase polymorphisms with sensorimotor gating, working memory and personality in healthy males. Neuropsychopharmacology 2011, 36, 1677–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangaro, M.; Bosia, M.; Zanoletti, A.; Bechi, M.; Cocchi, F.; Pirovano, A.; Lorenzi, C.; Bramanti, P.; Benedetti, F.; Smeraldi, E.; et al. Cognitive dysfunction and glutamate reuptake: Effect of EAAT2 polymorphism in schizophrenia. Neurosci. Lett. 2012, 522, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shibata, H.; Takeuchi, N.; Rachi, S.; Sakai, M.; Ninomiya, H.; Iwata, N.; Ozaki, N.; Fukumaki, Y. Association study of polymorphisms in the glutamate transporter genes SLC1A1, SLC1A3, and SLC1A6 with schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 271–278. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Steiner, J.; Bogerts, B. Glial cells in schizophrenia: Pathophysiological significance and possible consequences for therapy. Expert Rev. Neurother. 2009, 9, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, Y.; Tang, J.; Guo, T.; Li, X.; Yang, Y.; Chen, Q.; Zhao, X.; He, G.; Feng, G.; et al. SNPs and haplotypes in the S100B gene reveal association with schizophrenia. Biochem. Biophys Res. Commun. 2005, 328, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Hohoff, C.; Ponath, G.; Freitag, C.M.; Kastner, F.; Krakowitzky, P.; Domschke, K.; Koelkebeck, K.; Kipp, F.; von Eiff, C.; Deckert, J.; et al. Risk variants in the S100B gene predict elevated S100B serum concentrations in healthy individuals. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Zhang, Q.; Cheng, L.; Chen, M.; Wang, K.; Liu, Y.; Deng, X.; Chen, X.; Shen, Q.; Xu, Z.; et al. Risk variants in the S100B gene, associated with elevated S100B levels, are also associated with visuospatial disability of schizophrenia. Behav. Brain Res. 2011, 217, 363–368. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, H.G.; Steiner, J.; Guest, P.C.; Dobrowolny, H.; Bogerts, B. Glial cells as key players in schizophrenia pathology: Recent insights and concepts of therapy. Schizophr. Res. 2015, 161, 4–18. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, S.K.; Kim, J.W.; Kang, W.S.; Chung, J.H. Association of thrombospondin 1 gene with schizophrenia in Korean population. Mol. Biol. Rep. 2012, 39, 6875–6880. [Google Scholar] [CrossRef]
- Morita, Y.; Ujike, H.; Tanaka, Y.; Otani, K.; Kishimoto, M.; Morio, A.; Kotaka, T.; Okahisa, Y.; Matsushita, M.; Morikawa, A.; et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol. Psychiatry 2007, 61, 1200–1203. [Google Scholar] [CrossRef] [Green Version]
- Catts, V.S.; Wong, J.; Fillman, S.G.; Fung, S.J.; Shannon Weickert, C. Increased expression of astrocyte markers in schizophrenia: Association with neuroinflammation. Aust. N Z J. Psychiatry 2014, 48, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.J.; O’Grady, J.; Kleinman, J.E.; Weickert, C.S. Glial fibrillary acidic protein mRNA levels in the cingulate cortex of individuals with depression, bipolar disorder and schizophrenia. Neuroscience 2005, 133, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Pearce, R.K.; Hirsch, S.R.; Ansorge, O.; Thom, M.; Maier, M. Fibrillary astrocytes are decreased in the subgenual cingulate in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 357–362. [Google Scholar] [CrossRef]
- Steiner, J.; Bernstein, H.G.; Bielau, H.; Farkas, N.; Winter, J.; Dobrowolny, H.; Brisch, R.; Gos, T.; Mawrin, C.; Myint, A.M.; et al. S100B-immunopositive glia is elevated in paranoid as compared to residual schizophrenia: A morphometric study. J. Psychiatr. Res. 2008, 42, 868–876. [Google Scholar] [CrossRef]
- Schumberg, K.; Polyakova, M.; Steiner, J.; Schroeter, M.L. Serum S100B Is Related to Illness Duration and Clinical Symptoms in Schizophrenia-A Meta-Regression Analysis. Front. Cell Neurosci. 2016, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, B.; Radomska, K.J.; Zimmermann, B.; Kettunen, P.; Jazin, E.; Emilsson, L.S. QKI6B mRNA levels are upregulated in schizophrenia and predict GFAP expression. Brain Res. 2017, 1669, 63–68. [Google Scholar] [CrossRef]
