
Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke


1
Brain Barriers Group, School of Biomedical Sciences and Pharmacy, University of Newcastle, Callaghan, NSW 2321, Australia
2
Priority Research Centre for Stroke and Brain Injury, and Priority Research Centre for Brain & Mental Health, University of Newcastle, Callaghan, NSW 2321, Australia
3
Hunter Medical Research Institute, New Lambton Heights, NSW 2305, Australia
4
Institute of Infection & Global Health, University of Liverpool, Liverpool L7 3EA, UK
5
School of Pharmacy and Bioengineering, Keele University, Staffordshire ST5 5BG, UK
6
School of Medicine, Keele University, Staffordshire ST5 5BG, UK
7
Neural Tissue Engineering: Keele (NTEK), Keele University, Staffordshire ST5 5BG, UK
8
Clinical Informatics and Neurosurgery Fellow, The Cleveland Clinic, 33 Grosvenor Square, London SW1X 7HY, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(8), 4280; https://doi.org/10.3390/ijms22084280
Submission received: 7 March 2021
/
Revised: 11 April 2021
/
Accepted: 15 April 2021
/
Published: 20 April 2021
(This article belongs to the Special Issue Neuroregeneration and Brain Repair after Stroke)
{kind=link}
{kind=link}
Abstract
:Transient or permanent loss of tissue perfusion due to ischaemic stroke can lead to damage to the neurovasculature, and disrupt brain homeostasis, causing long-term motor and cognitive deficits. Despite promising pre-clinical studies, clinically approved neuroprotective therapies are lacking. Most studies have focused on neurons while ignoring the important roles of other cells of the neurovascular unit, such as astrocytes and pericytes. Astrocytes are important for the development and maintenance of the blood–brain barrier, brain homeostasis, structural support, control of cerebral blood flow and secretion of neuroprotective factors. Emerging data suggest that astrocyte activation exerts both beneficial and detrimental effects following ischaemic stroke. Activated astrocytes provide neuroprotection and contribute to neurorestoration, but also secrete inflammatory modulators, leading to aggravation of the ischaemic lesion. Astrocytes are more resistant than other cell types to stroke pathology, and exert a regulative effect in response to ischaemia. These roles of astrocytes following ischaemic stroke remain incompletely understood, though they represent an appealing target for neurovascular protection following stroke. In this review, we summarise the astrocytic contributions to neurovascular damage and repair following ischaemic stroke, and explore mechanisms of neuroprotection that promote revascularisation and neurorestoration, which may be targeted for developing novel therapies for ischaemic stroke.
1. Introduction
Globally, stroke is a leading cause of death and disability, causing over 5.5 million deaths in 2016 [1]. Ischaemic stroke, caused by a blocked artery leading to the brain, is the most common form of stroke, accounting for approximately 87% of all strokes [1]. In cerebral ischaemia, two distinct zones can be identified in the affected brain region: the ischaemic core and the penumbra. In the ischaemic core, brain cells die due to a lack of glucose and oxygen because of the loss of blood flow. In the penumbra where the blood flow is reduced, peri-lesional neural tissue is biochemically and metabolically compromised and is at risk of further damage and/or necrosis, but it also represents a potentially salvageable region of tissue, if appropriate treatment measures are available [2,3]. Reperfusion remains the only immediate treatment option following ischaemic stroke. Thrombolysis by recombinant tissue plasminogen activator [4] and clot removal by mechanical thrombectomy [5,6] are effective in acute stroke management [7,8]. However, these reperfusion strategies are only applicable to a small percentage of patients due to a short therapeutic time window, contra-indications and the costs associated with establishing the infrastructure to deliver these treatments [9]. There lies an opportunity for further neuroprotective intervention [10]. Neuroprotection is not an alternative to the treatment techniques of thrombectomy and thrombolysis; rather, it seeks to restrict injury to the brain parenchyma following an ischaemic insult by preventing salvageable penumbral neurons from dying. The concept of neuroprotection has shown promise in experimental studies, but has failed to be translated into clinical success [10,11]. A recent notable example is that of the eicosapeptide nerinetide that interferes with post-synaptic density protein-95, an excitatory neuronal protein; this showed promise in pre-clinical studies, but failed to show any benefit in human stroke trials [11]. One of the reasons for the failures of acute neuroprotective strategies is that they have largely targeted neurons. However, the brain comprises various other cell types, such as glial cells (astrocytes, oligodendrocytes and microglia), endothelial cells and pericytes, all of which influence neuronal function and survival [12]. These cells have not been sufficiently studied as therapeutic targets in ischaemic stroke, but understanding of their post-ischaemic behaviours, both harmful and beneficial, is increasing.
Glia are more numerous than neurons, except within the cerebellum, where human brain neurons outnumber glia by 4.3:1 [13,14]. Throughout the rest of the central nervous system (CNS), the ratio of glia to neurons in humans [14,15] ranges from 1.7:1 in the cerebral cortex [12], to 11.4:1 in the midbrain and hindbrain [10]; it is 17:1 in the thalamus [16]. Glial cells have important roles in brain homeostasis as well as in the development and maintenance of the blood–brain barrier (BBB) [17]. They can have either protective or detrimental effects on neurons following ischaemic stroke [18,19,20,21,22,23,24].
