
In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Liver Structure and Function
3. Mechanisms of General Cytotoxicity
3.1. Initial Injury
3.2. Mitochondrial Dysfunction
3.3. Cell Death
4. General Cytotoxicity In Vitro Methods
4.1. Membrane Integrity Assays
4.1.1. LDH Leakage Assay
4.1.2. Calcein-AM Assay
4.1.3. Protease Activity Assay
4.1.4. Trypan Blue Exclusion Assay
4.2. Mitochondrial Functionality Assays
4.2.1. Tetrazolium Salt Assays
4.2.2. Resazurin Reduction Assay
4.2.3. ATP Content Assay
4.2.4. Mitochondrial Membrane Potential Evaluation: Fluorescent Probe-Based Assays
4.3. Oxidative Stress Assays
4.3.1. Intracellular ROS Quantification: DCFH2-DA Fluorescence Probe-Based Assay
4.3.2. Intracellular/Mitochondrial Superoxide Quantification: DHE/Mito-HE Fluorescence Probe-Based Assays
4.3.3. Lipid Peroxidation: MDA/TBARS Assay
4.3.4. Antioxidant Status Assays: Evaluation of Enzymatic Antioxidant Activity
4.4. Cell Death Assays
4.4.1. Annexin V Staining Assay
4.4.2. PI dye Uptake Assay
4.4.3. Caspase Activity Assays
4.4.4. TUNEL Assay
4.5. Miscellaneous Assays: Neutral Red Uptake
5. Mechanisms of Liver-Specific Toxicity
5.1. Cholestasis
5.2. Steatosis
5.3. Fibrosis
6. Liver-Specific Toxicity In Vitro Methods
6.1. Cholestasis Assays
6.1.1. Transporter Inhibition Assays
Tauro-nor-THCA-24-DBD
CLF
CDFDA
TCA
Estradiol-17β-Glucuronide and CCK8
6.1.2. Drug-Induced Cholestasis Assay
6.2. Steatosis Assays
6.2.1. Lipid Quantification Assays
Oil Red O Staining
Nile Red Staining
BODIPY 493/503 staining
Absolute Lipid Quantification Assays
6.2.2. FAO Assays
6.2.3. FA Efflux Assays
6.3. Fibrosis Assays
6.3.1. Collagen Quantification Assays
Sirius Red Staining
Hydroxyproline Assay
Quantification via Immunoassays
6.3.2. MMP and TIMP Quantification: Zymography
6.3.3. HSC Activation Assays
Contraction Assay
α-SMA Quantification
7. Critical Parameters for Practical In Vitro Liver Toxicity Testing
7.1. Selection of the In Vitro Models
7.2. Selection of the In Vitro Assays
7.3. Selection of the Test Conditions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goldberg, D.S.; Forde, K.A.; Carbonari, D.M.; Lewis, J.D.; Leidl, K.B.; Reddy, K.R.; Haynes, K.; Roy, J.; Sha, D.; Marks, A.R.; et al. Population-Representative Incidence of Drug-Induced Acute Liver Failure Based on an Analysis of an Integrated Health Care System. Gastroenterology 2015, 148, 1353–1361.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germani, G.; Theocharidou, E.; Adam, R.; Karam, V.; Wendon, J.; O’Grady, J.; Burra, P.; Senzolo, M.; Mirza, D.; Castaing, D.; et al. Liver transplantation for acute liver failure in Europe: Outcomes over 20years from the ELTR database. J. Hepatol. 2012, 57, 288–296. [Google Scholar] [CrossRef]
- Lee, W.M. Drug-induced Acute Liver Failure. Clin. Liver Dis. 2013, 17, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Vilas-Boas, V.; Gijbels, E.; Cooreman, A.; Van Campenhout, R.; Gustafson, E.; Leroy, K.; Vinken, M. Industrial, Biocide, and Cosmetic Chemical Inducers of Cholestasis. Chem. Res. Toxicol. 2019, 32, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Gijbels, E.; Jonckheer, J.; De Waele, E.; Vinken, M. Cholestatic liver injury induced by food additives, dietary supplements and parenteral nutrition. Environ. Int. 2020, 136, 105422. [Google Scholar] [CrossRef]
- Organization for Economic Co-Operation and Development (OECD). Available online: https://www.oecd.org/ (accessed on 25 February 2021).
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US National Academy of Sciences. Toxicity Testing in the 21st Century: A Vision and a Strategy; The National Academies Press: Washington, DC, USA, 2007; pp. 1–196. [Google Scholar]
- Riss, T.; Niles, A.; Moravec, R.; Karassina, N.; Vidugiriene, J. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. In Assay Guidance Manual; Markossian, S., Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Niles, A.L.; Moravec, R.A.; Riss, T.L. Update onin vitrocytotoxicity assays for drug development. Expert Opin. Drug Discov. 2008, 3, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.M.; Noble, C. In vitro inhibition of lactate dehydrogenases by kepone. J. Agric. Food Chem. 1977, 25, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Kendig, D.M.; Tarloff, J.B. Inactivation of lactate dehydrogenase by several chemicals: Implications for in vitro toxicology studies. Toxicol. Vitro 2007, 21, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Mariani, E.; Meneghetti, A.; Cattini, L.; Facchini, A. Calcein-Acetyoxymethyl Cytotoxicity Assay: Standardization of a Method Allowing Additional Analyses on Recovered Effector Cells and Supernatants. Clin. Diagn. Lab. Immunol. 2001, 8, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Miles, F.; Lynch, J.; Sikes, R. Cell-based assays using calcein acetoxymethyl ester show variation in fluorescence with treatment conditions. J. Biol. Methods 2015, 2, e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niles, A.L.; Moravec, R.A.; Hesselberth, P.E.; Scurria, M.A.; Daily, W.J.; Riss, T.L. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal. Biochem. 2007, 366, 197–206. [Google Scholar] [CrossRef]
- Nyles, A.L.; Scurria, M.A.; Bernad, L.; McNamara, B.; Rashka, K.; Lange, D.; Guthmiller, P.; Riss, T.; Corporation, P.; Biosciences, P. Using protease biomarkers to measure viability and cytotoxicity. Cell. Notes 2007, 9, 16–20. [Google Scholar]
- Piccinini, F.; Tesei, A.; Arienti, C.; Bevilacqua, A. Cell Counting and Viability Assessment of 2D and 3D Cell Cultures: Expected Reliability of the Trypan Blue Assay. Biol. Proced. Online 2017, 19, 8. [Google Scholar] [CrossRef]
- Yip, D.K.; Auersperg, N. The dye-exclusion test for cell viability: Persistence of differential staining following fixation. Vitro Cell. Dev. Biol. Anim. 1972, 7, 323–329. [Google Scholar] [CrossRef]
- Kim, S.I.; Kim, H.J.; Lee, H.-J.; Lee, K.; Hong, D.; Lim, H.; Cho, K.; Jung, N.; Yi, Y.W. Application of a non-hazardous vital dye for cell counting with automated cell counters. Anal. Biochem. 2016, 492, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Vistica, D.T.; Skehan, P.; Scudiero, D.; Monks, A.; Pittman, A.; Boyd, M.R. Tetrazolium-based assays for cellular viability: A critical examination of selected parameters affecting formazan production. Cancer Res. 1991, 51, 2515–2520. [Google Scholar]
- Van Tonder, A.; Joubert, A.M.; Cromarty, A.D. Limitations of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay when compared to three commonly used cell enumeration assays. BMC Res. Notes 2015, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.; Kundu, S.; Kumar, S.; Chakrabarti, R. Vitamin A as an enzyme that catalyzes the reduction of MTT to formazan by vitamin C. J. Cell Biochem. 2000, 80, 133–138. [Google Scholar] [CrossRef]
- Ulukaya, E.; Colakogullari, M.; Wood, E.J. Interference by Anti-Cancer Chemotherapeutic Agents in the MTT-Tumor Chemosensitivity Assay. Chemotherapy 2004, 50, 43–50. [Google Scholar] [CrossRef]
- Lü, L.; Zhang, L.; Wai, M.S.M.; Yew, D.T.W.; Xu, J. Exocytosis of MTT formazan could exacerbate cell injury. Toxicol. Vitro 2012, 26, 636–644. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Tolosa, L.; Donato, M.T.; Gómez-Lechón, M.J. General Cytotoxicity Assessment by Means of the MTT Assay. Methods Mol. Biol. 2014, 1250, 333–348. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple Applications of Alamar Blue as an Indicator of Metabolic Function and Cellular Health in Cell Viability Bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol. Vitro 2004, 18, 703–710. [Google Scholar] [CrossRef]
- Erikstein, B.S.; Hagland, H.R.; Nikolaisen, J.; Kulawiec, M.; Singh, K.K.; Gjertsen, B.T.; Tronstad, K.J. Cellular stress induced by resazurin leads to autophagy and cell death via production of reactive oxygen species and mitochondrial impairment. J. Cell. Biochem. 2010, 111, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Goegan, P.; Johnson, G.; Vincent, R. Effects of serum protein and colloid on the alamarBlue assay in cell cultures. Toxicol. Vitro 1995, 9, 257–266. [Google Scholar] [CrossRef]
- Lomakina, G.Y.; Modestova, Y.A.; Ugarova, N.N. Bioluminescence assay for cell viability. Biochemistry 2015, 80, 701–713. [Google Scholar] [CrossRef]
- Morciano, G.; Sarti, A.C.; Marchi, S.; Missiroli, S.; Falzoni, S.; Raffaghello, L.; Pistoia, V.; Giorgi, C.; Di Virgilio, F.; Pinton, P. Use of luciferase probes to measure ATP in living cells and animals. Nat. Protoc. 2017, 12, 1542–1562. [Google Scholar] [CrossRef]
- Petty, R.D.; Sutherland, L.A.; Hunter, E.M.; Cree, I.A. Comparison of MTT and ATP-based assays for the measurement of viable cell number. J. Biolumin. Chemilumin. 1995, 10, 29–34. [Google Scholar] [CrossRef]
- Kanemura, Y.; Mori, H.; Kobayashi, S.; Islam, O.; Kodama, E.; Yamamoto, A.; Nakanishi, Y.; Arita, N.; Yamasaki, M.; Okano, H.; et al. Evaluation of in vitro proliferative activity of human fetal neural stem/progenitor cells using indirect measurements of viable cells based on cellular metabolic activity. J. Neurosci. Res. 2002, 69, 869–879. [Google Scholar] [CrossRef]
- Kijanska, M.; Kelm, J. In vitro 3D Spheroids and Microtissues: ATP-based Cell Viability and Toxicity Assays. In Assay Guidance Manual; Markossian, S., Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Idrees, A.; Chiono, V.; Ciardelli, G.; Shah, S.; Viebahn, R.; Zhang, X.; Salber, J. Validation of in vitro assays in three-dimensional human dermal constructs. Int. J. Artif. Organs 2018, 41, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Sakamuru, S.; Attene-Ramos, M.S.; Xia, M. Mitochondrial Membrane Potential Assay. Methods Mol. Biol. 2016, 1473, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef] [PubMed]
- Sakamuru, S.; Li, X.; Attene-Ramos, M.S.; Huang, R.; Lu, J.; Shou, L.; Shen, M.; Tice, R.R.; Austin, C.P.; Xia, M. Application of a homogenous membrane potential assay to assess mitochondrial function. Physiol. Genom. 2012, 44, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhao, Y.; Yin, J.; Lin, W. Organic fluorescent probes for detecting mitochondrial membrane potential. Co-ord. Chem. Rev. 2020, 420, 213419. [Google Scholar] [CrossRef]
- Mitra, K.; Lippincott-Schwartz, J. Analysis of Mitochondrial Dynamics and Functions Using Imaging Approaches. Curr. Protoc. Cell Biol. 2010, 46, 4.25.1–4.25.21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free. Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free. Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkitt, M.J.; Wardman, P. Cytochrome c Is a Potent Catalyst of Dichlorofluorescin Oxidation: Implications for the Role of Reactive Oxygen Species in Apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 329–333. [Google Scholar] [CrossRef]
- Karlsson, M.; Kurz, T.; Brunk, U.T.; Nilsson, S.E.; Frennesson, C.I. What does the commonly used DCF test for oxidative stress really show? Biochem. J. 2010, 428, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Folkes, L.K.; Patel, K.B.; Wardman, P.; Wrona, M. Kinetics of reaction of nitrogen dioxide with dihydrorhodamine and the reaction of the dihydrorhodamine radical with oxygen: Implications for quantifying peroxynitrite formation in cells. Arch. Biochem. Biophys. 2009, 484, 122–126. [Google Scholar] [CrossRef]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free. Radic. Biol. Med. 1999, 27, 146–159. [Google Scholar] [CrossRef]
- Samuel, E.L.G.; Marcano, D.C.; Berka, V.; Bitner, B.R.; Wu, G.; Potter, A.; Fabian, R.H.; Pautler, R.G.; Kent, T.A.; Tsai, A.-L.; et al. Highly efficient conversion of superoxide to oxygen using hydrophilic carbon clusters. Proc. Natl. Acad. Sci. USA 2015, 112, 2343–2348. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Kalivendi, S.; Zhang, H.; Joseph, J.; Nithipatikom, K.; Vásquez-Vivar, J.; Kalyanaraman, B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide. Free. Radic. Biol. Med. 2003, 34, 1359–1368. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free. Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [Green Version]
- Robinson, K.M.; Janes, M.S.; Beckman, J.S. The selective detection of mitochondrial superoxide by live cell imaging. Nat. Protoc. 2008, 3, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Srinivasan, S.; Hardy, M.; Ouari, O.; Lopez, M.; Vasquez-Vivar, J.; Avadhani, N.G.; Kalyanaraman, B. Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: Identification of homo- and heterodimers. Free. Radic. Biol. Med. 2008, 44, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devasagayam, T.P.A.; Boloor, K.K.; Ramasarma, T. Methods for estimating lipid peroxidation: An analysis of merits and demerits. Indian J. Biochem. Biophys. 2003, 40, 300–308. [Google Scholar] [PubMed]