- Gonzalez-Penas, J.; Costas, J.; Villamayor, M.J.G.; Xu, B. Enrichment of rare genetic variants in astrocyte gene enriched co-expression modules altered in postmortem brain samples of schizophrenia. Neurobiol. Dis. 2019, 121, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Greene, C.; Hanley, N.; Campbell, M. Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders. Transl. Psychiatry 2020, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Ghetti, A.; Pinto-Duarte, A.; Wang, X.; Dziewczapolski, G.; Galimi, F.; Huitron-Resendiz, S.; Pina-Crespo, J.C.; Roberts, A.J.; Verma, I.M.; et al. Astrocytes contribute to gamma oscillations and recognition memory. Proc. Natl. Acad. Sci. USA 2014, 111, E3343–E3352. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Chen, M.; Wang, F.; Windrem, M.; Wang, S.; Shanz, S.; Xu, Q.; Oberheim, N.A.; Bekar, L.; Betstadt, S.; et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 2013, 12, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Kol, A.; Adamsky, A.; Groysman, M.; Kreisel, T.; London, M.; Goshen, I. Astrocytes contribute to remote memory formation by modulating hippocampal-cortical communication during learning. Nat. Neurosci. 2020, 23, 1229–1239. [Google Scholar] [CrossRef]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208.e196. [Google Scholar] [CrossRef]
- Marty, N.; Dallaporta, M.; Foretz, M.; Emery, M.; Tarussio, D.; Bady, I.; Binnert, C.; Beermann, F.; Thorens, B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J. Clin. Investig. 2005, 115, 3545–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [Green Version]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogi, M.; Horiuchi, M. Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ. J. 2011, 75, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Taheri, S.; Gasparovic, C.; Huisa, B.N.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Grossetete, M.; Shah, N.J.; Wills, J.; Qualls, C.; et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke 2011, 42, 2158–2163. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Gu, C.; Mandeville, E.T.; Dong, Y.; Esposito, E.; Zhang, Y.; Yang, G.; Shen, Y.; Fu, X.; Lo, E.H.; et al. Anesthesia and Surgery Impair Blood-Brain Barrier and Cognitive Function in Mice. Front. Immunol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Pan, Y.; Short, J.L.; Choy, K.H.; Zeng, A.X.; Marriott, P.J.; Owada, Y.; Scanlon, M.J.; Porter, C.J.; Nicolazzo, J.A. Fatty Acid-Binding Protein 5 at the Blood-Brain Barrier Regulates Endogenous Brain Docosahexaenoic Acid Levels and Cognitive Function. J. Neurosci. 2016, 36, 11755–11767. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.; Kornhuber, J. Blood-cerebrospinal fluid barrier in schizophrenic patients. Eur. Arch. Psychiatry Neurol. Sci. 1987, 236, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Kirch, D.G.; Kaufmann, C.A.; Papadopoulos, N.M.; Martin, B.; Weinberger, D.R. Abnormal cerebrospinal fluid protein indices in schizophrenia. Biol. Psychiatry 1985, 20, 1039–1046. [Google Scholar] [CrossRef]
- Kirch, D.G.; Alexander, R.C.; Suddath, R.L.; Papadopoulos, N.M.; Kaufmann, C.A.; Daniel, D.G.; Wyatt, R.J. Blood-CSF barrier permeability and central nervous system immunoglobulin G in schizophrenia. J. Neural. Transm. Gen. Sect. 1992, 89, 219–232. [Google Scholar] [CrossRef]
- Muller, N.; Ackenheil, M. Immunoglobulin and albumin content of cerebrospinal fluid in schizophrenic patients: Relationship to negative symptomatology. Schizophr. Res. 1995, 14, 223–228. [Google Scholar] [CrossRef]
- Orlovska-Waast, S.; Kohler-Forsberg, O.; Brix, S.W.; Nordentoft, M.; Kondziella, D.; Krogh, J.; Benros, M.E. Cerebrospinal fluid markers of inflammation and infections in schizophrenia and affective disorders: A systematic review and meta-analysis. Mol. Psychiatry 2019, 24, 869–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meixensberger, S.; Bechter, K.; Dersch, R.; Feige, B.; Maier, S.; Schiele, M.A.; Runge, K.; Denzel, D.; Nickel, K.; Spieler, D.; et al. Sex difference in cerebrospinal fluid/blood albumin quotients in patients with schizophreniform and affective psychosis. Fluids Barriers CNS 2020, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Lara, D.R.; Gama, C.S.; Belmonte-de-Abreu, P.; Portela, L.V.; Goncalves, C.A.; Fonseca, M.; Hauck, S.; Souza, D.O. Increased serum S100B protein in schizophrenia: A study in medication-free patients. J. Psychiatr. Res. 2001, 35, 11–14. [Google Scholar] [CrossRef]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Riedel, G.; Platt, B.; Micheau, J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003, 140, 1–47. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Quack, G. Glutamate in CNS disorders as a target for drug development: An update. Drug News Perspect 1998, 11, 523–569. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, R.; Meyer, T.M.; Coyle, J.T.; Greene, R.W. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc. Natl. Acad. Sci. USA 1998, 95, 15730–15734. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Muhlhauser, M.; Yang, C.R. Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J. Neurophysiol. 2003, 89, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Borowsky, B.; Mezey, E.; Hoffman, B.J. Two glycine transporter variants with distinct localization in the CNS and peripheral tissues are encoded by a common gene. Neuron 1993, 10, 851–863. [Google Scholar] [CrossRef]