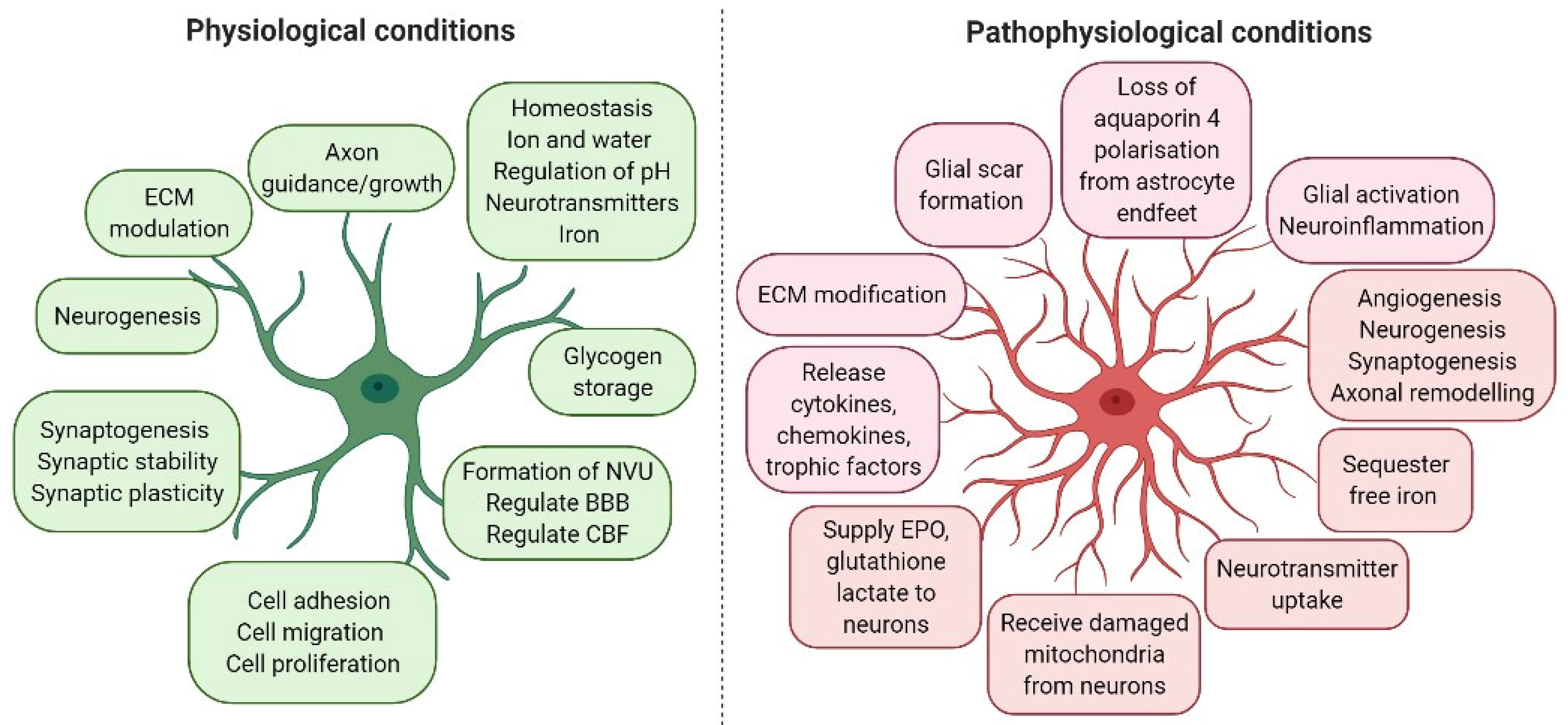
Astrocytes are the most common glial cell type, and probably the most common cell type in the CNS. A large number of astrocytes survive after a stroke and exert a regulatory effect in response to ischaemic stroke. Thus, astrocytes have become a potential treatment target for stroke [18,19,20,21]. Astrocytes fulfil many important roles in both healthy and injured brains (Figure 1). These include maintaining ion and pH homeostasis in the CNS; promoting the synthesis and removal of neurotransmitters; providing glucose supply and antioxidant defence; regulating synaptic activity by producing various cytokines, chemokines, growth factors and metabolites; supporting neurons; regulating cerebral blood flow (CBF); and supporting BBB formation and function [25].
Astrocytic morphological and functional characteristics are altered under pathological conditions, a process termed “reactive astrogliosis” (Figure 1). Astrogliosis is characterised by cellular hypertrophy, proliferation and increased expression of glial fibrillary acid protein (GFAP). It has been demonstrated that the increase in GFAP-positive cells in astrogliosis results not from the generation of new astrocytes, but rather from an increase in GFAP synthesis and a condensation of glial filaments in pre-existing cells, leading to more readily detectable GFAP by immunostaining [26]. In addition, S100β, a marker of astrocyte activation, is released from astrocytes during brain injury. More recent studies and genetic profiling of astrocytes led to the identification of another marker of astrocyte activation-aldehyde dehydrogenase 1 family member L1 (Aldh1L1) [27]. ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients [28]. Activated astrocytes also upregulate other structural and adhesion molecules, extracellular matrix (ECM) components and inflammatory chemokines and cytokines. This can result in a positive feed-forward loop, because the secretion of inflammatory cytokines further activates local cells.
Given the diverse functions of astrocytes, the purpose of this review is to summarise the role of astrocyte activation in the degeneration and regeneration of the neurovasculature following ischaemic stroke, and explore the roles of astrocytes in neuroprotection, neurotoxicity, neuroinflammation and neurorestoration following cerebral ischaemia.
2. Astrocyte Activation in Neuroprotection, Neurotoxicity and Neurorestoration
Astrocytes are essential for cell–cell communication in the neural tissue, being directly in contact with neurons, oligodendrocytes, microglia, endothelial cells and pericytes. In the human brain, astrocytes may have up to two million synapses within their domains. Their cellular processes enwrap synapse terminals and modulate neuronal activity [29]. Astrocytes form a “tripartite synapse” in the brain, and are a physical barrier to prevent neurotransmitters from diffusing away from the synapse [19]. They protect neurons during ischaemia by clearing glutamate from synaptic regions via glutamate transporters. Glutamate is converted by glutamine synthetase into glutamine that is shuttled back into the presynaptic terminal and re-used for glutamate synthesis [19]. Over 80% of glutamate transporters, especially excitatory amino acid transporter 2 (EAAT2, also known as glutamate transporter 1, GLT-1 in rodents), are located on astrocytes, making astrocytes the main site of glutamate uptake in the brain [30]. Glutamate uptake by astrocytes in ischaemia is essential for neuroprotection, as it terminates glutamate’s effects as a neurotransmitter, but also prevents extracellular glutamate levels from reaching excitotoxic levels [31].