- Ansarin, K.; Khoubnasabjafari, M.; Jouyban, A. Reliability of malondialdehyde as a biomarker of oxidative stress in psychological disorders. BioImpacts 2015, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Formation of a thiobarbituric-acid-reactive substance from deoxyribose in the presence of iron salts. FEBS Lett. 1981, 128, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free. Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- De Leon, J.A.D.; Borges, C.R. Evaluation of Oxidative Stress in Biological Samples Using the Thiobarbituric Acid Reactive Substances Assay. J. Vis. Exp. 2020, e61122. [Google Scholar] [CrossRef]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2009, 5, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Starkov, A.; Garcia-Arumi, E. Measurements of the Antioxidant Enzyme Activities of Superoxide Dismutase, Catalase, and Glutathione Peroxidase. Methods Cell. Biol. 2007, 80, 379–393. [Google Scholar] [CrossRef]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demchenko, A.P. Beyond annexin V: Fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013, 65, 157–172. [Google Scholar] [CrossRef] [Green Version]
- Kapty, J.; Banman, S.; Goping, I.S.; Mercer, J.R. Evaluation of Phosphatidylserine-Binding Peptides Targeting Apoptotic Cells. J. Biomol. Screen. 2012, 17, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Banfalvi, G. Methods to detect apoptotic cell death. Apoptosis 2016, 22, 306–323. [Google Scholar] [CrossRef]
- Rieger, A.M.; Nelson, K.L.; Konowalchuk, J.D.; Barreda, D.R. Modified Annexin V/Propidium Iodide Apoptosis Assay For Accurate Assessment of Cell Death. J. Vis. Exp. 2011, 2597, e2597. [Google Scholar] [CrossRef]
- Schutte, B.; Nuydens, R.; Geerts, H.; Ramaekers, F. Annexin V binding assay as a tool to measure apoptosis in differentiated neuronal cells. J. Neurosci. Methods 1998, 86, 63–69. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Batra, S. Protocol for Apoptosis Assay by Flow Cytometry Using Annexin V Staining Method. Bio. Protocol 2013, 3. [Google Scholar] [CrossRef]
- Kupcho, K.; Shultz, J.; Hurst, R.; Hartnett, J.; Zhou, W.; Machleidt, T.; Grailer, J.; Worzella, T.; Riss, T.; Lazar, D.; et al. A real-time, bioluminescent annexin V assay for the assessment of apoptosis. Apoptosis 2018, 24, 184–197. [Google Scholar] [CrossRef] [Green Version]
- Krämer, C.E.M.; Wiechert, W.; Kohlheyer, D. Time-resolved, single-cell analysis of induced and programmed cell death via non-invasive propidium iodide and counterstain perfusion. Sci. Rep. 2016, 6, 32104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Oczos, J.; Janowski, P.; Trembecka, D.; Dobrucki, J.; Darzynkiewicz, Z.; Wlodkowic, D. Rationale for the real-time and dynamic cell death assays using propidium iodide. Cytom. Part A 2010, 77, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Woolbright, B.L.; Jaeschke, H. Measuring Apoptosis and Necrosis in Cholestatic Liver Injury. Methods Mol. Biol. 2019, 1981, 133–147. [Google Scholar] [CrossRef]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and Beyond: Cytometry in Studies of Programmed Cell Death. Methods Cell Biol. 2011, 103, 55–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzynkiewicz, Z.; Pozarowski, P.; Lee, B.W.; Johnson, G.L. Fluorochrome-Labeled Inhibitors of Caspases: Convenient In Vitro and In Vivo Markers of Apoptotic Cells for Cytometric Analysis. Methods Mol. Biol. 2010, 682, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green? and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytom. A 2004, 61, 162–169. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Pozarowski, P. All that glitters is not gold: All that FLICA binds is not caspase. A caution in data interpretation—and new opportunities. Cytom. Part A 2007, 71, 536–537. [Google Scholar] [CrossRef]
- Martinez, M.M.; Reif, R.D.; Pappas, D. Detection of apoptosis: A review of conventional and novel techniques. Anal. Methods 2010, 2, 996–1004. [Google Scholar] [CrossRef]
- Moleiro, A.F.; Conceição, G.; Leite-Moreira, A.F.; Rocha-Sousa, A. A Critical Analysis of the AvailableIn VitroandEx VivoMethods to Study Retinal Angiogenesis. J. Ophthalmol. 2017, 2017, 3034953. [Google Scholar] [CrossRef] [Green Version]
- Majtnerová, P.; Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 2018, 45, 1469–1478. [Google Scholar] [CrossRef] [Green Version]
- Darzynkiewicz, Z.; Galkowski, D.; Zhao, H. Analysis of apoptosis by cytometry using TUNEL assay. Methods 2008, 44, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Hitomi, M.; Van Der Wee, K.; Rothenberg, F.; Fisher, S.A.; Zucker, R.; Svoboda, K.K.H.; Goldsmith, E.C.; Heiskanen, K.M.; Nieminen, A.-L. The Pros and Cons of Apoptosis Assays for Use in the Study of Cells, Tissues, and Organs. Microsc. Microanal. 2002, 8, 375–391. [Google Scholar] [CrossRef]
- Repetto, G.; Del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Ates, G.; Vanhaecke, T.; Rogiers, V.; Rodrigues, R.M. Assaying Cellular Viability Using the Neutral Red Uptake Assay. Methods Mol. Biol. 2017, 1601, 19–26. [Google Scholar] [CrossRef]
- Babich, H.; Borenfreund, E. Applications of the Neutral Red Cytotoxicity Assay to In Vitro Toxicology. Altern. Lab. Anim. 1990, 18, 129–144. [Google Scholar] [CrossRef]
- Löwik, C.; Alblas, M.; Van De Ruit, M.; Papapoulos, S.; Van Der Pluijm, G. Quantification of Adherent and Nonadherent Cells Cultured in 96-Well Plates Using the Supravital Stain Neutral Red. Anal. Biochem. 1993, 213, 426–433. [Google Scholar] [CrossRef]
- Bouhifd, M.; Bories, G.; Casado, J.; Coecke, S.; Norlén, H.; Parissis, N.; Rodrigues, R.M.; Whelan, M.P. Automation of an in vitro cytotoxicity assay used to estimate starting doses in acute oral systemic toxicity tests. Food Chem. Toxicol. 2012, 50, 2084–2096. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Bouhifd, M.; Bories, G.; Sacco, M.-G.; Gribaldo, L.; Fabbri, M.; Coecke, S.; Whelan, M.P. Assessment of an automated in vitro basal cytotoxicity test system based on metabolically-competent cells. Toxicol. Vitro 2013, 27, 760–767. [Google Scholar] [CrossRef]
- Borenfreund, E.; Babich, H.; Martin-Alguacil, N. Comparisons of two in vitro cytotoxicity assays—The neutral red (NR) and tetrazolium MTT tests. Toxicol. Vitro 1988, 2, 1–6. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, P.F., Jr.; Moskwa, P.S.; Donish, W.H.; Pera, P.J.; Richardson, D.; Andrese, A.P. A semi-automated neutral red based chemosensitivity assay for drug screening. Investig. New Drugs 1990, 8, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Zuang, V. The Neutral Red Release Assay: A Review. Altern. Lab. Anim. 2001, 29, 575–599. [Google Scholar] [CrossRef] [PubMed]
- Zurita, J.L.; Jos, A.; del Peso, A.; Salguero, M.; Lopez-Artiguez, M.; Repetto, G. Ecotoxicological evaluation of the antimalarial drug chloroquine. Aquat. Toxicol. 2005, 75, 97–107. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, T.; Sempels, W.; Snoeys, J.; Holmstock, N.; Chatterjee, S.; Stieger, B.; Augustijns, P.; Hofkens, J.; Mizuno, H.; Annaert, P. Confocal Imaging with a Fluorescent Bile Acid Analogue Closely Mimicking Hepatic Taurocholate Disposition. J. Pharm. Sci. 2014, 103, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Milkiewicz, P.; Saksena, S.; Cardenas, T.; Mills, C.O.; Elias, E. Plasma elimination of cholyl-lysyl-fluorescein (CLF): A pilot study in patients with liver cirrhosis. Liver Int. 2000, 20, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Barber, J.A.; Stahl, S.H.; Summers, C.; Barrett, G.; Park, B.K.; Foster, J.R.; Kenna, J.G. Quantification of Drug-Induced Inhibition of Canalicular Cholyl-l-Lysyl-Fluorescein Excretion From Hepatocytes by High Content Cell Imaging. Toxicol. Sci. 2015, 148, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herédi-Szabó, K.; Kis, E.; Molnar, E.; Gyorfi, A.; Krajcsi, P. Characterization of 5(6)-Carboxy-2,′7′-Dichlorofluorescein Transport by MRP2 and Utilization of this Substrate as a Fluorescent Surrogate for LTC4. J. Biomol. Screen. 2008, 13, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Ansede, J.H.; Smith, W.R.; Perry, C.H.; Claire, R.L.S.; Brouwer, K.R. An In Vitro Assay to Assess Transporter-Based Cholestatic Hepatotoxicity Using Sandwich-Cultured Rat Hepatocytes. Drug Metab. Dispos. 2009, 38, 276–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, P.; Weinheimer, S.; Michalewicz, M.; Taub, M.E. Prediction and Quantification of Hepatic Transporter-Mediated Uptake of Pitavastatin Utilizing a Combination of the Relative Activity Factor Approach and Mechanistic Modeling. Drug Metab. Dispos. 2018, 46, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Deferm, N.; De Vocht, T.; Qi, B.; Van Brantegem, P.; Gijbels, E.; Vinken, M.; De Witte, P.; Bouillon, T.; Annaert, P. Current insights in the complexities underlying drug-induced cholestasis. Crit. Rev. Toxicol. 2019, 49, 520–548. [Google Scholar] [CrossRef]
- Van Brantegem, P.; Chatterjee, S.; De Bruyn, T.; Annaert, P.; Deferm, N. Drug-induced cholestasis assay in primary hepatocytes. MethodsX 2020, 7, 101080. [Google Scholar] [CrossRef]
- De Bruyn, T.; Chatterjee, S.; Fattah, S.; Keemink, J.; Nicolaï, J.; Augustijns, P.; Annaert, P. Sandwich-cultured hepatocytes: Utility forin vitroexploration of hepatobiliary drug disposition and drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2013, 9, 589–616. [Google Scholar] [CrossRef]
- Cui, W.; Chen, S.L.; Hu, K.-Q. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am. J. Transl. Res. 2010, 2, 95–104. [Google Scholar]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, R.; Schaart, G.; Hesselink, M.K. Optimisation of oil red O staining permits combination with immunofluorescence and automated quantification of lipids. Histochem. Cell Biol. 2001, 116, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kraus, N.A.; Ehebauer, F.; Zapp, B.; Rudolphi, B.; Kraus, B.J.; Kraus, D. Quantitative assessment of adipocyte differentiation in cell culture. Adipocyte 2016, 5, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, M.J.; Schriever, S.C.; Roscher, A.A.; Ensenauer, R. Digital image analysis approach for lipid droplet size quantitation of Oil Red O-stained cultured cells. Anal. Biochem. 2014, 445, 87–89. [Google Scholar] [CrossRef]
- Cholewiak, R.W.; Butcher, L.; Kettlewell, N.M. Oil red O and hematoxylin: A rapid histologic technic. Physiol. Behav. 1968, 3, 585–586, IN5–IN6. [Google Scholar] [CrossRef]
- Fowler, S.D.; Greenspan, P. Application of Nile red, a fluorescent hydrophobic probe, for the detection of neutral lipid deposits in tissue sections: Comparison with oil red O. J. Histochem. Cytochem. 1985, 33, 833–836. [Google Scholar] [CrossRef]
- Fukumoto, S.; Fujimoto, T. Deformation of lipid droplets in fixed samples. Histochem. Cell Biol. 2002, 118, 423–428. [Google Scholar] [CrossRef]
- Carriel, V.; Campos, F.; Aneiros-Fernández, J.; Kiernan, J.A. Tissue Fixation and Processing for the Histological Identification of Lipids. Methods Mol. Biol. 2017, 1560, 197–206. [Google Scholar] [CrossRef]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile red: A selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef] [Green Version]
- McMILLIAN, M.K.; Grant, E.R.; Zhong, Z.; Parker, J.B.; Li, L.; Zivin, R.A.; Burczynski, M.E.; Johnson, M.D. Nile Red Binding to HepG2 Cells: An Improved Assay forIn VitroStudies of Hepatosteatosis. Vitro Mol. Toxicol. 2001, 14, 177–190. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.-E. A human hepatocellular in vitro model to investigate steatosis. Chem. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Cohen, M.; Nahmias, Y. In Vitro Cell Culture Models of Hepatic Steatosis. Methods Mol. Biol. 2014, 1250, 377–390. [Google Scholar] [CrossRef]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human hepatic 3D spheroids as a model for steatosis and insulin resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Mirejovsky, D.; Patel, A.S.; Rodriguez, D.D.; Hunt, T.J. Lipid Adsorption onto Hydrogel Contact Lens Materials. Advantages of Nile Red over Oil Red O in Visualization of Lipids. Optom. Vis. Sci. 1991, 68, 858–864. [Google Scholar] [CrossRef]