- Papouin, T.; Dunphy, J.; Tolman, M.; Foley, J.C.; Haydon, P.G. Astrocytic control of synaptic function. Philos Trans. R Soc. Lond B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lozada, Z.; Ortega, A. Glutamatergic Transmission: A Matter of Three. Neural Plast. 2015, 2015, 787396. [Google Scholar] [CrossRef] [Green Version]
- Xin, W.; Mironova, Y.A.; Shen, H.; Marino, R.A.M.; Waisman, A.; Lamers, W.H.; Bergles, D.E.; Bonci, A. Oligodendrocytes Support Neuronal Glutamatergic Transmission via Expression of Glutamine Synthetase. Cell Rep. 2019, 27, 2262–2271. [Google Scholar] [CrossRef] [Green Version]
- Ehmsen, J.T.; Ma, T.M.; Sason, H.; Rosenberg, D.; Ogo, T.; Furuya, S.; Snyder, S.H.; Wolosker, H. D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J. Neurosci. 2013, 33, 12464–12469. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Yamada, K.; Furuya, S.; Mitoma, J.; Hirabayashi, Y.; Watanabe, M. 3-Phosphoglycerate dehydrogenase, a key enzyme for l-serine biosynthesis, is preferentially expressed in the radial glia/astrocyte lineage and olfactory ensheathing glia in the mouse brain. J. Neurosci. 2001, 21, 7691–7704. [Google Scholar] [CrossRef] [Green Version]
- Neame, S.; Safory, H.; Radzishevsky, I.; Touitou, A.; Marchesani, F.; Marchetti, M.; Kellner, S.; Berlin, S.; Foltyn, V.N.; Engelender, S.; et al. The NMDA receptor activation by d-serine and glycine is controlled by an astrocytic Phgdh-dependent serine shuttle. Proc. Natl. Acad. Sci. USA 2019, 116, 20736–20742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Wada, A.; Yoshida, K.; Miyoshi, Y.; Sayano, T.; Esaki, K.; Kinoshita, M.O.; Tomonaga, S.; Azuma, N.; Watanabe, M.; et al. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. J. Biol Chem. 2010, 285, 41380–41390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulenburg, V.; Armsen, W.; Betz, H.; Gomeza, J. Glycine transporters: Essential regulators of neurotransmission. Trends Biochem. Sci. 2005, 30, 325–333. [Google Scholar] [CrossRef]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.; Shen, H.Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.F. Deletion of adenosine A2A receptors from astrocytes disrupts glutamate homeostasis leading to psychomotor and cognitive impairment: Relevance to schizophrenia. Biol. Psychiatry 2015, 78, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros-Yáñez, I.; Castillo, C.A.; Merighi, S.; Gessi, S. The role of adenosine receptors in psychostimulant addiction. Front. Pharmacol. 2018, 8, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halberstadt, A.L. The phencyclidine-glutamate model of schizophrenia. Clin. Neuropharmacol. 1995, 18, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Bickel, S.; Javitt, D.C. Neurophysiological and neurochemical animal models of schizophrenia: Focus on glutamate. Behav. Brain Res. 2009, 204, 352–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsel, P.; Byne, W.; Roussos, P.; Tan, W.; Siever, L.; Haroutunian, V. Astrocyte and glutamate markers in the superficial, deep, and white matter layers of the anterior cingulate gyrus in schizophrenia. Neuropsychopharmacology 2011, 36, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Steffek, A.E.; McCullumsmith, R.E.; Haroutunian, V.; Meador-Woodruff, J.H. Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr. Res. 2008, 103, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullumsmith, R.E.; O’Donovan, S.M.; Drummond, J.B.; Benesh, F.S.; Simmons, M.; Roberts, R.; Lauriat, T.; Haroutunian, V.; Meador-Woodruff, J.H. Cell-specific abnormalities of glutamate transporters in schizophrenia: Sick astrocytes and compensating relay neurons? Mol. Psychiatry 2016, 21, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Arguello, P.A.; Gogos, J.A. Modeling madness in mice: One piece at a time. Neuron 2006, 52, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.M.; Miyakawa, T. Schizophrenia-relevant behavioral testing in rodent models: A uniquely human disorder? Biol. Psychiatry 2006, 59, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellendonk, C.; Simpson, E.H.; Kandel, E.R. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci. 2009, 32, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J.; Hyman, S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, J.L.; Corvin, A.P.; Donohoe, G.; O’Tuathaigh, C.M.; Mitchell, K.J.; Gill, M. Functional genomics and schizophrenia: Endophenotypes and mutant models. Psychiatr. Clin. North. Am. 2007, 30, 365–399. [Google Scholar] [CrossRef]