Pellerin and Magistretti [32] reported that glutamate additionally stimulated glycolysis (i.e., glucose utilisation and lactate production) in astrocytes, and postulated the astrocyte-neuron lactate shuttle (ANLS) hypothesis which defines the strong metabolic association between astrocytes and neurons. The ANLS hypothesis states that astrocytes serve as a “lactate source”, whereas neurons serve as a “lactate sink” [32]. Lactate shuttling consists of astrocytic production of lactate through glycolysis or glycogenolysis; the lactate is then exported via the astrocyte-specific monocarboxylate transporters (MCT1 and MCT4) and taken up by neurons via MCT2 [33]. However, Bak et al. [34] claimed that neurons were well equipped to metabolise glucose in an activity-dependent manner, and preferred glucose over lactate. The emerging role of astrocytes has helped in settling this debate in favour for ANLS hypothesis [35,36].
In the brain, glycogen is stored mainly in astrocytes, the utilisation of which can sustain periods of high neuronal activity during hypoglycaemia [37]. Astrocyte glycogen plays an important role in maintaining neuronal survival during conditions of hypoglycaemia in vitro [38] and in vivo [39]. A part of the glucose that enters the astrocyte is converted into glycogen before entering the glycolytic pathway, despite this being energetically unfavourable compared to classical glycolysis [40]. In addition, astrocytes receive damaged mitochondria from neurons for mitophagy, and deliver healthy mitochondria to neurons [41]. Enhancing mitochondrial energy production is key to re-establishing the function of brain cells after stroke.
Astrocytes have been shown to promote neuronal survival by enhanced synthesis and release of antioxidants such as glutathione (GSH), and increased expression of transcription factors such as NF-E2-related factor 2 (Nrf2) [42]. Astrocytes are the main source of GSH in the CNS, and the concentration of GSH in astrocytes is twice that of neurons [43]. Neurons co-cultured with activated astrocytes had a 1.7-fold fold increase in GSH concentration compared with neurons cultured alone [44], and the viability of neurons was significantly enhanced by Nrf2-dependent enhancement of glial GSH synthesis and release [45], strongly supporting the profound antioxidant activity of astrocytes, which protects brain cells from death during strokes.
Astrocytes form highly interconnected networks via gap junctions, which are clusters of channels made from connexins (Cx30 and Cx43) located in astrocyte endfeet. Gap junctions mediate intercellular communication and solute movement between astrocytes [46], and regulate extracellular potassium concentration [19]. Cx43 expression has been shown to increase following hypoxia/ischaemia injury [47], and abnormal opening of these channels can lead to excitotoxicity and increased inflammation due to uncontrolled release of glutamate and ATP, and a Ca2+ overload [48,49]. Inhibiting gap junctions either protects neurons from death by restricting the flow of neurotoxic metabolites or increases the susceptibility of co-cultured neurons to glutamate cytotoxicity [50,51,52]. Li et al. [47] have demonstrated the potential to target Cx43 as a treatment strategy for improving stroke outcomes. Inhibition of Cx43 in a neonatal rat model reduced active astrogliosis and cerebral infarct volume, and improved functional recovery [47].
Liddelow et al. [53] termed the two subtypes of reactive astrocytes “A1” and “A2”. They found that A1 reactive astrocytes were induced by IL-1α, tumour necrosis factor (TNFα) and C1q secreted by activated microglia. These astrocytes have few physiological functions but contribute to the death of neurons and oligodendrocytes. One of the most upregulated genes in A1 reactive astrocytes is complement component C3. In contrast, this is not upregulated in A2 reactive astrocytes, which upregulate many neurotrophic factors and thrombospondins, and promote neuronal survival and tissue repair [54]. Therefore, C3 can be used as a marker to differentiate reactive astrocyte types. Reactive astrogliosis can generate neurotoxic mediators such as S100β. S100β at low doses can be neuroprotective, but during ischaemia, high levels released by astrocytes can be neurotoxic. A clinical trial targeted reducing secretion of S100β from astrocytes in ischaemic stroke patients produced a favourable trend in reduction of the National Institutes of Health Stroke Scale (NIHSS) that should be confirmed in a future clinical trial [55].
In subacute and chronic stages, cerebral ischaemia recovery relies on profound neurorestoration processes, including angiogenesis, neurogenesis and synaptogenesis [19]. Astrocytes promote the secretion of various neurotrophic factors, such as nerve growth factor (NGF), brain-derived growth factor (BDNF), neurotrophin 3 (NT-3), erythropoietin (EPO), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), insulin growth factor (IGF), glial derived neurotrophic factor (GDNF) and basic fibroblast growth factor (bFGF). These trophic factors promote angiogenesis, neurogenesis, axonal remodelling and the growth/survival of neurons and oligodendrocytes during ischaemia [56,57]. Astrocytes may also function as support cells in the subventricular zone where neural progenitor cells reside [58].
Astrocytes express growth factors and signalling molecules (such as members of the Jagged/Notch and WNT signalling pathways) that regulate stem cell proliferation and differentiation, and modulate neuroinflammation. Notch signalling influences astrocyte morphology, and in ischaemic lesions, regulates the proliferation of reactive astrocytes [59]. In human astrocytes, hypoxia has been shown to upregulate the WNT signalling pathway [60]. For human astrocytes cultured in hypoxic conditions, hypothermic treatment inhibited WNT signalling, possibly indicating a reason why hypothermia has not reliably demonstrated efficacy as a stroke therapy [60]. The composition of the ECM in the CNS, much of which is astrocyte derived, also plays a critical role at certain sites in controlling stem cell fate, maturation and survival [61].
3. Astrocytes Modulate Cerebral Blood Flow, Angiogenesis and the Blood–Brain Barrier
Astrocytes make extensive contact with blood vessels and play an important role in regulating CBF, in addition to primary CBF regulators such as small arteries and arterioles, which either dilate or contract under the influences of multiple complex physiological control systems (i.e., cerebral autoregulation). Glutamate, a major neurotoxic neurotransmitter in ischaemic stroke, activates metabotropic glutamate receptors (mGluR) in astrocytes, leading to an increase in intracellular Ca2+ and the synthesis of arachidonic acid (AA). AA, along with the metabolites derived from AA generation in astrocytes such as prostaglandin E2 (PGE2) and epoxyeicosatrienoic acids (EETs), dilate blood vessels [62]. In addition, the release of K+ from astrocytes may also contribute to vasodilation [63], whereas secretion of 20-hydroxyeicosatetreanoic acid (20-HETE) from astrocytes constricts vessels [64,65].