- Diaz, G.; Melis, M.; Batetta, B.; Angius, F.; Falchi, A.M. Hydrophobic characterization of intracellular lipids in situ by Nile Red red/yellow emission ratio. Micron 2008, 39, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, P.; Fowler, S.D. Spectrofluorometric studies of the lipid probe, nile red. J. Lipid Res. 1985, 26, 781–789. [Google Scholar] [CrossRef]
- Rumin, J.; Bonnefond, H.; Saint-Jean, B.; Rouxel, C.; Sciandra, A.; Bernard, O.; Cadoret, J.-P.; Bougaran, G. The use of fluorescent Nile red and BODIPY for lipid measurement in microalgae. Biotechnol. Biofuels 2015, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, N. Photophysics of Nile red in solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2000, 56, 1003–1010. [Google Scholar] [CrossRef]
- Brown, W.J.; Sullivan, T.R.; Greenspan, P. Nile red staining of lysosomal phospholipid inclusions. Histochem. Cell Biol. 1992, 97, 349–354. [Google Scholar] [CrossRef]
- Fam, T.K.; Klymchenko, A.S.; Collot, M. Recent Advances in Fluorescent Probes for Lipid Droplets. Materials 2018, 11, 1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Negro, A.; Herrera, G.; Castell, J.V.; O’Connor, J.-E.; Gómez-Lechón, M.J. Cytometric analysis for drug-induced steatosis in HepG2 cells. Chem. Interact. 2009, 181, 417–423. [Google Scholar] [CrossRef]
- Qiu, B.; Simon, M.C. BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio Protocol 2016, 6, e1912. [Google Scholar] [CrossRef] [Green Version]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Vanhaecke, T.; Rogiers, V.; Rodrigues, R.M.; De Kock, J. Flow cytometric quantification of neutral lipids in a human skin stem cell-derived model of NASH. MethodsX 2020, 7, 101068. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Shinohara, Y.; Suzuki, M.; Fujimoto, T. A pitfall in using BODIPY dyes to label lipid droplets for fluorescence microscopy. Histochem. Cell Biol. 2010, 133, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Studer, A.M.; Brown, D.A.; Wolins, N.E. Fluorescent Detection of Lipid Droplets and Associated Proteins. Curr. Protoc. Cell Biol. 2016, 71, 4.31.1–4.31.14. [Google Scholar] [CrossRef]
- Koreivienė, J. Microalgae Lipid Staining with Fluorescent BODIPY Dye. Methods Mol. Biol. 2017, 1980, 47–53. [Google Scholar] [CrossRef]
- Suzuki, M.; Shinohara, Y.; Fujimoto, T. Histochemical Detection of Lipid Droplets in Cultured Cells. Methods Mol. Biol. 2012, 931, 483–491. [Google Scholar] [CrossRef]
- Collot, M.; Fam, T.K.; AshokKumar, P.; Faklaris, O.; Galli, T.; Danglot, L.; Klymchenko, A.S. Ultrabright and Fluorogenic Probes for Multicolor Imaging and Tracking of Lipid Droplets in Cells and Tissues. J. Am. Chem. Soc. 2018, 140, 5401–5411. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.; Süss, R.; Teuber, K.; Eibisch, M.; Schiller, J. Lipid analysis by thin-layer chromatography—A review of the current state. J. Chromatogr. A 2011, 1218, 2754–2774. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Zheng, Y.; VanderGheynst, J.S. Rapid Quantitative Analysis of Lipids Using a Colorimetric Method in a Microplate Format. Lipids 2011, 46, 95–103. [Google Scholar] [CrossRef]
- Li, L.; Han, J.; Wang, Z.; Liu, J.; Wei, J.; Xiong, S.; Zhao, Z. Mass Spectrometry Methodology in Lipid Analysis. Int. J. Mol. Sci. 2014, 15, 10492–10507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.A.; Anderson, S.; Rawle, J.M. Chemical Basis of the Sulfo-phospho-vanillin Reaction for Estimating Total Serum Lipids. Clin. Chem. 1972, 18, 199–202. [Google Scholar] [CrossRef]
- Fossati, P.; Prencipe, L. Serum triglycerides determined colorimetrically with an enzyme that produces hydrogen peroxide. Clin. Chem. 1982, 28, 2077–2080. [Google Scholar] [CrossRef]
- Meiattini, F.; Prencipe, L.; Bardelli, F.; Giannini, G.; Tarli, P. The 4-hydroxybenzoate/4-aminophenazone chromogenic system used in the enzymic determination of serum cholesterol. Clin. Chem. 1978, 24, 2161–2165. [Google Scholar] [CrossRef]
- Huynh, F.K.; Green, M.F.; Koves, T.R.; Hirschey, M.D. Measurement of Fatty Acid Oxidation Rates in Animal Tissues and Cell Lines. Complex Carbohydr. Part D 2014, 542, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.-Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef] [PubMed]
- Angrish, M.M.; McQueen, C.A.; Cohen-Hubal, E.; Bruno, M.; Ge, Y.; Chorley, B.N. Editor’s Highlight: Mechanistic Toxicity Tests Based on an Adverse Outcome Pathway Network for Hepatic Steatosis. Toxicol. Sci. 2017, 159, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkin, A.H.; Ortegon, A.M.; Cho, S.; Shen, W.-J.; Falcon, A.; Kraemer, F.B.; Lee, S.-J.; Stahl, A. Evidence for protein-mediated fatty acid efflux by adipocytes. Acta Physiol. 2011, 204, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasbargen, K.B.; Shen, W.-J.; Zhang, Y.; Hou, X.; Wang, W.; Shuo, Q.; Bernlohr, D.A.; Azhar, S.; Kraemer, F.B. Slc43a3 is a regulator of free fatty acid flux. J. Lipid Res. 2020, 61, 734–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junqueira, L.C.U.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. J. Mol. Histol. 1979, 11, 447–455. [Google Scholar] [CrossRef]
- Lattouf, R.; Younes, R.; Lutomski, D.; Naaman, N.; Godeau, G.; Senni, K.; Changotade, S. Picrosirius Red Staining. J. Histochem. Cytochem. 2014, 62, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, B.; Siebert, H.; Hofmann, U.; Frantz, S. Determination of collagen content within picrosirius red stained paraffin-embedded tissue sections using fluorescence microscopy. MethodsX 2015, 2, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Wegner, K.A.; Keikhosravi, A.; Eliceiri, K.W.; Vezina, C.M. Fluorescence of Picrosirius Red Multiplexed With Immunohistochemistry for the Quantitative Assessment of Collagen in Tissue Sections. J. Histochem. Cytochem. 2017, 65, 479–490. [Google Scholar] [CrossRef]
- Cissell, D.D.; Link, J.M.; Hu, J.C.; Athanasiou, K.A. A Modified Hydroxyproline Assay Based on Hydrochloric Acid in Ehrlich’s Solution Accurately Measures Tissue Collagen Content. Tissue Eng. Part C Methods 2017, 23, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Segnani, C.; Ippolito, C.; Antonioli, L.; Pellegrini, C.; Blandizzi, C.; Dolfi, A.; Bernardini, N. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PLoS ONE 2015, 10, e0144630. [Google Scholar] [CrossRef] [PubMed]
- Lareu, R.R.; Zeugolis, D.I.; Abu-Rub, M.; Pandit, A.; Raghunath, M. Essential modification of the Sircol Collagen Assay for the accurate quantification of collagen content in complex protein solutions. Acta Biomater. 2010, 6, 3146–3151. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Englert, J.M.; Crum, L.P.; Oury, T.D. A novel method for accurate collagen and biochemical assessment of pulmonary tissue utilizing one animal. Int. J. Clin. Exp. Pathol. 2011, 4, 349–355. [Google Scholar] [PubMed]
- Rich, L.; Whittaker, P. Collagen and picrosirius red staining: A polarized light assessment of fibrillar hue and spatial distribu-tion. Braz. J. Morphol. Sci. 2005, 22, 97–104. [Google Scholar]
- Walsh, B.J.; Thornton, S.C.; Penny, R.; Breit, S.N.; Walsh, B.J.; Walsh, B. Microplate reader-based quantitation of collagens. Anal. Biochem. 1992, 203, 187–190. [Google Scholar] [CrossRef]
- Reddy, G.K.; Enwemeka, C.S. A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 1996, 29, 225–229. [Google Scholar] [CrossRef]
- Stoilov, I.; Starcher, B.C.; Mecham, R.P.; Broekelmann, T.J. Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol. 2018, 143, 133–146. [Google Scholar] [CrossRef]
- Rennard, S.; Berg, R.; Martin, G.; Foidart, J.; Robey, P. Enzyme-linked immunoassay (ELISA) for connective tissue components. Anal. Biochem. 1980, 104, 205–214. [Google Scholar] [CrossRef]
- Osmekhina, E.; Neubauer, A.; Klinzing, K.; Myllyharju, J.; Neubauer, P. Sandwich ELISA for quantitative detection of human collagen prolyl 4-hydroxylase. Microb. Cell Factories 2010, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, O.S.; Bon, H.; Twomey, B.; Holdsworth, G.; Ford, K.; Bergin, M.; Huang, L.; Muzylak, M.; Healy, L.J.; Hurdowar, V.; et al. An immunofluorescence assay for extracellular matrix components highlights the role of epithelial cells in producing a stable, fibrillar extracellular matrix. Biol. Open 2017, 6, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Quasnichka, H.L.; Tarlton, J.F.; Anderson-MacKenzie, J.M.; Billingham, M.E.; Bailey, A.J.; Pickford, A.R. Development of an assay for the quantification of type I collagen synthesis in the guinea pig. J. Immunol. Methods 2005, 297, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Bielajew, B.J.; Hu, J.C.; Athanasiou, K.A. Collagen: Quantification, biomechanics and role of minor subtypes in cartilage. Nat. Rev. Mater. 2020, 5, 730–747. [Google Scholar] [CrossRef]
- Chen, C.Z.; Raghunath, M. Focus on collagen: In vitro systems to study fibrogenesis and antifibrosis_state of the art. Fibrogenesis Tissue Repair 2009, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Vandooren, J.; Geurts, N.; Martens, E.; Steen, P.E.V.D.; Opdenakker, G. Zymography methods for visualizing hydrolytic enzymes. Nat. Methods 2013, 10, 211–220. [Google Scholar] [CrossRef]
- Cheng, X.-C.; Fang, H.; Xu, W.-F. Advances in assays of matrix metalloproteinases (MMPs) and their inhibitors. J. Enzym. Inhib. Med. Chem. 2008, 23, 154–167. [Google Scholar] [CrossRef]
- Sharma, K.; Bhattacharyya, D. Reverse Zymography: Overview and Pitfalls. Breast Cancer 2017, 1626, 125–132. [Google Scholar] [CrossRef]
- Min, D.; Lyons, J.G.; Jia, J.; Lo, L.; McLennan, S.V. 2-Methoxy-2,4-diphenyl-3(2H)-furanone-labeled gelatin zymography and reverse zymography: A rapid real-time method for quantification of matrix metalloproteinases-2 and -9 and tissue inhibitors of metalloproteinases. Electrophoresis 2006, 27, 357–364. [Google Scholar] [CrossRef]
- Kawada, N.; Klein, H.; Decker, K. Eicosanoid-mediated contractility of hepatic stellate cells. Biochem. J. 1992, 285, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Kawada, N.; Inoue, M. Effect of adrenomedullin on hepatic pericytes (stellate cells) of the rat. FEBS Lett. 1994, 356, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Kawada, N.; Kuroki, T.; Kobayashi, K.; Inoue, M.; Kaneda, K.; Decker, K. Action of endothelins on hepatic stellate cells. J. Gastroenterol. 1995, 30, 731–738. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; Rogers, D.D.; Wyatt, T.A.; Sorrell, M.F.; Tuma, D.J. Transforming growth factor-β induces contraction of activated hepatic stellate cells. J. Hepatol. 2004, 41, 60–66. [Google Scholar] [CrossRef]
- Melton, A.C.; Datta, A.; Yee, H.F. [Ca2+]i-independent contractile force generation by rat hepatic stellate cells in response to endothelin-1. Am. J. Physiol. Liver Physiol. 2006, 290, G7–G13. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth Muscle α Actin (Acta2) and Myofibroblast Function during Hepatic Wound Healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ding, Q.; Ling, L.-P.; Wu, Y.; Meng, D.-X.; Li, X.; Zhang, C.-Q. Metformin attenuates motility, contraction, and fibrogenic response of hepatic stellate cells in vivo and in vitro by activating AMP-activated protein kinase. World J. Gastroenterol. 2018, 24, 819–832. [Google Scholar] [CrossRef]
- Kim, S.-W.; Roh, J.; Park, C.-S. Immunohistochemistry for Pathologists: Protocols, Pitfalls, and Tips. J. Pathol. Transl. Med. 2016, 50, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bismuth, H. Surgical anatomy and anatomical surgery of the liver. World J. Surg. 1982, 6, 3–9. [Google Scholar] [CrossRef]
- Rutkauskas, S.; Gedrimas, V.; Pundzius, J.; Barauskas, G.; Basevicius, A. Clinical and anatomical basis for the classification of the structural parts of liver. Medicina 2006, 42, 98–106. [Google Scholar]
- Abdel-Misih, S.R.Z.; Bloomston, M. Liver Anatomy. Surg. Clin. North Am. 2010, 90, 643–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malarkey, D.E.; Johnson, K.; Ryan, L.; Boorman, G.; Maronpot, R.R. New Insights into Functional Aspects of Liver Morphology. Toxicol. Pathol. 2005, 33, 27–34. [Google Scholar] [CrossRef]
- Ishibashi, H.; Nakamura, M.; Komori, A.; Migita, K.; Shimoda, S. Liver architecture, cell function, and disease. Semin. Immunopathol. 2009, 31, 399–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungermann, K. Functional Heterogeneity of Periportal and Perivenous Hepatocytes. Enzym 1986, 35, 161–180. [Google Scholar] [CrossRef]