- Xia, M.; Abazyan, S.; Jouroukhin, Y.; Pletnikov, M. Behavioral sequelae of astrocyte dysfunction: Focus on animal models of schizophrenia. Schizophr. Res. 2016, 176, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Burbaeva, G.; Boksha, I.S.; Turishcheva, M.S.; Vorobyeva, E.A.; Savushkina, O.K.; Tereshkina, E.B. Glutamine synthetase and glutamate dehydrogenase in the prefrontal cortex of patients with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 675–680. [Google Scholar] [CrossRef]
- Matute, C.; Melone, M.; Vallejo-Illarramendi, A.; Conti, F. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia 2005, 49, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, M.; Melone, M.; Gubbini, A.; Battistacci, S.; Conti, F. GLT-1 upregulation impairs prepulse inhibition of the startle reflex in adult rats. Glia 2009, 57, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Matos-Ocasio, F.; Hernandez-Lopez, A.; Thompson, K.J. Ceftriaxone, a GLT-1 transporter activator, disrupts hippocampal learning in rats. Pharmacol. Biochem. Behav. 2014, 122, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Son, H.; Kim, S.; Jung, D.H.; Baek, J.H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Lee, D.K.; et al. Insufficient glutamine synthetase activity during synaptogenesis causes spatial memory impairment in adult mice. Sci. Rep. 2019, 9, 252. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Li, Y.; Huang, Y.; Zhao, H.; Gui, L. Adenosine receptor A1-A2a heteromers regulate EAAT2 expression and glutamate uptake via YY1-induced repression of PPARgamma transcription. PPAR Res. 2020, 2020, 2410264. [Google Scholar] [CrossRef] [Green Version]
- Balu, D.T.; Coyle, J.T. The NMDA receptor ‘glycine modulatory site’ in schizophrenia: D-serine, glycine, and beyond. Curr. Opin. Pharmacol. 2015, 20, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov. 2013, 12, 866–885. [Google Scholar] [CrossRef]
- Marques, B.L.; Oliveira-Lima, O.C.; Carvalho, G.A.; de Almeida Chiarelli, R.; Ribeiro, R.I.; Parreira, R.C.; da Madeira Freitas, E.M.; Resende, R.R.; Klempin, F.; Ulrich, H.; et al. Neurobiology of glycine transporters: From molecules to behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef]
- Mihali, A.; Subramani, S.; Kaunitz, G.; Rayport, S.; Gaisler-Salomon, I. Modeling resilience to schizophrenia in genetically modified mice: A novel approach to drug discovery. Expert Rev. Neurother. 2012, 12, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, H.; Boison, D.; Singer, P.; Feldon, J.; Pauly-Evers, M.; Yee, B.K. Glycine transporter 1 as a potential therapeutic target for schizophrenia-related symptoms: Evidence from genetically modified mouse models and pharmacological inhibition. Biochem. Pharmacol. 2011, 81, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.; Ralph-Williams, R.J.; Martina, M.; Bergeron, R.; Berger-Sweeney, J.; Dunham, K.S.; Jiang, Z.; Caine, S.B.; Coyle, J.T. Gene knockout of glycine transporter 1: Characterization of the behavioral phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Murtas, G.; Marcone, G.L.; Sacchi, S.; Pollegioni, L. L-serine synthesis via the phosphorylated pathway in humans. Cell Mol. Life Sci. 2020, 77, 5131–5148. [Google Scholar] [CrossRef]
- Wolosker, H.; Blackshaw, S.; Snyder, S.H. Serine racemase: A glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc. Natl. Acad. Sci. USA 1999, 96, 13409–13414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.K.; Shishido, Y.; Ichise-Shishido, S.; Kawazoe, T.; Ono, K.; Iwana, S.; Tomita, Y.; Yorita, K.; Sakai, T.; Fukui, K. Potential role for astroglial D-amino acid oxidase in extracellular D-serine metabolism and cytotoxicity. J. Biochem. 2006, 139, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Furuya, S.; Yoshida, K.; Kawakami, Y.; Yang, J.H.; Sayano, T.; Azuma, N.; Tanaka, H.; Kuhara, S.; Hirabayashi, Y. Inactivation of the 3-phosphoglycerate dehydrogenase gene in mice: Changes in gene expression and associated regulatory networks resulting from serine deficiency. Funct. Integr. Genom. 2008, 8, 235–249. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jego, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e508. [Google Scholar] [CrossRef]