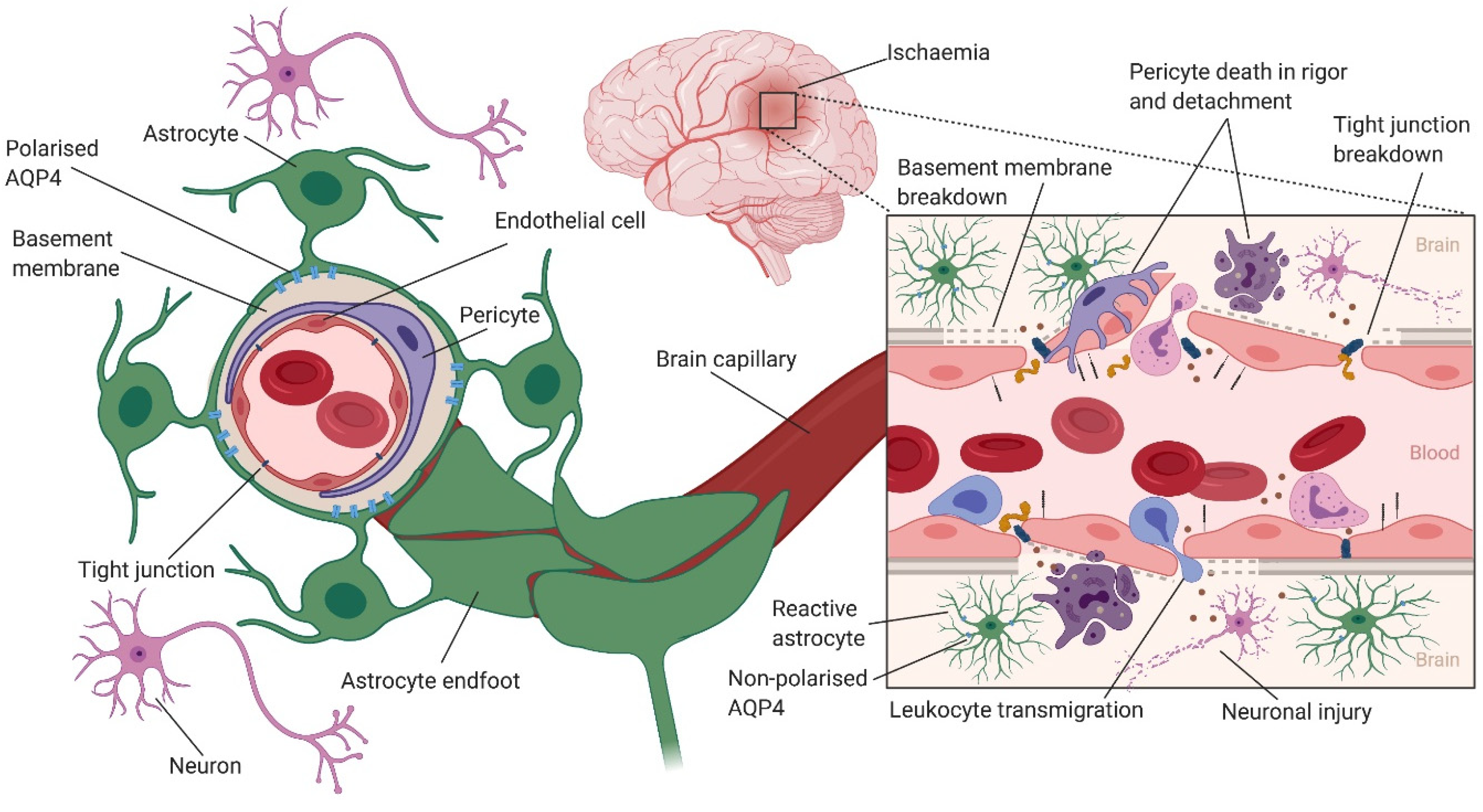
Following cerebral ischaemia, astrocyte swelling is one of the earliest responses, which is mainly due to (i) translocation of aquaporin 4 (AQP4) to the cell surface, facilitating water influx down an osmotic gradient (ionic oedema) [66], and (ii) increased uptake of glutamate and lactate [67]. AQP4 knockout mice subjected to ischaemia showed a 35% reduction in brain oedema [66]. Hypoxia-induced translocation of AQP4 is influenced by Ca2+, calmodulin (CaM) and protein kinase A (PKA), and oedema-related pathology can be reduced through pharmacological inhibition of Ca2+, CaM or PKA [66]. In the ischaemic brain regions, swollen astrocytes compress cerebral vessels, leading to further reduction of CBF [68].
Hypoxia inducible factor (HIF), an important transcriptional factor that regulates hypoxia-responsive genes, is implicated in the ischaemic brain [69]. HIF activation in the penumbra promotes angiogenesis, which enhances the transport of oxygen and glucose to the brain in subacute and chronic phases. However, it does not contribute to the recovery in the acute phase, as capillary restructuring requires at least a week [70]. VEGF, a HIF downstream gene, is upregulated in the penumbra upon onset of cerebral ischaemia [71]. In mammals, the VEGF gene consists of five subtypes, VEGF-A/B/C/D/F, including placental growth factor. VEGF-B mediates embryonic angiogenesis in myocardial tissue, and VEGF-C mediates lymphangiogenesis. The primary molecule that is associated with endothelial cell proliferation in cerebral ischemia is VEGF-A. VEGFs bind to VEGF receptors, namely, VEGFR-1, 2 and 3 [71]. The increase in VEGF-A and VEGF-receptors begins as early as 2–4 h after the onset of stroke and lasts for at least 28 days [72].