- Jungermann, K.; Keitzmann, T. Zonation of Parenchymal and Nonparenchymal Metabolism in Liver. Annu. Rev. Nutr. 1996, 16, 179–203. [Google Scholar] [CrossRef]
- Weibel, E.R.; Staubli, W.; Gnagi, H.R.; Hess, F.A. Correlated Morphometric and Biochemical Studies on The Liver Cell. J. Cell Biol. 1969, 42, 68–91. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, A.M.; Brouwer, A.; Knook, D.L. Sinusoidal endothelial cells of the liver: Fine structure and function in relation to age. J. Electron Microsc. Tech. 1990, 14, 218–236. [Google Scholar] [CrossRef]
- Shang, L.; Hosseini, M.; Liu, X.; Kisseleva, T.; Brenner, D.A. Human hepatic stellate cell isolation and characterization. J. Gastroenterol. 2018, 53, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, T.T.S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Smedsrod, B.; De Bleser, P.J.; Braet, F.; Lovisetti, P.; Vanderkerken, K.; Wisse, E.; Geerts, A. Cell biology of liver endothelial and Kupffer cells. Gut 1994, 35, 1509–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2020, 10, 3112. [Google Scholar] [CrossRef] [Green Version]
- Ekwall, B. The basal cytotoxicity concept. In Alternative Methods in Toxicology and the Life Sciences: Education, Researchers, Testing; Goldberg, A.M., Van Zutphen, L.F.M., Principe, M.L., Eds.; Mary Ann Liebert: New York, NY, USA, 1995; pp. 721–725. [Google Scholar]
- Eisenbrand, G.; Pool-Zobel, B.; Baker, V.; Balls, M.; Blaauboer, B.; Boobis, A.; Carere, A.; Kevekordes, S.; Lhuguenot, J.-C.; Pieters, R.; et al. Methods of in vitro toxicology. Food Chem. Toxicol. 2002, 40, 193–236. [Google Scholar] [CrossRef]
- Schoonen, W.G.E.J.; Westerink, W.M.A.; Horbach, G.J. High-throughput screening for analysis of in vitro toxicity. Galanin 2009, 99, 401–452. [Google Scholar] [CrossRef]
- Vinken, M.; Blaauboer, B.J. In vitro testing of basal cytotoxicity: Establishment of an adverse outcome pathway from chemical insult to cell death. Toxicol. Vitro 2017, 39, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escher, B.I.; Eggen, R.I.L.; Schreiber, U.; Schreiber, Z.; Vye, E.; Wisner, B.; Schwarzenbach, R.P. Baseline Toxicity (Narcosis) of Organic Chemicals Determined by In Vitro Membrane Potential Measurements in Energy-Transducing Membranes. Environ. Sci. Technol. 2002, 36, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, I.E. Mitochondria, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2011; pp. 1–480. [Google Scholar]
- Jones, D.P.; Lemasters, J.J.; Han, D.; Boelsterli, U.A.; Kaplowitz, N. Mechanisms of Pathogenesis in Drug Hepatotoxicity Putting the Stress on Mitochondria. Mol. Interv. 2010, 10, 98–111. [Google Scholar] [CrossRef]
- Pessayre, D.; Mansouri, A.; Berson, A.; Fromenty, B. Mitochondrial Involvement in Drug-Induced Liver Injury. Handb. Exp. Pharmacol. 2010, 311–365. [Google Scholar] [CrossRef]
- Pessayre, D.; Fromenty, B.; Berson, A.; Robin, M.-A.; Lettéron, P.; Moreau, R.; Mansouri, A. Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev. 2012, 44, 34–87. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhaszova, M.; Wang, S.; Zorov, D.B.; Nuss, H.B.; Gleichmann, M.; Mattson, M.P.; Sollott, S.J. The Identity and Regulation of the Mitochondrial Permeability Transition Pore. Ann. N. Y. Acad. Sci. 2008, 1123, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Guicciardi, M.E. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Gores, G.J.; Lemasters, J.J. Apoptosis and necrosis in the liver: A tale of two deaths? Hepatology 2006, 43, S31–S44. [Google Scholar] [CrossRef]
- Schulze-Bergkamen, H.; Schuchmann, M.; Fleischer, B.; Galle, P.R. The role of apoptosis versus oncotic necrosis in liver injury: Facts or faith? J. Hepatol. 2006, 44, 984–993. [Google Scholar] [CrossRef]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Au, J.S.; Navarro, V.J.; Rossi, S. Review article: Drug-induced liver injury-its pathophysiology and evolving diagnostic tools. Aliment. Pharmacol. Ther. 2011, 34, 11–20. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Dufour, J.-F. Biomarkers for Hepatocellular Apoptosis in the Management of Liver Diseases. Curr. Pharm. Biotechnol. 2012, 13, 2221–2227. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Grattagliano, I.; Bonfrate, L.; Diogo, C.V.; Wang, H.H.; Wang, D.Q.H.; Portincasa, P. Biochemical mechanisms in drug-induced liver injury: Certainties and doubts. World J. Gastroenterol. 2009, 15, 4865–4876. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, C.; Callewaert, D.M. An enzyme-release assay for natural cytotoxicity. J. Immunol. Methods 1983, 64, 313–320. [Google Scholar] [CrossRef]
- Decker, T.; Lohmann-Matthes, M.-L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1988, 115, 61–69. [Google Scholar] [CrossRef]
- Kaja, S.; Payne, A.J.; Singh, T.; Ghuman, J.K.; Sieck, E.G.; Koulen, P. An optimized lactate dehydrogenase release assay for screening of drug candidates in neuroscience. J. Pharmacol. Toxicol. Methods 2015, 73, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, N.G.; Dedoussis, G.V.; Spanakos, G.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. An improved fluorescence assay for the determination of lymphocyte-mediated cytotoxicity using flow cytometry. J. Immunol. Methods 1994, 177, 101–111. [Google Scholar] [CrossRef]
- Cho, M.-H.; Niles, A.; Huang, R.; Inglese, J.; Austin, C.P.; Riss, T.; Xia, M. A bioluminescent cytotoxicity assay for assessment of membrane integrity using a proteolytic biomarker. Toxicol. Vitro 2008, 22, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2001, 21, A.3B.1–A.3B.2. [Google Scholar] [CrossRef]
- Jain, A.; Singh, D.; Dubey, K.; Maurya, R.; Mittal, S.; Pandey, A. Models and methods for in vitro toxicology. In In Vitro Toxicology; Dhawan, A., Kwon, S., Eds.; Elsevier Academic Press: Massachusetts, MA, USA, 2018; pp. 45–65. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Gonzalez, R.; Tarloff, J. Evaluation of hepatic subcellular fractions for Alamar blue and MTT reductase activity. Toxicol. Vitro 2001, 15, 257–259. [Google Scholar] [CrossRef]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an Aqueous Soluble Tetrazolium/Formazan Assay for Cell Growth Assays in Culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef]
- Ishiyama, M.; Shiga, M.; Sasamoto, K.; Mizoguchi, M.; He, P.-G. A New Sulfonated Tetrazolium Salt That Produces a Highly Water-Soluble Formazan Dye. Chem. Pharm. Bull. 1993, 41, 1118–1122. [Google Scholar] [CrossRef] [Green Version]
- Crouch, S.; Kozlowski, R.; Slater, K.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Qian, T.; He, L.; Kim, J.-S.; Elmore, S.P.; Cascio, W.E.; Brenner, D.A. Role of Mitochondrial Inner Membrane Permeabilization in Necrotic Cell Death, Apoptosis, and Autophagy. Antioxid. Redox Signal. 2002, 4, 769–781. [Google Scholar] [CrossRef]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef]
- Chen, L.B. Mitochondrial Membrane Potential in Living Cells. Annu. Rev. Cell Biol. 1988, 4, 155–181. [Google Scholar] [CrossRef]
- Scaduto, R.C.; Grotyohann, L.W. Measurement of Mitochondrial Membrane Potential Using Fluorescent Rhodamine Derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-Y.; Huang, H.-C.; Huang, T.-C.; Yang, P.-C.; Wang, Y.-C.; Juan, H.-F. Flow Cytometric Detection of Mitochondrial Membrane Potential. Bio Protocol 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring Mitochondrial Transmembrane Potential by TMRE Staining. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protocol 2019, 9, e3128. [Google Scholar] [CrossRef]
- Dziubla, T.; Butterfield, D.A. Oxidative Stress and Biomaterials; Academic Press: Massachusetts, MA, USA, 2016; pp. 1–404. [Google Scholar]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2007, 3, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Joseph, J.; Fales, H.M.; Sokoloski, E.A.; Levine, R.L.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. USA 2005, 102, 5727–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zou, M.-H. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. Methods Mol. Biol. 2018, 1732, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Ghani, A.; Barril, C.; Bedgood, D.R.; Prenzler, P.D. Measurement of antioxidant activity with the thiobarbituric acid reactive substances assay. Food Chem. 2017, 230, 195–207. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Tsikas, D.; Rothmann, S.; Schneider, J.Y.; Suchy, M.-T.; Trettin, A.; Modun, D.; Stuke, N.; Maassen, N.; Frölich, J.C. Development, validation and biomedical applications of stable-isotope dilution GC–MS and GC–MS/MS techniques for circulating malondialdehyde (MDA) after pentafluorobenzyl bromide derivatization: MDA as a biomarker of oxidative stress and its relation to 15( S )-8- iso -prostaglandin F 2α and nitric oxide (NO). J. Chromatogr. B 2016, 1019, 95–111. [Google Scholar] [CrossRef]
- Dong, X.; Tang, J.; Chen, X. Sensitive determination of malondialdehyde in rat prostate by high performance liquid chromatography with fluorescence detection. Sci. Rep. 2020, 10, 3990. [Google Scholar] [CrossRef] [Green Version]
- Lord, H.L.; Rosenfeld, J.; Volovich, V.; Kumbhare, D.; Parkinson, B. Determination of malondialdehyde in human plasma by fully automated solid phase analytical derivatization. J. Chromatogr. B 2009, 877, 1292–1298. [Google Scholar] [CrossRef]
- Marshall, P.J.; Warso, M.A.; Lands, W.E. Selective microdetermination of lipid hydroperoxides. Anal. Biochem. 1985, 145, 192–199. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluo-rescence detection: Comparison with ultraviolet-visible spectrophotometry. Clin. Chem. 2001, 47, 1725–1727. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Cullen, J.J.; Mitros, F.A.; Oberley, L.W. Expression of Antioxidant Enzymes in Diseases of the Human Pancreas: Another Link Between Chronic Pancreatitis and Pancreatic Cancer. Pancreas 2003, 26, 23–27. [Google Scholar] [CrossRef]
- Beers, R.F.; Sizer, I.W. A Spectrophotometric Method For Measuring The Breakdown Of Hydrogen Peroxide By Catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Nebot, C.; Moutet, M.; Huet, P.; Xu, J.; Yadan, J.; Chaudière, J. Spectrophotometric Assay of Superoxide Dismutase Activity Based on the Activated Autoxidation of a Tetracyclic Catechol. Anal. Biochem. 1993, 214, 442–451. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide Dismutase. J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Spitz, D.R.; Oberley, L.W. Measurement of Mn SOD and Cu Zn SOD Activity in Mammalian Tissue Homogenates. Curr. Protoc. Toxicol. 2001, 8, 7.5.1–7.5.11. [Google Scholar] [CrossRef]
- Coles, P. Raids cause French workers to take stock. Nat. Cell Biol. 1989, 339, 407. [Google Scholar] [CrossRef]
- Mariño, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013, 23, 1247–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Majumder, P.; Sanyal, S.; Singh, J.; Jana, K.; Das, C.; Dasgupta, D. The DNA intercalators ethidium bromide and propidium iodide also bind to core histones. FEBS Open Bio 2014, 4, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Boughaba, J.A.; Chojnowski, G.; Christensen, M.E.; Waterhouse, N.J. Dead Cert: Measuring Cell Death. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Arndt-Jovin, D.J.; Jovin, T.M. Chapter 16 Fluorescence Labeling and Microscopy of DNA. Methods Cell Biol. 1989, 30, 417–448. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Huang, X.; Okafuji, M.; King, M.A. Cytometric Methods to Detect Apoptosis. Methods Cell Biol. 2004, 75, 307–341. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Barnard, E.A.; Dar, Z. Specific Proteases of the Rat Mast Cell. Nat. Cell Biol. 1967, 213, 1198–1202. [Google Scholar] [CrossRef]
- Winckler, J. Vitalfärbung von Lysosomen und anderen Zellorganellen der Ratte mit Neutralrot Vital Staining of Lysosomes and Other Cell Organelles of the Rat with Neutral Red. Prog. Histochem. Cytochem. 1974, 6, 1–91. [Google Scholar] [CrossRef]
- Filman, D.; Brawn, R.; Dandliker, W. Intracellular supravital stain delocalization as an assay for antibody-dependent complement-mediated cell damage. J. Immunol. Methods 1975, 6, 189–207. [Google Scholar] [CrossRef]
- European Commission Joint Research Center. Recommendation on the 3T3 NRU Assay for Supporting the Identification of Substances Not Requiring Classification for Acute Oral Toxicity. Available online: https://ec.europa.eu/jrc/en/eurl/ecvam (accessed on 25 February 2021).