- Balu, D.T.; Takagi, S.; Puhl, M.D.; Benneyworth, M.A.; Coyle, J.T. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol. Neurobiol. 2014, 34, 419–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benneyworth, M.A.; Li, Y.; Basu, A.C.; Bolshakov, V.Y.; Coyle, J.T. Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol. Neurobiol. 2012, 32, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Verrall, L.; Burnet, P.W.; Betts, J.F.; Harrison, P.J. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol. Psychiatry 2010, 15, 122–137. [Google Scholar] [CrossRef]
- Schell, M.J.; Molliver, M.E.; Snyder, S.H. D-serine, an endogenous synaptic modulator: Localization to astrocytes and glutamate-stimulated release. Proc. Natl. Acad. Sci. USA 1995, 92, 3948–3952. [Google Scholar] [CrossRef] [Green Version]
- Balu, D.T.; Li, Y.; Puhl, M.D.; Benneyworth, M.A.; Basu, A.C.; Takagi, S.; Bolshakov, V.Y.; Coyle, J.T. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. USA 2013, 110, E2400–E2409. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.C.; Tsai, G.E.; Ma, C.L.; Ehmsen, J.T.; Mustafa, A.K.; Han, L.; Jiang, Z.I.; Benneyworth, M.A.; Froimowitz, M.P.; Lange, N.; et al. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol. Psychiatry 2009, 14, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVito, L.M.; Balu, D.T.; Kanter, B.R.; Lykken, C.; Basu, A.C.; Coyle, J.T.; Eichenbaum, H. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology. Genes Brain Behav. 2011, 10, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, T.M.; Pisansky, M.T.; Young, A.; Miller, R.F.; Gewirtz, J.C. Sociality deficits in serine racemase knockout mice. Brain Behav. 2019, 9, e01383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.C.; Hung, W.L.; Lin, B.X.; Shih, M.H.; Lu, L.Y.; Luo, D.Z.; Tai, H.C.; Studer, V.; Min, M.Y.; Lai, W.S. Therapeutic potential and underlying mechanism of sarcosine (N-methylglycine) in N-methyl-D-aspartate (NMDA) receptor hypofunction models of schizophrenia. J. Psychopharmacol. 2019, 33, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Labrie, V.; Wong, A.H.; Roder, J.C. Contributions of the D-serine pathway to schizophrenia. Neuropharmacology 2012, 62, 1484–1503. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; Bernasconi, M.; Martineau, M.; Mothet, J.P.; Ruzzene, M.; Pilone, M.S.; Pollegioni, L.; Molla, G. pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: Effect on schizophrenia susceptibility. J. Biol. Chem. 2008, 283, 22244–22256. [Google Scholar] [CrossRef] [Green Version]
- Hambsch, B.; Keyworth, H.; Lind, J.; Otte, D.M.; Racz, I.; Kitchen, I.; Bailey, A.; Zimmer, A. Chronic nicotine improves short-term memory selectively in a G72 mouse model of schizophrenia. Br. J. Pharmacol. 2014, 171, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Otte, D.M.; Bilkei-Gorzo, A.; Filiou, M.D.; Turck, C.W.; Yilmaz, O.; Holst, M.I.; Schilling, K.; Abou-Jamra, R.; Schumacher, J.; Benzel, I.; et al. Behavioral changes in G72/G30 transgenic mice. Eur Neuropsychopharmacol. 2009, 19, 339–348. [Google Scholar] [CrossRef]
- Otte, D.M.; Sommersberg, B.; Kudin, A.; Guerrero, C.; Albayram, O.; Filiou, M.D.; Frisch, P.; Yilmaz, O.; Drews, E.; Turck, C.W.; et al. N-acetyl cysteine treatment rescues cognitive deficits induced by mitochondrial dysfunction in G72/G30 transgenic mice. Neuropsychopharmacology 2011, 36, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Erta, M.; Ferrer, B.; Comes, G.; Giralt, M.; Hidalgo, J. Astrocyte-specific deficiency of interleukin-6 and its receptor reveal specific roles in survival, body weight and behavior. Brain Behav. Immun. 2013, 27, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.S.; Chang, C.Y.; Wong, W.R.; Pei, J.C.; Chen, Y.S.; Hong, W.L. Assessing schizophrenia-relevant cognitive and social deficits in mice: A selection of mouse behavioral tasks and potential therapeutic compounds. Curr. Pharm. Des. 2014, 20, 5139–5150. [Google Scholar] [CrossRef] [PubMed]