Angiogenesis is the growth of new blood vessels from existing vessels, typically in response to hypoxia. Angiogenesis encompasses coordinated remodelling of the basal lamina matrix with endothelial cells to generate new blood vessels. The extent of angiogenesis within the penumbra of ischaemic stroke correlates with the patients’ survival time [73]. VEGF’s role in neuroprotection is less understood, but its ability to influence neuron survival is noted [71]. Increasing VEGF has been shown to increase neural proliferation markers such as 5-bromo-2′-deoxyuridine (BrdU) in the hippocampus [73]. Zhang et al. [74] showed that VEGF mediated an increase in CBF that maintained the penumbral blood supply. Nevertheless, VEGF can activate matrix metalloproteinases (MMPs) such as MMP9 and downregulate tight junction proteins such as claudin-5 and occludin, leading to BBB disruption during the acute phase of ischaemic stroke [75], leading to haemorrhagic transformation and further brain cell death [76].
The critical importance of astrocytes in the induction and maintenance of BBB structure and function has long been established [77,78,79,80,81]. Confocal microscopy studies have shown that brain endothelial cells are surrounded by the perivascular endfeet of astrocytes that contain rosette-like whorls of filaments. Multiple endfeet from the same astrocyte can interact with several endothelial cells, and an endothelial cell may be surrounded by endfeet from several astrocytes [82]. Several studies have provided a great deal of information to support the role of astrocytes in upregulating many BBB features, including low paracellular permeability/functional tight junctions [83,84,85,86], transporters [87,88,89] and enzymes [90]. In addition to astrocytes, pericytes seem to play an important role in orchestrating the proper formation of the BBB and the NVU. Pericytes extend their processes along and around pre-capillary arterioles, capillaries and post-capillary venules, and may have different morphological and functional features depending on their positions along the vascular tree [91].
Increased BBB permeability following ischaemic stroke has been shown to have a complex biphasic profile [92,93]. An early opening of the BBB within hours of ischemia is followed by a refractory phase, and then a second opening in 72 h [92,94]. Using transgenic mouse models and two-photon time-lapse microscopy, Knowland et al. [95] visualised structural tight junction protein localisation and BBB permeability following transient middle cerebral artery occlusion (MCAO). They demonstrated that an early (within 4–6 h after stroke) increase in BBB permeability was associated with upregulation of endothelial transcytosis, while the delayed (2–3 days after stroke) increase in BBB permeability was due to the remodelling and disassembly of tight junction proteins [95] (Figure 2).
BBB breakdown leads to cerebral oedema [96,97] and haemorrhagic transformation [98,99], which are common complications of ischemic stroke that can impact the outcomes of these patients with potentially serious and life-threatening consequences [100]. BBB breakdown is the main cause of vasogenic oedema, where water and plasma proteins enter the brain interstitial space, leading to brain tissue swelling as the brain capillaries behave like fenestrated capillaries. However, cytotoxic oedema does not lead to brain tissue swelling. Rather, cells of the CNS, particularly astrocytes swell following CNS injury (e.g., ischaemic stroke). During ischaemic stroke, a fall in cellular ATP levels leads to the inhibition of ATP-dependent transporters (e.g., Na+/K+ ATPase). The resulting influx of osmolytes such as Na+, which generates an osmotic force, drives an influx of water into the cells, causing cellular swelling [101,102]. Astrocytes in particular play a major role in cytotoxic oedema via the water channel AQP4, as discussed in detail in the next section.
4. The Role of Astrocytic Aquaporin 4 in Cerebral Oedema Formation
Aquaporins (AQPs) are small integral membrane proteins (MW ∼ 30,000) that mainly facilitate water transport across cell membranes in response to osmotic gradients, and are found in several tissues, including the brain, kidneys and lungs [103]. Fourteen AQPs have been identified in humans and rodents, with at least eight of these having been shown to be directly involved in water transport [104]. AQP1, 4 and 9 are expressed in the brain, but only AQP1 (located in the apical membrane of choroid plexus epithelial cells) and AQP4 (highly expressed on astrocyte endfeet; in the subpial astrocyte processes and the basolateral membranes of ependymal cells; and in subependymal astrocyte processes), are critically important in regulating brain water flux [104,105]. In addition to water transport, a subgroup of AQPs known as aquaglyceroporins (AQP3, 7 and 9) transport glycerol, along with small polar solutes, ions and various gases [106,107,108]. Furthermore, AQP4 has been shown to be involved in astrocyte migration, glial scar formation, neuroinflammation and extracellular K+ uptake [109].
Oedema is a major and often life-threatening consequence of stroke [101]. Cytotoxic oedema has been shown to be primarily mediated by AQP4, as the water is transported through AQP4 located in astrocyte endfeet into the CNS with the BBB intact [110,111]. However, vasogenic oedema is independent of AQP4, and water enters the CNS through intercellular spaces due to BBB breakdown [111]. Frydenlund et al. [112] have shown that the expression level of perivascular AQP4 pool is subject to regional-specific temporary changes after transient MCAO in mice. This has important implications for cytotoxic oedema formation and dissolution, as perivascular AQP4 allows bidirectional water flow, and therefore is most likely to be the rate-limiting step for both water influx and efflux after ischaemic stroke [112]. In support of this, AQP4-deficient mice had reduced cytotoxic brain oedema and improved neurological outcomes 24 h after permanent MCAO [113]. Further work has shown that AQP4 knockout mice had decreased infarct volume, neuronal cell death and neuroinflammation, and had improved long-term outcomes after transient MCAO compared with wild type mice [114]. However, AQP4 knockout mice had increased water accumulation at three and seven days after transient MCAO compared with wild type mice [114]. This suggests that vasogenic oedema due to reperfusion rather than cytotoxic oedema is responsible for this increase in water content and impaired clearance. Therefore, AQP4 inhibitors may be a therapeutic option for reducing cytotoxic oedema after ischaemic stroke.