- Organization for Economic Co-operation and Development (OECD). Test No. 432: In Vitro 3T3 NRU Phototoxicity Test. In OECD Guidelines for the Testing of Chemicals; Section 4; OECD Publishing: Paris, France, 2019; pp. 1–20. [Google Scholar]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Vinken, M.; Maes, M.; Vanhaecke, T.; Rogiers, V. Drug-Induced Liver Injury: Mechanisms, Types and Biomarkers. Curr. Med. Chem. 2013, 20, 3011–3021. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Trauner, M. Molecular aspects of bile formation and cholestasis. Trends Mol. Med. 2003, 9, 558–564. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Heathcote, E.J.; Gershwin, M.E. Pathogenesis of Cholestatic Liver Disease and Therapeutic Approaches. Gastroenterology 2010, 139, 1481–1496. [Google Scholar] [CrossRef] [PubMed]
- Zollner, G.; Wagner, M.; Trauner, M. Nuclear receptors as drug targets in cholestasis and drug-induced hepatotoxicity. Pharmacol. Ther. 2010, 126, 228–243. [Google Scholar] [CrossRef]
- Wenniger, L.M.D.B.; Beuers, U. Bile salts and cholestasis. Dig. Liver Dis. 2010, 42, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Padda, M.S.; Sanchez, M.; Akhtar, A.J.; Boyer, J.L. Drug-induced cholestasis. Hepatology 2011, 53, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Khan, G.J.; Wu, Z.; Wang, X.; Zhang, L.; Jiang, Z. Bile acid homeostasis paradigm and its connotation with cholestatic liver diseases. Drug Discov. Today 2019, 24, 112–128. [Google Scholar] [CrossRef]
- Amacher, D.E. A toxicologist’s guide to biomarkers of hepatic response. Hum. Exp. Toxicol. 2002, 21, 253–262. [Google Scholar] [CrossRef]
- Gijbels, E.; Vilas-Boas, V.; Deferm, N.; Devisscher, L.; Jaeschke, H.; Annaert, P.; Vinken, M. Mechanisms and in vitro models of drug-induced cholestasis. Arch. Toxicol. 2019, 93, 1169–1186. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kakar, S. Histological patterns in drug-induced liver disease. J. Clin. Pathol. 2009, 62, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative Mechanisms of Environmental Chemical–Induced Steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willebrords, J.; Pereira, I.V.A.; Maes, M.; Yanguas, S.C.; Colle, I.; Bossche, B.V.D.; Da Silva, T.C.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.; et al. Strategies, models and biomarkers in experimental non-alcoholic fatty liver disease research. Prog. Lipid Res. 2015, 59, 106–125. [Google Scholar] [CrossRef] [Green Version]
- Angrish, M.M.; Kaiser, J.P.; McQueen, C.A.; Chorley, B.N. Tipping the Balance: Hepatotoxicity and the 4 Apical Key Events of Hepatic Steatosis. Toxicol. Sci. 2016, 150, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; Bossche, B.V.D.; De Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Kis, E.; Ioja, E.; Rajnai, Z.; Jani, M.; Méhn, D.; Herédi-Szabó, K.; Krajcsi, P. BSEP inhibition—In vitro screens to assess cholestatic potential of drugs. Toxicol. Vitro 2012, 26, 1294–1299. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Murai, T.; Yabuuchi, H.; Hui, S.-P.; Kurosawa, T. Measurement of Bile Salt Export Pump Transport Activities using a Fluorescent Bile Acid Derivative. Drug Metab. Pharmacokinet. 2010, 25, 214–219. [Google Scholar] [CrossRef]
- Mills, C.; Rahman, K.; Coleman, R.; Elias, E. Cholyl-lysylfluorescein: Synthesis, biliary excretion in vivo and during single-pass perfusion of isolated perfused rat liver. Biochim. Biophys. Acta BBA Gen. Subj. 1991, 1115, 151–156. [Google Scholar] [CrossRef]
- De Waart, D.R.; Häusler, S.; Vlaming, M.L.H.; Kunne, C.; Hänggi, E.; Gruss, H.-J.; Elferink, R.P.J.O.; Stieger, B. Hepatic Transport Mechanisms of Cholyl-l-Lysyl-Fluorescein. J. Pharmacol. Exp. Ther. 2010, 334, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohara, H.; Bajaj, P.; Yamanaka, K.; Miyawaki, A.; Harada, K.; Miyamoto, K.; Matsui, T.; Okai, Y.; Wagoner, M.; Shinozawa, T. High-Throughput Screening to Evaluate Inhibition of Bile Acid Transporters Using Human Hepatocytes Isolated From Chimeric Mice. Toxicol. Sci. 2019, 173, 347–361. [Google Scholar] [CrossRef]
- Roma, M.G.; Orsler, D.J.; Coleman, R. Canalicular Retention as anin VitroAssay of Tight Junctional Permeability in Isolated Hepatocyte Couplets: Effects of Protein Kinase Modulation and Cholestatic Agents. Fundam. Appl. Toxicol. 1997, 37, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Laupèze, B.; Amiot, L.; Courtois, A.; Vernhet, L.; Drénou, B.; Fauchet, R.; Fardel, O. Use of the anionic dye carboxy-2′,7′-dichlorofluorescein for sensitive flow cytometric detection of multidrug resistance-associated protein activity. Int. J. Oncol. 1999, 15, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Cantrill, C.; Houston, J.B. Understanding the Interplay Between Uptake and Efflux Transporters Within In Vitro Systems in Defining Hepatocellular Drug Concentrations. J. Pharm. Sci. 2017, 106, 2815–2825. [Google Scholar] [CrossRef]
- Swift, B.; Pfeifer, N.D.; Brouwer, K.L. Sandwich-cultured hepatocytes: Anin vitromodel to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev. 2010, 42, 446–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Köck, K.; Sedykh, A.; Tropsha, A.; Brouwer, K.L. An updated review on drug-induced cholestasis: Mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J. Pharm. Sci. 2013, 102, 3037–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakawa, K.; Matsunaga, S.; Yamaoka, Y.; Dairaku, M.; Fukano, K.; Kimura, H.; Chimuro, T.; Nishitsuji, H.; Watashi, K.; Shimotohno, K.; et al. Development of a cell-based assay to identify hepatitis B virus entry inhibitors targeting the sodium taurocholate cotransporting polypeptide. Oncotarget 2018, 9, 23681–23694. [Google Scholar] [CrossRef] [Green Version]
- Ni, X.; Gao, Y.; Wu, Z.; Ma, L.; Chen, C.; Wang, L.; Lin, Y.; Hui, L.; Pan, G. Functional human induced hepatocytes (hiHeps) with bile acid synthesis and transport capacities: A novel in vitro cholestatic model. Sci. Rep. 2016, 6, 38694. [Google Scholar] [CrossRef] [Green Version]
- Donkers, J.M.; Zehnder, B.; Van Westen, G.J.P.; Kwakkenbos, M.J.; Ijzerman, A.P.; Elferink, R.P.J.O.; Beuers, U.; Urban, S.; Van De Graaf, S.F.J. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 15307. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-Y.; Ouyang, X.; Chen, Y.; Soroka, C.J.; Wang, J.; Mennone, A.; Wang, Y.; Mehal, W.Z.; Jain, D.; Boyer, J.L. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017, 2, e90780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.D.; de Morais, S.M.; Xu, J.J. Inhibition of human organic anion transporting polypeptide OATP 1B1 as a mechanism of drug-induced hyperbilirubinemia. Chem. Interact. 2004, 150, 179–187. [Google Scholar] [CrossRef]
- Izumi, S.; Nozaki, Y.; Komori, T.; Maeda, K.; Takenaka, O.; Kusano, K.; Yoshimura, T.; Kusuhara, H.; Sugiyama, Y. Substrate-Dependent Inhibition of Organic Anion Transporting Polypeptide 1B1: Comparative Analysis with Prototypical Probe Substrates Estradiol-17β-Glucuronide, Estrone-3-Sulfate, and Sulfobromophthalein. Drug Metab. Dispos. 2013, 41, 1859–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, S.; Nozaki, Y.; Maeda, K.; Komori, T.; Takenaka, O.; Kusuhara, H.; Sugiyama, Y. Investigation of the Impact of Substrate Selection on In Vitro Organic Anion Transporting Polypeptide 1B1 Inhibition Profiles for the Prediction of Drug-Drug Interactions. Drug Metab. Dispos. 2014, 43, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letschert, K.; Komatsu, M.; Hummel-Eisenbeiss, J.; Keppler, D.; Pitarque, M.; Rodríguez-Antona, C.; Oscarson, M.; Ingelman-Sundberg, M. Vectorial Transport of the Peptide CCK-8 by Double-Transfected MDCKII Cells Stably Expressing the Organic Anion Transporter OATP1B3 (OATP8) and the Export Pump ABCC2. J. Pharmacol. Exp. Ther. 2005, 313, 549–556. [Google Scholar] [CrossRef]