- Young, J.W.; Powell, S.B.; Risbrough, V.; Marston, H.M.; Geyer, M.A. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol. Ther. 2009, 122, 150–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barch, D.M.; Carter, C.S.; Arnsten, A.; Buchanan, R.W.; Cohen, J.D.; Geyer, M.; Green, M.F.; Krystal, J.H.; Nuechterlein, K.; Robbins, T.; et al. Selecting paradigms from cognitive neuroscience for translation into use in clinical trials: Proceedings of the third CNTRICS meeting. Schizophr. Bull. 2009, 35, 109–114. [Google Scholar] [CrossRef]
- Velligan, D.I.; Kern, R.S.; Gold, J.M. Cognitive rehabilitation for schizophrenia and the putative role of motivation and expectancies. Schizophr. Bull. 2006, 32, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, A.; Barlati, S.; Ceraso, A.; Nibbio, G.; Ariu, C.; Deste, G.; Wykes, T. Effectiveness, Core Elements, and Moderators of Response of Cognitive Remediation for Schizophrenia: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Elvevåg, B.; Goldberg, T.E. Cognitive impairment in schizophrenia is the core of the disorder. Crit. Rev. Neurobiol. 2000, 14, 1–21. [Google Scholar] [CrossRef]
- Crinella, F.M.; Yu, J. Brain mechanisms in problem solving and intelligence: A replication and extension. Intelligence 1995, 21, 225–246. [Google Scholar] [CrossRef]
- Galsworthy, M.J.; Paya-Cano, J.L.; Liu, L.; Monleon, S.; Gregoryan, G.; Fernandes, C.; Schalkwyk, L.C.; Plomin, R. Assessing reliability, heritability and general cognitive ability in a battery of cognitive tasks for laboratory mice. Behav. Genet. 2005, 35, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Galsworthy, M.J.; Paya-Cano, J.L.; Monleón, S.; Plomin, R. Evidence for general cognitive ability (g) in heterogeneous stock mice and an analysis of potential confounds. Genes Brain Behav. 2002, 1, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Ben Abdallah, N.M.; Fuss, J.; Trusel, M.; Galsworthy, M.J.; Bobsin, K.; Colacicco, G.; Deacon, R.M.; Riva, M.A.; Kellendonk, C.; Sprengel, R.; et al. The puzzle box as a simple and efficient behavioral test for exploring impairments of general cognition and executive functions in mouse models of schizophrenia. Exp. Neurol. 2011, 227, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, M.; Mielnik, C.A.; Ramsey, A.J. NMDA receptor-deficient mice display sexual dimorphism in the onset and severity of behavioural abnormalities. Genes Brain. Behav. 2014, 13, 850–862. [Google Scholar] [CrossRef]
- Cornblatt, B.A.; Keilp, J.G. Impaired attention, genetics, and the pathophysiology of schizophrenia. Schizophr. Bull. 1994, 20, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, Y.; Robbins, T.W. Psychopharmacological approaches to modulating attention in the five-choice serial reaction time task: Implications for schizophrenia. Psychopharmacology 2004, 174, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Tucci, V.; Hardy, A.; Nolan, P.M. A comparison of physiological and behavioural parameters in C57BL/6J mice undergoing food or water restriction regimes. Behav. Brain Res. 2006, 173, 22–29. [Google Scholar] [CrossRef]
- Alkam, T.; Hiramatsu, M.; Mamiya, T.; Aoyama, Y.; Nitta, A.; Yamada, K.; Kim, H.C.; Nabeshima, T. Evaluation of object-based attention in mice. Behav. Brain Res. 2011, 220, 185–193. [Google Scholar] [CrossRef]
- Wulaer, B.; Kunisawa, K.; Kubota, H.; Suento, W.J.; Saito, K.; Mouri, A.; Nabeshima, T. Prefrontal cortex, dorsomedial striatum, and dentate gyrus are necessary in the object-based attention test in mice. Mol. Brain 2020, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Salamone, J.D.; Correa, M.; Ferrigno, S.; Yang, J.H.; Rotolo, R.A.; Presby, R.E. The Psychopharmacology of Effort-Related Decision Making: Dopamine, Adenosine, and Insights into the Neurochemistry of Motivation. Pharmacol. Rev. 2018, 70, 747–762. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Sweet, R.A. Schizophrenia from a neural circuitry perspective: Advancing toward rational pharmacological therapies. J. Clin. Investig. 2009, 119, 706–716. [Google Scholar] [CrossRef]
- Salamone, J.D.; Steinpreis, R.E.; McCullough, L.D.; Smith, P.; Grebel, D.; Mahan, K. Haloperidol and nucleus accumbens dopamine depletion suppress lever pressing for food but increase free food consumption in a novel food choice procedure. Psychopharmacology 1991, 104, 515–521. [Google Scholar] [CrossRef]
- Sommer, S.; Danysz, W.; Russ, H.; Valastro, B.; Flik, G.; Hauber, W. The dopamine reuptake inhibitor MRZ-9547 increases progressive ratio responding in rats. Int J. Neuropsychopharmacol. 2014, 17, 2045–2056. [Google Scholar] [CrossRef] [Green Version]