The potential of AQPs as a therapeutic target for reducing oedema has been demonstrated in several studies [109,115,116]. For example, work by Kourghi et al. [117] using in vitro models of choroid plexus and oocytes demonstrated the potential of several loop-diuretic derivatives to inhibit AQP1 and 4 functions. In addition, treating mice subjected to stroke with the loop diuretic bumetanide led to reduced infarct volume and ipsilateral hemispheric water content, as well as a significant reduction in AQP4 protein expression compared with untreated animals [118,119]. In addition to the loop diuretics and their derivatives, antiepileptic drugs and other small molecule inhibitors such as tetraethylammonium [120] and TGN-020 [121] have been shown to inhibit water transport, but with limited progress. Nevertheless, no drug has been approved to target AQPs to reduce brain oedema in stroke. Recently, Sylvain et al. [122] have shown that an FDA-approved drug, trifluoperazine, can reduce cerebral oedema during the early acute phase in mice subjected to photothrombotic stroke. Trifluoperazine is a phenothiazine derivative and a dopamine antagonist with antipsychotic and antiemetic activities, but also inhibits AQP4 expression at both gene and protein levels, and leads to an increase in glycogen levels, suggesting trifluoperazine treatment can be beneficial for brain energy metabolism [122]. Treatment with trifluoperazine inhibited AQP4 localisation to the blood–spinal cord barrier, and led to a reduction in CNS oedema and the acceleration of functional recovery in a rat spinal cord injury model [66].
Several previous studies have also demonstrated that changes in astrocyte AQP4 localisation/polarisation can result in responses to ischaemia and other insults [123,124,125,126], which may not be accompanied by any change in AQP4 expression levels [112,123,127]. This change in AQP4 polarisation might be a potential protective mechanism that counteracts early oedema formation to minimise brain damage [125]. Therefore, inhibiting changes in AQP4 localisation might be a better therapeutic strategy than complete blocking of AQP4, which has an important role in brain fluid homeostasis.
5. The Role of Astrocyte Activation in Neuroinflammation after Ischaemic Stroke
Microglia and astrocytes are the major immunocompetent cells of the CNS, although astrocytes are frequently overlooked in this regard [53]. Astrocytes contain a number of receptors that are involved in innate immunity, such as Toll-like receptors (TLR), nucleotide-binding oligomerisation domains, double-stranded RNA-dependent protein kinase, scavenger receptors, mannose receptor and components of the complement system [128]. Both complement component C1q and C3 are associated with astrocyte A1 phenotype [129]. Complement peptide C3a promotes astrocyte survival in response to ischaemic stress, and the protective effect can be reversed by C3a-receptor deficiency [130]. Astrocytes can modulate immune response by inhibiting T cells and monocyte activation [131]. After ischaemic stroke, some reactive astrocytes transform into nonprofessional phagocytes and “clean up” the infarct area [132,133].
Both microglia and astrocytes respond to damage-associated molecule patterns (DAMPs) in ischaemic stroke [134]. DAMPs elicit a strong inflammatory response by activating pattern recognition receptors, such as TLR2 and TLR4 that are crucial inflammatory mediators after stroke. Through activating TLR4, MMP9 is upregulated in neurons and astrocytes [135]. TLR2 plays a detrimental role in the haemorrhagic mouse brain by activating MMP9 in astrocytes, compromising the BBB and enhancing neutrophil infiltration and proinflammatory cytokine gene expression [136]. Suppression of TLR2 and TLR4 reduces nuclear factor-kappa B (NF-κB) activity, which is involved in proinflammatory and redox-active pathways [137]. NF-κB signalling pathway regulates the secretion of cytokines and chemokines in astrocytes under physiological and pathophysiological conditions. The commonly produced cytokines by active astrocytes are interleukins (IL-6, IL-10 and IL-1β), IFN-γ and transforming growth factor β (TGF-β). Studies have previously pointed out that depending on the extent of ischaemic injury, the cytokines secreted by astrocytes may be neurodegenerative or neuroprotective [56]. TGF-β was seen to protect neurons against excitotoxicity by inhibiting the tissue plasminogen activator (tPA) potentiated NMDA-induced neuronal death through a mechanism involving the upregulation of the type-1 plasminogen activator inhibitor (PAI-1) in astrocytes. Additionally, TGF-β was reported to prevent neuronal apoptosis via ERK1/2 pathway [56,57]. The inhibition of the NF-κB pathways leads to reductions in the expression of proinflammatory genes, such as TNF-α, vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), and the infiltration of CD11b+ leukocytes [138]. Glycolysis induced by tumour suppressor protein 53 reduces the degradation of IκBα and inhibits NF-κB translocation in astrocytes, thereby ameliorating neuroinflammation [139]. Ischaemic preconditioning stimuli ameliorate the inflammatory response after stroke via the TLR/cytokine pathway [140]. Reactive oxygen species (ROS) formation after ischaemic stroke activates signal transducer and activator of transcription 3 (STAT3). A ROS scavenger dimethylthiourea has been shown to inhibit the activation of the STAT3 pathway and attenuates neuroinflammatory and neuronal injury [141]. In addition, the activation of Notch-1 pathway after stroke facilitates the proliferation of reactive astrocytes and restricts infiltration by immune cells [142].
The local inflammation leads to the upregulation of adhesion molecules such as VCAM-1, ICAM-1 and P-selectin in endothelial cells, and the secretion of MMPs such as MMP2 and MMP9 [143,144]. The enhanced expression of adhesion molecules in brain endothelial cells attracts leukocytes and platelets, and promotes the tethering/rolling, firm adhesion and transmigration of these cells across the BBB [145]. Adhesion of leukocytes causes further damage due to the activation of signalling pathways and the release of ROS and inflammatory cytokines, and MMPs from other cells of the NVU. This creates a proinflammatory environment, which further activates the endothelium and leads to increased BBB permeability (Figure 2). This increased permeability due to inflammation has been demonstrated using magnetic resonance imaging (MRI) at both the acute and chronic phases of stroke in patients with ischaemic stroke [146,147] and in animal models [148,149].