- Patik, I.; Szekely, V.; Nemet, O.; Szepesi, A.; Kucsma, N.; Varady, G.; Szakacs, G.; Bakos, E.; Ozvegy-Laczka, C. Identification of novel cell-impermeant fluorescent substrates for testing the function and drug interaction of Organic Anion-Transporting Polypeptides, OATP1B1/1B3 and 2B1. Sci. Rep. 2018, 8, 2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, J.; Xu, S.; Ni, C.; Fang, Z.; Hong, M.; Ni, C. Amino-terminal region of human organic anion transporting polypeptide 1B1 dictates transporter stability and substrate interaction. Toxicol. Appl. Pharmacol. 2019, 378, 114642. [Google Scholar] [CrossRef]
- Chang, J.H.; Plise, E.; Cheong, J.; Ho, Q.; Lin, M. Evaluating theIn VitroInhibition of UGT1A1, OATP1B1, OATP1B3, MRP2, and BSEP in Predicting Drug-Induced Hyperbilirubinemia. Mol. Pharm. 2013, 10, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, T.; Van Westen, G.J.P.; Ijzerman, A.P.; Stieger, B.; De Witte, P.; Augustijns, P.F.; Annaert, P.P. Structure-Based Identification of OATP1B1/3 Inhibitors. Mol. Pharmacol. 2013, 83, 1257–1267. [Google Scholar] [CrossRef]
- Lillie, R.D.; Ashburn, L.L. Supersaturated solutions of fat stains in dilute isopropanol for demonstration of acute fatty degen-eration not shown by Herxheimer’s technique. Arch. Pathol 1943, 36, 432–440. [Google Scholar]
- Kuri-Harcuch, W.; Ramírez-Zacarías, J.L.; Castro-Muñozledo, F. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with oil red O. Histochem. Cell Biol. 1992, 97, 493–497. [Google Scholar] [CrossRef]
- Levene, A.P.; Kudo, H.; Thursz, M.R.; Anstee, Q.M.; Goldin, R.D. Is oil red-O staining and digital image analysis the gold standard for quantifying steatosis in the liver? Hepatology 2010, 51, 1859. [Google Scholar] [CrossRef] [PubMed]
- Catta-Preta, M.; Mendonca, L.S.; Fraulob-Aquino, J.; Aguila, M.B.; Mandarim-De-Lacerda, C.A. A critical analysis of three quantitative methods of assessment of hepatic steatosis in liver biopsies. Virchows Archiv 2011, 459, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Levene, A.P.; Kudo, H.; Armstrong, M.J.; Thursz, M.R.; Gedroyc, W.M.; Anstee, Q.M.; Goldin, R.D. Quantifying hepatic steatosis-more than meets the eye. Histopathology 2012, 60, 971–981. [Google Scholar] [CrossRef]
- Subramaniam, H.N.; Chaubal, K.A. Evaluation of intracellular lipids by standardized staining with a Sudan black B fraction. J. Biochem. Biophys. Methods 1990, 21, 9–16. [Google Scholar] [CrossRef]
- Aoki, T.; Hagiwara, H.; Fujimoto, T. Peculiar Distribution of Fodrin in Fat-Storing Cells. Exp. Cell Res. 1997, 234, 313–320. [Google Scholar] [CrossRef]
- Horobin, R.W.; Kiernan, J.A. Conn’s Biological Stains: A Handbook of Dyes, Stains and Fluorochromes for Use in Biology and Medicine; Taylor & Francis: London, UK, 2002. [Google Scholar]
- Wiederschain, G.Y. The Molecular Probes handbook. A guide to fluorescent probes and labeling technologies. Biochemistry 2011, 76, 1276. [Google Scholar] [CrossRef] [Green Version]
- Gocze, P.M.; Freeman, D.A. Factors underlying the variability of lipid droplet fluorescence in MA-10 leydig tumor cells. Cytometry 1994, 17, 151–158. [Google Scholar] [CrossRef]
- Daemen, S.; Van Zandvoort, M.; Parekh, S.H.; Hesselink, M.K.C. Microscopy tools for the investigation of intracellular lipid storage and dynamics. Mol. Metab. 2016, 5, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Amacher, D.E. Tetracycline-Induced Steatosis in Primary Canine Hepatocyte Cultures. Fundam. Appl. Toxicol. 1997, 40, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Janorkar, A.V.; King, K.R.; Megeed, Z.; Yarmush, M.L. Development of an in vitro cell culture model of hepatic steatosis using hepatocyte-derived reporter cells. Biotechnol. Bioeng. 2008, 102, 1466–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romualdo, G.R.; Da Silva, T.C.; de Albuquerque Landi, M.F.; Morais, J.A.; Barbisan, L.F.; Vinken, M.; Oliveira, C.P.; Cogliati, B. Sorafenib reduces steatosis-induced fibrogenesis in a human 3D co-culture model of non-alcoholic fatty liver disease. Environ. Toxicol. 2021, 36, 168–176. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.-B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Reis, A.; Rudnitskaya, A.; Blackburn, G.J.; Fauzi, N.M.; Pitt, A.R.; Spickett, C.M. A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 2013, 54, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Löfgren, L.; Forsberg, G.-B.; Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extracti on of animal tissue. Sci. Rep. 2016, 6, 27688. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, Y.; Sun, J.; Fan, S.; Yao, X.; Ran, X.; Zheng, C.; Huang, M.; Bi, H. Optimization of lipid extraction and analytical protocols for UHPLC-ESI-HRMS-based lipidomic analysis of adherent mammalian cancer cells. Anal. Bioanal. Chem. 2017, 409, 5349–5358. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.W.; Nadanaciva, S.; Swiss, R.; Divakaruni, A.S.; Will, Y. Assessment of Fatty Acid Beta Oxidation in Cells and Isolated Mitochondria. Curr. Protoc. Toxicol. 2014, 60, 25.3.1–25.3.19. [Google Scholar] [CrossRef]
- Olofsson, S.-O.; Boren, J. Apolipoprotein B: A clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J. Intern. Med. 2005, 258, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Dominiczak, M.H.; Caslake, M.J. Apolipoproteins: Metabolic role and clinical biochemistry applications. Ann. Clin. Biochem. Int. J. Lab. Med. 2011, 48, 498–515. [Google Scholar] [CrossRef]
- Sweat, F.; Puchtler, H.; Rosenthal, S.I. Sirius Red F3ba As A Stain for Connective Tissue. Arch. Pathol. 1964, 78, 69–72. [Google Scholar]
- Junqueira, L.C.U.; Cossermelli, W.; Brentani, R. Differential Staining of Collagens Type I, II and III by Sirius Red and Polarization Microscopy. Arch. Histol. Jpn. 1978, 41, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, R.E.; Logan, M.A. The Determination of Hydroxyproline. J. Biol. Chem. 1950, 184, 299–306. [Google Scholar] [CrossRef]
- Stegemann, H.; Stalder, K. Determination of hydroxyproline. Clin. Chim. Acta 1967, 18, 267–273. [Google Scholar] [CrossRef]
- Pataridis, S.; Eckhardt, A.; Mikulíková, K.; Sedláková, P.; Miksik, I. Identification of collagen types in tissues using HPLC-MS/MS. J. Sep. Sci. 2008, 31, 3483–3488. [Google Scholar] [CrossRef]
- Qiu, B.; Wei, F.; Sun, X.; Wang, X.; Duan, B.; Shi, C.; Zhang, J.; Zhang, J.; Qiu, W.; Mu, W. Measurement of hydroxyproline in collagen with three different methods. Mol. Med. Rep. 2014, 10, 1157–1163. [Google Scholar] [CrossRef] [Green Version]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, S.; D’Esposito, V.; Oriente, F.; Formisano, P.; Di Carlo, A. Substrate-zymography: A still worthwhile method for gelatinases analysis in biological samples. Clin. Chem. Lab. Med. 2015, 54, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Wilkesman, J.; Kurz, L. Zymography Principles. Breast Cancer 2017, 1626, 3–10. [Google Scholar] [CrossRef]
- Prescimone, T.; Tognotti, D.; Caselli, C.; Cabiati, M.; D’Amico, A.; Del Ry, S.; Giannessi, D. Reappraisal of Quantitative Gel Zymography for Matrix Metalloproteinases. J. Clin. Lab. Anal. 2014, 28, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Soon, R.K.; Yee, H.F. Stellate Cell Contraction: Role, Regulation, and Potential Therapeutic Target. Clin. Liver Dis. 2008, 12, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Pinzani, M.; Failli, P.; Ruocco, C.; Casini, A.; Milani, S.; Baldi, E.; Giotti, A.; Gentilini, P. Fat-storing cells as liver-specific pericytes. Spatial dynamics of agonist-stimulated intracellular calcium transients. J. Clin. Investig. 1992, 90, 642–646. [Google Scholar] [CrossRef]
- Bataller, R.; Gasull, X.; Ginès, P.; Hellemans, K.; Görbig, M.N.; Nicolás, J.M.; Sancho-Bru, P.; Heras, D.D.L.; Gual, A.; Geerts, A.; et al. In vitro and in vivo activation of rat hepatic stellate cells results in de novo expression of L-type voltage-operated calcium channels. Hepatology 2001, 33, 956–962. [Google Scholar] [CrossRef]