- Salamone, J.D.; Cousins, M.S.; Bucher, S. Anhedonia or anergia? Effects of haloperidol and nucleus accumbens dopamine depletion on instrumental response selection in a T-maze cost/benefit procedure. Behav Brain Res. 1994, 65, 221–229. [Google Scholar] [CrossRef]
- Yohn, S.E.; Thompson, C.; Randall, P.A.; Lee, C.A.; Muller, C.E.; Baqi, Y.; Correa, M.; Salamone, J.D. The VMAT-2 inhibitor tetrabenazine alters effort-related decision making as measured by the T-maze barrier choice task: Reversal with the adenosine A2A antagonist MSX-3 and the catecholamine uptake blocker bupropion. Psychopharmacology 2015, 232, 1313–1323. [Google Scholar] [CrossRef]
- Farrar, A.M.; Font, L.; Pereira, M.; Mingote, S.; Bunce, J.G.; Chrobak, J.J.; Salamone, J.D. Forebrain circuitry involved in effort-related choice: Injections of the GABAA agonist muscimol into ventral pallidum alter response allocation in food-seeking behavior. Neuroscience 2008, 152, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Floresco, S.B.; Tse, M.T.; Ghods-Sharifi, S. Dopaminergic and glutamatergic regulation of effort- and delay-based decision making. Neuropsychopharmacology 2008, 33, 1966–1979. [Google Scholar] [CrossRef] [PubMed]
- Dallerac, G.; Chever, O.; Rouach, N. How do astrocytes shape synaptic transmission? Insights from electrophysiology. Front. Cell Neurosci. 2013, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, A.M.; Spellman, T.; Gordon, J.A. Electrophysiological endophenotypes in rodent models of schizophrenia and psychosis. Biol. Psychiatry 2015, 77, 1041–1049. [Google Scholar] [CrossRef] [Green Version]
- Pirttimaki, T.M.; Sims, R.E.; Saunders, G.; Antonio, S.A.; Codadu, N.K.; Parri, H.R. Astrocyte-Mediated Neuronal Synchronization Properties Revealed by False Gliotransmitter Release. J. Neurosci. 2017, 37, 9859–9870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, I.; Vazquez-Juarez, E.; Lindskog, M. Reducing Glutamate Uptake in Rat Hippocampal Slices Enhances Astrocytic Membrane Depolarization While Down-Regulating CA3-CA1 Synaptic Response. Front. Synaptic Neurosci. 2020, 12, 37. [Google Scholar] [CrossRef]
- Mishima, T.; Sakatani, S.; Hirase, H. Intracellular labeling of single cortical astrocytes in vivo. J. Neurosci. Methods 2007, 166, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Poskanzer, K.E.; Yuste, R. Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sc.i USA 2016, 113, E2675–E2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.W.; Kao, H.Y.; Min, M.Y.; Lai, W.S. A sex- and region-specific role of akt1 in the modulation of methamphetamine-induced hyperlocomotion and striatal neuronal activity: Implications in schizophrenia and methamphetamine-induced psychosis. Schizophr. Bull. 2014, 40, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.H.; Pei, J.C.; Luo, D.Z.; Chen, C.; Chen, Y.W.; Lai, W.S. Investigation of gene effects and epistatic interactions between Akt1 and neuregulin 1 in the regulation of behavioral phenotypes and social functions in genetic mouse models of schizophrenia. Front. Behav. Neurosci. 2014, 8, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.H.; Vyas, N.S.; Puri, B.K.; Nijran, K.S.; Al-Nahhas, A. Positron emission tomography in schizophrenia: A new perspective. J. Nucl. Med. 2010, 51, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Virdee, K.; Cumming, P.; Caprioli, D.; Jupp, B.; Rominger, A.; Aigbirhio, F.I.; Fryer, T.D.; Riss, P.J.; Dalley, J.W. Applications of positron emission tomography in animal models of neurological and neuropsychiatric disorders. Neurosci. Biobehav. Rev. 2012, 36, 1188–1216. [Google Scholar] [CrossRef]
- Zimmer, E.R.; Parent, M.J.; Souza, D.G.; Leuzy, A.; Lecrux, C.; Kim, H.I.; Gauthier, S.; Pellerin, L.; Hamel, E.; Rosa-Neto, P. [(18)F]FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci. 2017, 20, 393–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.F.; Scholl, M.; Almkvist, O.; Wall, A.; Engler, H.; Langstrom, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vieitez, E.; Ni, R.; Gulyas, B.; Toth, M.; Haggkvist, J.; Halldin, C.; Voytenko, L.; Marutle, A.; Nordberg, A. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: A correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Owen, F.; Crow, T.J.; Frith, C.D.; Johnson, J.A.; Johnstone, E.C.; Lofthouse, R.; Owens, D.G.; Poulter, M. Selective decreases in MAO-B activity in post-mortem brains from schizophrenic patients with type II syndrome. Br. J. Psychiatry J. Ment. Sci. 1987, 151, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.L.; Li, C.X.; Li, S.B.; Liu, Y.; Hu, L. Association study of monoamine oxidase A/B genes and schizophrenia in Han Chinese. Behav. Brain Funct. 2011, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merali, Z.; Wong, T.; Leung, J.; Gao, M.M.; Mikulis, D.; Kassner, A. Dynamic contrast-enhanced MRI and CT provide comparable measurement of blood-brain barrier permeability in a rodent stroke model. Magn. Reson. Imaging 2015, 33, 1007–1012. [Google Scholar] [CrossRef]