Transcriptome analyses show that reactive astrocytes after ischaemic stroke exert both proinflammatory and neuroprotective functions. Zamanian et al. [150] found that Lcn2 (which may directly promote neuronal death) was induced 228-fold, and Serpina3n was induced 9.1-fold in reactive astrocytes one day after experimental ischaemic stroke. Rakers et al. [151] also found that markers of reactive astrocytes, Lcn2, GFAP, vimentin and Timp1, were highly expressed, and contribute to inflammation (e.g., Spp1, Cd52, Lcn2 and Ifi202b), cell division and migration (e.g., Cdk1, Myo1f and Anxa3). A2-specific transcripts were intriguingly predominant at 72 h after transient MCAO [151]. Furthermore, reactive astrocytes produce and release proinflammatory mediators (e.g., IL-6, TNF-α, IL-1α, IL-1β and IFN-γ), and free radicals (e.g., NO, superoxide, peroxynitrite), which can lead to neuronal death, infarct progression and increased BBB permeability [19].
6. Astrocyte Activation and Glial Scar Formation
The activation of astrocytes in stroke highlights the climax of structural and chemical modifications that act together to form inveterate scars. Elevated synthesis of GFAP and other complementary filaments such as nestin and vimentin occurs at the site of injury in cerebral ischaemia [152]. TGF-β signalling is increased in astrocytes, which regulates glial scar formation and the immune response to stroke [153]. A glial scar is formed around dying brain tissue after ischaemic stroke, which consists predominately of reactive astrocytes, microglia and ECM. Prominent biochemical components of the gliotic scar include secreted macromolecules such as chondroitin sulfate proteoglycans (CSPGs), e.g., aggrecan, brevican, neurocan and phosphacan [154]. Astrocytes in acute lesions upregulate CSPGs, whereas astrocytes in chronic multiple sclerosis lesions express high levels of a protease called “a disintegrin and metalloproteinase with thrombospondin motifs” (ADAMTS) that metabolises CSPGs [155].
The scar has traditionally been viewed to inhibit neurite outgrowth and axonal regeneration. The characteristic of growth inhibitory factor secretion by reactive astrocytes serves as a hindrance to axonal extensions, which deters CNS function recovery at a chronic stage [156,157]. A number of studies consider glial scar formation and reactive gliosis as maladaptive responses; therefore blocking its formation may be beneficial. Targeting glial scar formation or reactive gliosis occurs as an interesting strategy for the treatment of stroke and other neurological disorders. Administration of lipoic acid averts the formation of the glial scar, and promotes angiogenic effects as a vital mechanism for neural rejuvenation [158]. Cyclosporine A (CsA), the most widely used immunosuppressive agent, has been shown to be neuroprotective due to significantly reducing astrogliosis and glial scar formation rather than reducing infarct volume in a rat model of stroke [159].
Nevertheless, reactive gliosis and glial scar formation can serve as neuroprotection in certain stages and regulate neural plasticity inclusive of axonal sprouting, neuron generation, synapse control and function; and maintain CNS homeostasis and restrict neuroinflammation [160]. In addition, glial scar formation prevents the secondary degeneration that occurs at the underlying site of injury where the damage extends to the surrounding environment [161].
7. The Role of Astrocyte Secretions in Ischaemic Stroke
Astrocytes are highly secretory cells in the CNS [162]. By secreting neurotrophic factors such as secreted protein acidic and rich in cysteine (SPARC) family proteins and the Hevin glycoprotein, astrocytes promote growth and survival of neurons, control synapse formation and regulate synaptogenesis [25,163]. Other neurotrophic factors such as NGF, BDNF, NT-3, EPO, VEGF, EGF, IGF, GDNF and bFGF secreted by astrocytes promote neuorestoration following stroke (Section 2) [56,57]. Netrins, ephrins and semaphorins are astrocyte-derived proteins that are known to govern axon guidance [25]. Astrocytes also secrete matrix proteins such as connective tissue growth factor (CTGF)/CCN family proteins, MMP family proteins and tenascins. CCN proteins regulate a broad range of cellular activities, such as adhesion, migration and proliferation [19,163]. MMPs are involved in the degradation of ECM, and are engaged with neuronal injury [25,163]. Many components of iron transport are observed in the astrocyte secretome, including transferrin, hephaestin and ceruloplasmin, thereby regulating iron homeostasis [164,165].
The brain has the second-highest expression level of apolipoprotein E (ApoE), which is a well-reported astrocyte secreted protein and is primarily synthesised locally by astrocytes [166,167]. ApoE was originally known to be involved in lipid transport, but also is reported to modulate neurotransmitter release/sequestration, and regulates brain homeostasis [168]. APOE4, but not APOE3, activates the CypA-MMP9 pathway, and may lead to accelerated BBB breakdown causing neuronal and synaptic dysfunction [169,170]. In APOE4 carriers, BBB breakdown contributes to cognitive decline independent of Alzheimer’s disease pathology [170].
The close proximity and the ability of astrocytes to secrete soluble factors allow them to induce BBB phenotype in brain endothelial cells [171]. Astrocytes have been shown to release VEGF, GDNF, bFGF and angiopoietin-1, which influence BBB function. For example, most of these factors are usually added to endothelial growth media in culture to induce BBB phenotype in brain endothelial cells [172]. Therefore, most in vitro BBB models use astrocytes in co-cultures to take advantage of the astrocyte-secreted factors to increase the tightness of brain endothelial cells-characterised by measurement of the transendothelial electrical resistance (TEER), or by assaying paracellular permeability markers across the monolayer [173,174]. Astrocytes are a source of MMPs and VEGF that damage the BBB after ischaemia [175,176]. Other chemical mediators released by astrocytes, such as sonic hedgehog protein (Shh), GDNF, prostaglandins, NO and AA, also regulate tight junctions, blood vessel diameter and blood flow [177,178]. Astrocytes also secrete cytokines and chemokines, such as IL-6, IL-10, IL-1β, IFN-γ and TGF-β. These cytokines can be either neurodegenerative or neuroprotective (Section 5).
Dhandapani et al. [179] reported that soluble factors in astrocyte-conditioned medium (ACM) protect murine neurons from serum-deprivation induced cell death by releasing TGF-β, which activates the activator protein-1 (AP-1) protective pathway and prevents apoptosis. A study has also reported that ACM constituents such as interleukins (IL-6, IL-10 and IL-1β) and TGF-β play vital roles in ACM-induced ischaemic tolerance in neurons [180]. Song et al. [181] demonstrated that ACM exerts neuroprotective effects in ischaemic stroke, through modulation of NT-3, GDNF and TNF-α secretion. Another study reported that ACM provides a neuroprotective effect by regulating apoptosis-related protein expression [182]. Overall, the results from these studies suggest that important factors secreted by astrocytes are vital for neuronal protection during ischaemic injury.