- Perea, L.; Coll, M.; Sancho-Bru, P. Assessment of Liver Fibrotic Insults In Vitro. Methods Mol. Biol. 2014, 1250, 391–401. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Ginannicorradini, S.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Onettimuda, A.; Berloco, P.; Rossi, M. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Rice, A.; Pinato, D.; Rombouts, K.; Hernandez, A.D.R. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci. Rep. 2019, 9, 7299. [Google Scholar] [CrossRef]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Djerir, N.E.H.; Danckaert, A.; Fernandes, J.; Roux, P.; Charrueau, C.; Lachagès, A.-M.; Charlotte, F.; Brocheriou, I.; Clément, K.; et al. Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis. Sci. Rep. 2020, 10, 3850. [Google Scholar] [CrossRef]
- Vinken, M.; Hengstler, J.G. Characterization of hepatocyte-based in vitro systems for reliable toxicity testing. Arch. Toxicol. 2018, 92, 2981–2986. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, J.X.; Lauschke, V.M. Comprehensive Evaluation of Organotypic and Microphysiological Liver Models for Prediction of Drug-Induced Liver Injury. Front. Pharmacol. 2019, 10, 1093. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Cooreman, A.; Gijbels, E.; Van Campenhout, R.; Gustafson, E.; Ballet, S.; Annaert, P.; Cogliati, B.; Vinken, M. Primary hepatocytes and their cultures for the testing of drug-induced liver injury. Adv. Pharmacol. 2019, 85, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Vanhaecke, T.; Rogiers, V. Primary hepatocyte cultures as in vitro tools for toxicity testing: Quo vadis? Toxicol. Vitro 2012, 26, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.J.; Huang, R.; Austin, C.P.; Xia, M. The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today 2010, 15, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Lechon, M.; Lahoz, A.; Gombau, L.; Castell, J.; Donato, M. In Vitro Evaluation of Potential Hepatotoxicity Induced by Drugs. Curr. Pharm. Des. 2010, 16, 1963–1977. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J. High-Content Analysis in Toxicology: Screening Substances for Human Toxicity Potential, Elucidating Subcellular Mechanisms andIn VivoUse as Translational Safety Biomarkers. Basic Clin. Pharmacol. Toxicol. 2014, 115, 4–17. [Google Scholar] [CrossRef]
- Fraczek, J.; Bolleyn, J.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Primary hepatocyte cultures for pharmaco-toxicological studies: At the busy crossroad of various anti-dedifferentiation strategies. Arch. Toxicol. 2012, 87, 577–610. [Google Scholar] [CrossRef]
- Vinken, M.; Maes, M.; Oliveira, A.G.; Cogliati, B.; Marques, P.E.; Menezes, G.B.; Dagli, M.L.Z.; Vanhaecke, T.; Rogiers, V. Primary hepatocytes and their cultures in liver apoptosis research. Arch. Toxicol. 2013, 88, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Vinken, M.; Decrock, E.; Doktorova, T.; Ramboer, E.; De Vuyst, E.; Vanhaecke, T.; Leybaert, L.; Rogiers, V. Characterization of spontaneous cell death in monolayer cultures of primary hepatocytes. Arch. Toxicol. 2011, 85, 1589–1596. [Google Scholar] [CrossRef]
- Yang, K.; Guo, C.; Woodhead, J.L.; Claire, R.L.S.; Watkins, P.B.; Siler, S.Q.; Howell, B.A.; Brouwer, K.L. Sandwich-Cultured Hepatocytes as a Tool to Study Drug Disposition and Drug-Induced Liver Injury. J. Pharm. Sci. 2016, 105, 443–459. [Google Scholar] [CrossRef] [Green Version]
- Andersson, T.B. Evolution of Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Basic Clin. Pharmacol. Toxicol. 2017, 121, 234–238. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Gehart, H.; Artegiani, B.; Löpez-Iglesias, C.; Dekkers, F.; Basak, O.; Van Es, J.; Lopes, S.M.C.D.S.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, D.F.G.; Puigvert, L.F.; Messner, S.; Mortiz, W.; Ingelman-Sundberg, M. Hepatic 3D spheroid models for the detection and study of compounds with cholestatic liability. Sci. Rep. 2016, 6, 35434. [Google Scholar] [CrossRef]
- Parmentier, C.; Hendriks, D.F.; Heyd, B.; Bachellier, P.; Ingelman-Sundberg, M.; Richert, L. Inter-individual differences in the susceptibility of primary human hepatocytes towards drug-induced cholestasis are compound and time dependent. Toxicol. Lett. 2018, 295, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Ghodsizadeh, A.; Taei, A.; Totonchi, M.; Seifinejad, A.; Gourabi, H.; Pournasr, B.; Aghdami, N.; Malekzadeh, R.; Almadani, N.; Salekdeh, G.H.; et al. Generation of Liver Disease-Specific Induced Pluripotent Stem Cells Along with Efficient Differentiation to Functional Hepatocyte-Like Cells. Stem Cell Rev. Rep. 2010, 6, 622–632. [Google Scholar] [CrossRef]
- Lynch, S.; Pridgeon, C.S.; Duckworth, C.A.; Sharma, P.; Park, B.K.; Goldring, C.E. Stem cell models as an in vitro model for predictive toxicology. Biochem. J. 2019, 476, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell–Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Farrell, G.C. The effects of cell density, attachment substratum and dexamethasone on spontaneous apoptosis of rat hepatocytes in primary culture. Vitro Cell. Dev. Biol. Anim. 1999, 35, 417–424. [Google Scholar] [CrossRef]
- Vanhaecke, T.; Henkens, T.; Kass, G.E.; Rogiers, V. Effect of the histone deacetylase inhibitor trichostatin A on spontaneous apoptosis in various types of adult rat hepatocyte cultures. Biochem. Pharmacol. 2004, 68, 753–760. [Google Scholar] [CrossRef]
- Elaut, G.; Vanhaecke, T.; Heyden, Y.V.; Rogiers, V. Spontaneous apoptosis, necrosis, energy status, glutathione levels and biotransformation capacities of isolated rat hepatocytes in suspension: Effect of the incubation medium. Biochem. Pharmacol. 2005, 69, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Tuschl, G.; Hrach, J.; Walter, Y.; Hewitt, P.G.; Mueller, S.O. Serum-free collagen sandwich cultures of adult rat hepatocytes maintain liver-like properties long term: A valuable model for in vitro toxicity and drug–drug interaction studies. Chem. Interact. 2009, 181, 124–137. [Google Scholar] [CrossRef]
- Bailly-Maitre, B.; De Sousa, G.; Zucchini, N.; Gugenheim, J.; Boulukos, K.E.; Rahmani, R. Spontaneous apoptosis in primary cultures of human and rat hepatocytes: Molecular mechanisms and regulation by dexamethasone. Cell Death Differ. 2002, 9, 945–955. [Google Scholar] [CrossRef] [Green Version]
- McKim, J.M., Jr. Building a Tiered Approach to In Vitro Predictive Toxicity Screening: A Focus on Assays with In Vivo Relevance. Comb. Chem. High. Throughput Screen 2010, 13, 188–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.; Vanhaecke, T.; Cogliati, B.; Yanguas, S.C.; Willebrords, J.; Rogiers, V.; Vinken, M. Measurement of Apoptotic and Necrotic Cell Death in Primary Hepatocyte Cultures. Methods Mol. Biol. 2015, 1250, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, V.; Stöber, R.; Nemade, H.; Sotiriadou, I.; Hescheler, J.; Hengstler, J.; Sachinidis, A. Transcriptomics of Hepatocytes Treated with Toxicants for Investigating Molecular Mechanisms Underlying Hepatotoxicity. Breast Cancer 2014, 1250, 225–240. [Google Scholar] [CrossRef]
- Vaes, W.H.J.; Ramos, E.U.; Hamwijk, C.; Van Holsteijn, I.; Blaauboer, B.J.; Seinen, W.; Verhaar, H.J.M.; Hermens, J.L.M. Solid Phase Microextraction as a Tool To Determine Membrane/Water Partition Coefficients and Bioavailable Concentrations inin VitroSystems. Chem. Res. Toxicol. 1997, 10, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.I.; Di Consiglio, E.; Blaauboer, B.J.; Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicol. Vitro 2015, 30, 217–224. [Google Scholar] [CrossRef]