- Yu, M.; Yang, D.; Wang, M.; Wei, X.; Li, W. Early stage of diffusional kurtosis imaging and dynamic contrast-enhanced magnetic resonance imaging correlated with long-term neurocognitive function after experimental traumatic brain injury. Neurosci. Lett. 2019, 705, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Dickie, B.R.; Vandesquille, M.; Ulloa, J.; Boutin, H.; Parkes, L.M.; Parker, G.J.M. Water-exchange MRI detects subtle blood-brain barrier breakdown in Alzheimer’s disease rats. Neuroimage 2019, 184, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benveniste, H.; Lee, H.; Ozturk, B.; Chen, X.; Koundal, S.; Vaska, P.; Tannenbaum, A.; Volkow, N.D. Glymphatic Cerebrospinal Fluid and Solute Transport Quantified by MRI and PET Imaging. Neuroscience 2020. [Google Scholar] [CrossRef] [PubMed]
- Saxton, M.J. Single-particle tracking: Connecting the dots. Nat. Methods 2008, 5, 671–672. [Google Scholar] [CrossRef]
- Shen, H.; Tauzin, L.J.; Baiyasi, R.; Wang, W.; Moringo, N.; Shuang, B.; Landes, C.F. Single Particle Tracking: From Theory to Biophysical Applications. Chem. Rev. 2017, 117, 7331–7376. [Google Scholar] [CrossRef]
- Al Awabdh, S.; Gupta-Agarwal, S.; Sheehan, D.F.; Muir, J.; Norkett, R.; Twelvetrees, A.E.; Griffin, L.D.; Kittler, J.T. Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 2016, 64, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Arizono, M.; Bannai, H.; Nakamura, K.; Niwa, F.; Enomoto, M.; Matsu-Ura, T.; Miyamoto, A.; Sherwood, M.W.; Nakamura, T.; Mikoshiba, K. Receptor-selective diffusion barrier enhances sensitivity of astrocytic processes to metabotropic glutamate receptor stimulation. Sci. Signal. 2012, 5, ra27. [Google Scholar] [CrossRef]
- Crane, J.M.; Van Hoek, A.N.; Skach, W.R.; Verkman, A.S. Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by quantum dot single particle tracking. Mol. Biol. Cell 2008, 19, 3369–3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, H. Molecular membrane dynamics: Insights into synaptic function and neuropathological disease. Neurosci. Res. 2018, 129, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ballas, N.; Lioy, D.T.; Grunseich, C.; Mandel, G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 2009, 12, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Doering, L.C. Astrocytes prevent abnormal neuronal development in the fragile x mouse. J. Neurosci. 2010, 30, 4508–4514. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Li, B.; Verkhratsky, A. Targeting astrocytes in bipolar disorder. Expert Rev. Neurother. 2016, 16, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Bochukova, E.G.; Lawler, K.; Croizier, S.; Keogh, J.M.; Patel, N.; Strohbehn, G.; Lo, K.K.; Humphrey, J.; Hokken-Koelega, A.; Damen, L.; et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018, 22, 3401–3408. [Google Scholar] [CrossRef] [Green Version]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-Y.; Luo, D.-Z.; Pei, J.-C.; Kuo, M.-C.; Hsieh, Y.-C.; Lai, W.-S. Not Just a Bystander: The Emerging Role of Astrocytes and Research Tools in Studying Cognitive Dysfunctions in Schizophrenia. Int. J. Mol. Sci. 2021, 22, 5343. https://doi.org/10.3390/ijms22105343
Chang C-Y, Luo D-Z, Pei J-C, Kuo M-C, Hsieh Y-C, Lai W-S. Not Just a Bystander: The Emerging Role of Astrocytes and Research Tools in Studying Cognitive Dysfunctions in Schizophrenia. International Journal of Molecular Sciences. 2021; 22(10):5343. https://doi.org/10.3390/ijms22105343
Chicago/Turabian StyleChang, Chia-Yuan, Da-Zhong Luo, Ju-Chun Pei, Ming-Che Kuo, Yi-Chen Hsieh, and Wen-Sung Lai. 2021. "Not Just a Bystander: The Emerging Role of Astrocytes and Research Tools in Studying Cognitive Dysfunctions in Schizophrenia" International Journal of Molecular Sciences 22, no. 10: 5343. https://doi.org/10.3390/ijms22105343