- Punt, A.; Bouwmeester, H.; Blaauboer, B.J.; Coecke, S.; Hakkert, B.; Hendriks, D.F.G.; Jennings, P.; Kramer, N.I.; Neuhoff, S.; Masereeuw, R.; et al. New approach methodologies (NAMs) for human-relevant biokinetics predictions. ALTEX 2020, 37, 607–622. [Google Scholar] [CrossRef]
- Jamalzadeh, L.; Ghafoori, H.; Sariri, R.; Rabuti, H.; Nasirzade, J.; Hasani, H.; Aghamaali, M.R. Cytotoxic Effects of Some Common Organic Solvents on MCF-7, RAW-264.7 and Human Umbilical Vein Endothelial Cells. Avicenna J. Med Biochem. 2016, 4, 10–33453. [Google Scholar] [CrossRef] [Green Version]
- Vinken, M. Adverse Outcome Pathways and Drug-Induced Liver Injury Testing. Chem. Res. Toxicol. 2015, 28, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Gijbels, E.; Vilas-Boas, V.; Annaert, P.; Vanhaecke, T.; Devisscher, L.; Vinken, M. Robustness testing and optimization of an adverse outcome pathway on cholestatic liver injury. Arch. Toxicol. 2020, 94, 1151–1172. [Google Scholar] [CrossRef] [PubMed]
- Burbank, M.G.; Sharanek, A.; Burban, A.; Mialanne, H.; Aerts, H.; Guguen-Guillouzo, C.; Weaver, R.J.; Guillouzo, A. From the Cover: MechanisticInsights in Cytotoxic and Cholestatic Potential of the Endothelial Receptor Antagonists Using HepaRG Cells. Toxicol. Sci. 2017, 157, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. The adverse outcome pathway concept: A pragmatic tool in toxicology. Toxicology 2013, 312, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Delrue, N.; Sachana, M.; Sakuratani, Y.; Gourmelon, A.; Leinala, E.; Diderich, R. The Adverse Outcome Pathway Concept: A Basis for Developing Regulatory Decision-making Tools. Altern. Lab. Anim. 2016, 44, 417–429. [Google Scholar] [CrossRef]
- Vinken, M.; Knapen, D.; Vergauwen, L.; Hengstler, J.G.; Angrish, M.; Whelan, M. Adverse outcome pathways: A concise introduction for toxicologists. Arch. Toxicol. 2017, 91, 3697–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinken, M.; Kramer, N.; Allen, T.E.H.; Hoffmans, Y.; Thatcher, N.; Levorato, S.; Traussnig, H.; Schulte, S.; Boobis, A.; Thiel, A.; et al. The use of adverse outcome pathways in the safety evaluation of food additives. Arch. Toxicol. 2020, 94, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Horvat, T.; Landesmann, B.; Lostia, A.; Vinken, M.; Munn, S.; Whelan, M. Adverse outcome pathway development from protein alkylation to liver fibrosis. Arch. Toxicol. 2016, 91, 1523–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burden, N.; Sewell, F.; Andersen, M.E.; Boobis, A.; Chipman, J.K.; Cronin, M.T.D.; Hutchinson, T.H.; Kimber, I.; Whelan, M. Adverse Outcome Pathways can drive non-animal approaches for safety assessment. J. Appl. Toxicol. 2015, 35, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Bale, S.S.; Vernetti, L.; Senutovitch, N.; Jindal, R.; Hegde, M.; Gough, A.; McCarty, W.J.; Bakan, A.; Bhushan, A.; Shun, T.Y.; et al. In vitro platforms for evaluating liver toxicity. Exp. Biol. Med. 2014, 239, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Bin Raies, A.; Bajic, V.B. In silicotoxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef] [Green Version]
- Tollefsen, K.E.; Scholz, S.; Cronin, M.T.; Edwards, S.W.; de Knecht, J.; Crofton, K.; Garcia-Reyero, N.; Hartung, T.; Worth, A.; Patlewicz, G. Applying Adverse Outcome Pathways (AOPs) to support Integrated Approaches to Testing and Assessment (IATA). Regul. Toxicol. Pharmacol. 2014, 70, 629–640. [Google Scholar] [CrossRef]
- Rodrigues, R.M.; Kollipara, L.; Chaudhari, U.; Sachinidis, A.; Zahedi, R.P.; Sickmann, A.; Kopp-Schneider, A.; Jiang, X.; Keun, H.; Hengstler, J.; et al. Omics-based responses induced by bosentan in human hepatoma HepaRG cell cultures. Arch. Toxicol. 2018, 92, 1939–1952. [Google Scholar] [CrossRef]
- Jiang, J.; Pieterman, C.D.; Ertaylan, G.; Peeters, R.L.M.; De Kok, T.M.C.M. The application of omics-based human liver platforms for investigating the mechanism of drug-induced hepatotoxicity in vitro. Arch. Toxicol. 2019, 93, 3067–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinken, M. Omics-based input and output in the development and use of adverse outcome pathways. Curr. Opin. Toxicol. 2019, 18, 8–12. [Google Scholar] [CrossRef]
- Luechtefeld, T.; Hartung, T. Computational approaches to chemical hazard assessment. ALTEX 2017, 34, 459–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luechtefeld, T.; Marsh, D.; Hartung, T. Missing the Difference Between Big Data and Artificial Intelligence in RASAR Versus Traditional QSAR. Toxicol. Sci. 2019, 167, 4–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]





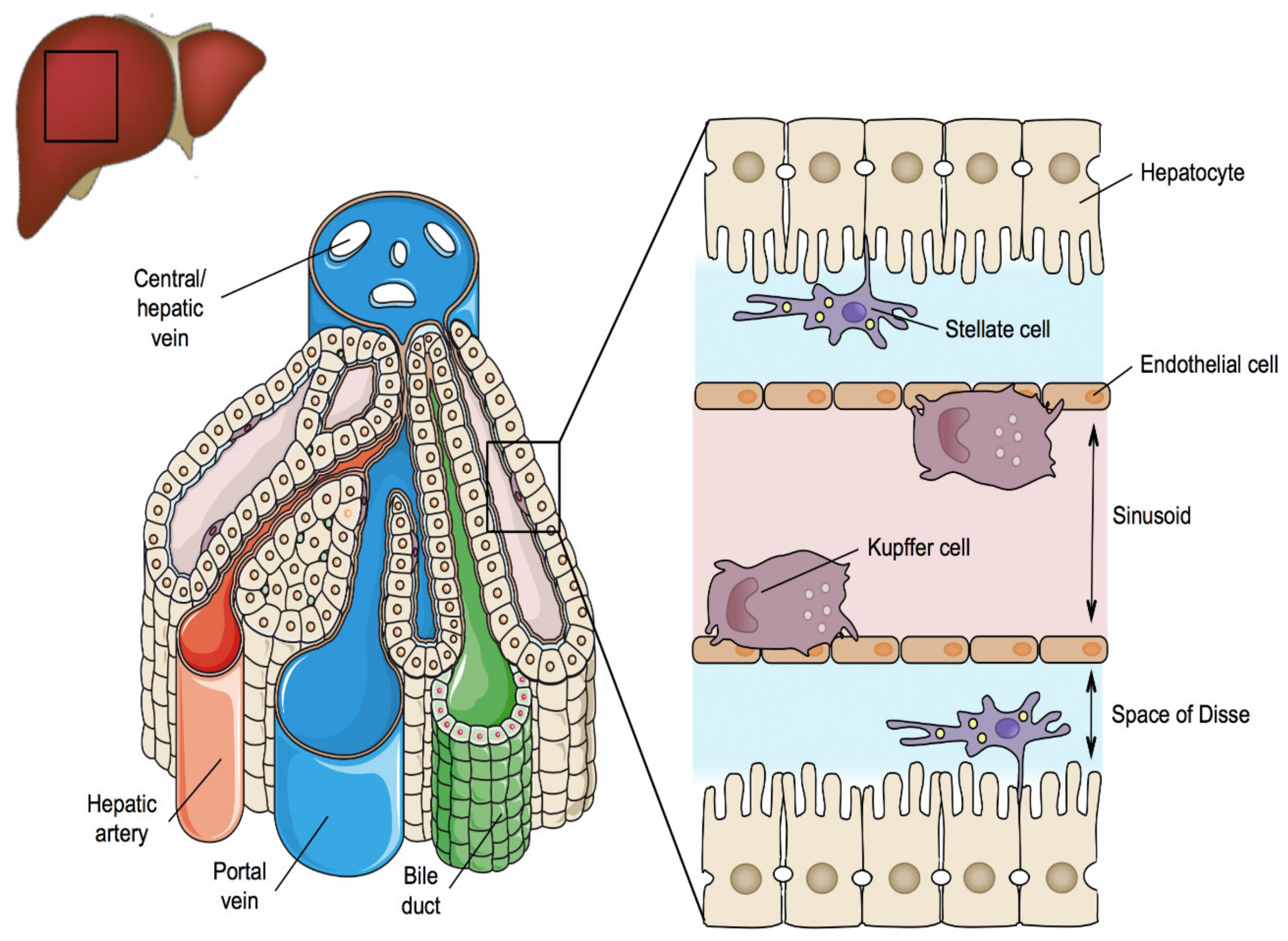{kind=link}
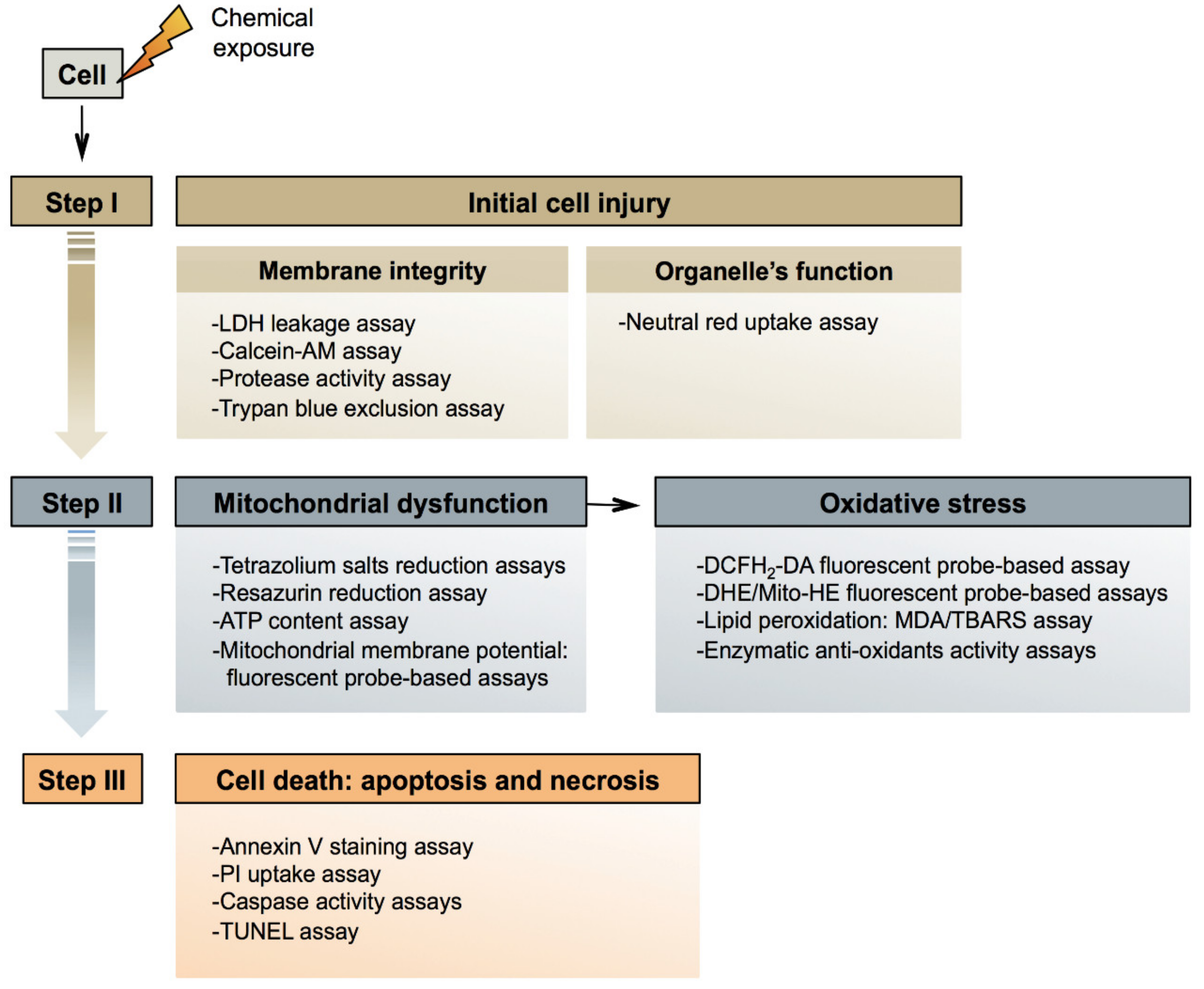{kind=link}
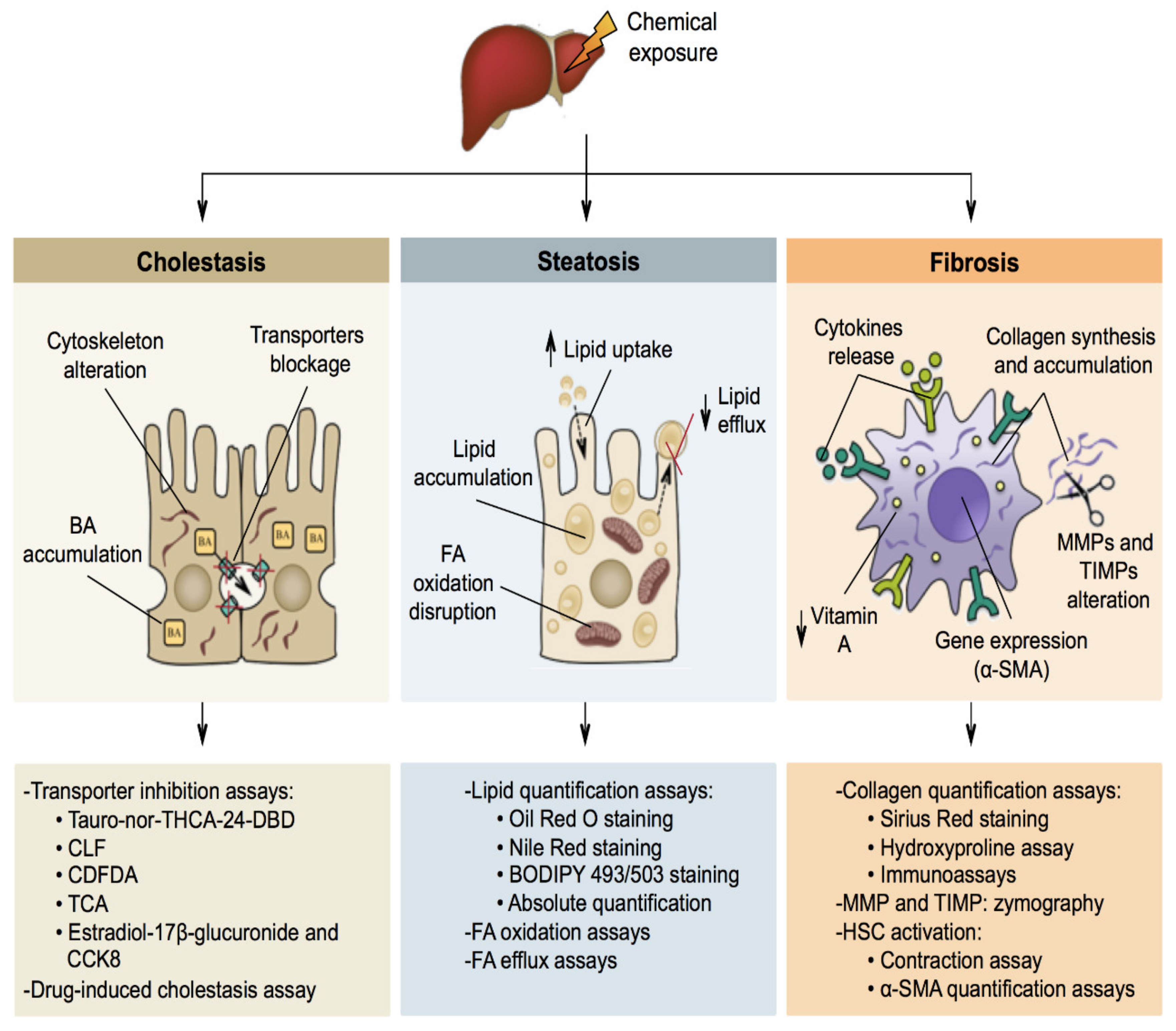{kind=link}
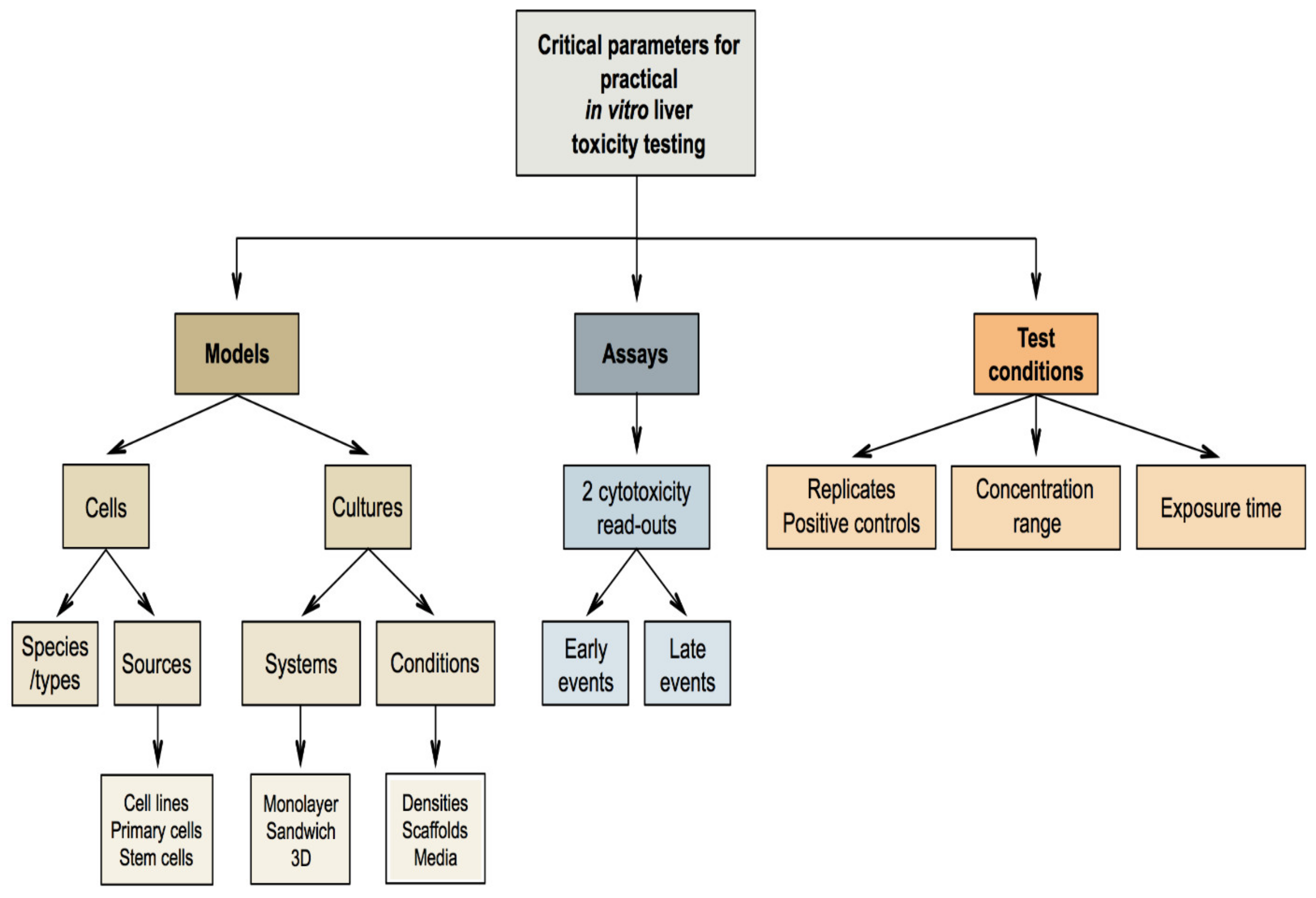{kind=link}
| Assay | Advantages | Limitations | References | |
|---|---|---|---|---|
| Membrane integrity | LDH leakage assay |
|
| [9,10,11,12,13] |
| Calcein-AM assay |
|
| [14,15,16] | |
| Protease activity assay |
|
| [17,18] | |
| Trypan blue exclusion assay |
|
| [19,20,21] | |
| Mitochondrial functionality | Tetrazolium salt assays |
|
| [9,22,23,24,25,26] |
| Resazurin reduction assay |
|
| [14,27,28,29,30,31,32] | |
| ATP content assay |
|
| [10,27,33,34,35,36,37,38] | |
| Mitochondrial membrane potential evaluation: fluorescent probe-based assays |
|
| [39,40,41,42,43] | |
| Oxidative stress | DCFH2-DA fluorescence probe-based assay |
|
| [44,45,46,47,48,49,50,51,52] |
| DHE/Mito-HE fluorescence probe-based assays |
|
| [53,54,55,56,57] | |
| Lipid peroxidation: MDA/TBARS assay |
|
| [58,59,60,61,62] | |
| Enzymatic antioxidants activity assays |
|
| [63,64,65] | |
| Cell death | Annexin V staining assay |
|
| [66,67,68,69,70,71,72] |
| PI dye uptake assay |
|
| [69,73,74] | |
| Caspase activity assays |
|
| [75,76,77,78,79] | |
| TUNEL assay |
|
| [68,80,81,82,83,84] | |
| Miscellaneous | Neutral red uptake assay |
|
| [85,86,87,88,89,90,91,92,93,94,95] |
| Assay | Advantages | Limitations | References | |
|---|---|---|---|---|
| Cholestasis | Transporter inhibition assays |
|
| [96,97,98,99,100,101] |
| Drug-induced cholestasis assay |
|
| [102,103,104] | |
| Steatosis | Oil Red O staining |
|
| [105,106,107,108,109,110,111,112,113] |
| Nile Red staining |
|
| [114,115,116,117,118,119,120,121,122,123] [124,125] | |
| BODIPY 493/503 staining |
|
| [125,126,127,128,129,130,131,132,133] | |
| Absolute lipid quantification assays |
|
| [134,135,136,137,138,139] | |
| FA oxidation assays |
|
| [140,141,142] | |
| FA efflux assays |
|
| [143,144,145] | |
| Fibrosis | Sirius Red staining |
|
| [146,147,148,149,150,151,152,153,154,155] |
| Hydroxyproline assay |
|
| [150,153,156,157] | |
| Collagen quantification via immunoassays |
|
| [158,159,160,161,162,163] | |
| MMP and TIMP quantification: zymography |
|
| [164,165,166,167] | |
| HSC activation assays |
|
| [168,169,170,171,172,173,174,175] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabernilla, A.; dos Santos Rodrigues, B.; Pieters, A.; Caufriez, A.; Leroy, K.; Van Campenhout, R.; Cooreman, A.; Gomes, A.R.; Arnesdotter, E.; Gijbels, E.; et al. In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach. Int. J. Mol. Sci. 2021, 22, 5038. https://doi.org/10.3390/ijms22095038
Tabernilla A, dos Santos Rodrigues B, Pieters A, Caufriez A, Leroy K, Van Campenhout R, Cooreman A, Gomes AR, Arnesdotter E, Gijbels E, et al. In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach. International Journal of Molecular Sciences. 2021; 22(9):5038. https://doi.org/10.3390/ijms22095038
Chicago/Turabian StyleTabernilla, Andrés, Bruna dos Santos Rodrigues, Alanah Pieters, Anne Caufriez, Kaat Leroy, Raf Van Campenhout, Axelle Cooreman, Ana Rita Gomes, Emma Arnesdotter, Eva Gijbels, and et al. 2021. "In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach" International Journal of Molecular Sciences 22, no. 9: 5038. https://doi.org/10.3390/ijms22095038