On the other hand, the secretory activity of reactive astrocytes can exacerbate tissue injury. For example, an increased concentration of TNF-α can inhibit neurite outgrowth [183]. Astrocytes also release several pro-inflammatory cytokines (TNF-α, IL-1β, IL-6 and IFN-γ) in response to acute ischaemia, which trigger the production of secondary mediators such as AA metabolites, NO, ROS and MMPs that promote neuronal degeneration and axonal demyelination [56]. The factors secreted by astrocytes can act in an autocrine/paracrine fashion, thereby resulting in amplification of secretion, contributing to sustained astrogliosis and neurotoxicity [184]. Activated astrocytes also secrete compounds with potentially direct toxic effects on neurons/axons and oligodendrocytes/myelin, such as reactive oxygen and nitrogen species, glutamate and ATP [185].
In conjunction with soluble proteins, ACM contains extracellular vesicles (EVs), which can be broadly divided into exosomes (less than 200 nm), microvesicles (200–1000 nm) and apoptotic bodies (larger than 1000 nm). Exosomes are being widely pursued due to their abilities to cross the BBB and interact with target cells [186]. Studies suggest that cells take up exosomes by endocytosis via receptor-mediated adhesion, direct fusion, or via ligand-receptor interactions. Exosomes may contain proteins, lipids, metabolites, miRNA/sRNA, DNA, enzymes, growth factors and cytokines depending on their origins and targets [187]. A study reported that preconditioning neurons with exosomes obtained from glial cells were protective during acute ischaemia [188]. It has been reported that glial cells transfer miRNA to neurons via exosomes targeting signalling pathways such as PI3K/AKT pathway, Hippo, MAPK or mTOR. Exosomal content (such as miRNA) is reported to promote neurogenesis, axonal remodelling, vascular remodelling, and to reduce neuroinflammation [187]. Pei et al. [189] reported that astrocyte-derived exosomes suppressed autophagy and ameliorated neuronal damage during ischaemic stroke. Another study by Hira et al. [190] reported that astrocyte-derived exosomes treated with semaphorin 3a inhibitor enhanced stroke recovery via prostaglandin d2 synthase. Taylor et al. [191] reported that in response to oxidative stress, cultured astrocytes released elevated amounts of heat-shock protein 70 (HSP70) and synapsin 1 in association with exosomes. miR-92b-3p released from astrocytes subjected to oxygen glucose deprivation (OGD) was associated with activation of the PI3K/AKT pathway and ameliorated OGD-induced injury in neurons [192]. Viral vectors expressing miR-124 were found to increase neurogenesis and to promote neuroprotection against cerebral ischaemia in vivo [193]. Astrocytes also release the miR-17-92 cluster that promotes neurite elongation [194]. Taken together, emerging evidence suggest that extracellular vesicles are an important part of the astrocyte secretome and can have important therapeutic implications for ischaemic stroke [195].
8. Targeting Astrocytes as a New Therapy for Stroke
The aim of astrocyte targeted strategies is to maximise the potential for astrocyte survival following ischaemic stroke, which may lead to an increase in neuronal survival in the penumbra, essential for favourable post-stroke outcomes [196]. Several promising targets have been identified in preclinical studies that could increase astrocyte survival. For example, overexpressing heat HSP72 and dismutase 2 can increase astrocytes’ resistance under ischaemic stress [52], and ceftriaxone, an antibiotic, has been shown to upregulate expression of GLT-1 in astrocytes, leading to neuronal protection in stroke [197,198]. Furthermore, Deng et al. [199] showed bone marrow-derived mesenchymal stem cell (BMSC)-derived exosomal miR-138-5p promoted the proliferation of mouse primary astrocytes after OGD. BMSC-derived miR-138-5p delivered to astrocytes via exosomes alleviated neuron injury in mice following MCAO.
S100β, a calcium dependent protein which is produced primarily by astrocytes in the CNS, is released into the serum and CSF 24–96 h after ischaemic stroke onset [200,201]. S100β can be neuroprotective at low concentrations but can lead to neuron and astrocyte death at high concentrations [201]. It has been proposed that targeting astrocytes to reduce S100β level could be neuroprotective [202]. Arundic acid (ONO-2506), which reduces the synthesis of S100β in astrocytes [203], has been shown to decrease stroke volume and improve functional outcomes [204]. The protective effect of the arundic acid was greater when administered 24 h after the stroke onset compared with immediate administration [204]. Based on these results, Pettigrew et al. [55] conducted a multi-centre, dose-escalating, randomised, double-blind, placebo-controlled phase 1 trial to examine the effect of arundic acid in ischaemic stroke patients, and showed increasing the infusion of arundic acid in multiple-dose can reduce S100β protein level in serum after acute stroke. However, clinical trials have not been able to show efficacy of arundic acid in stroke [202].
As reviewed by Liu and Chopp [19], in addition to affecting cell survival, astrocytes contribute to angiogenesis, neurogenesis, synaptogenesis and axonal remodelling, leading to improved function in the days to weeks following stroke. Astrocytes can be targeted in these periods to improve stroke rehabilitation, thereby making a significant contribution to improving stroke recovery.
Several studies have suggested that genetically modified astrocytes can be used as a therapeutic target based on their regulation of proteins relevant to immune response and cytotoxicity. For example, Tau hyperphosphorylation (a neurodegeneration hallmark and is closely linked to cognitive function deficiency) occurs following focal brain ischaemia [205]. Therefore, proteins that control microtubular mounts (e.g., glycogen kinase 3 (GSK3) and cyclin-dependent kinase 5 (CDK5)) and remodelling of the actin cytoskeleton can be potential targets for neuroregeneration following stroke [206]. In the CDK5 scenario, Gutiérrez-Vargas et al. [207] designed a CDK5-targeted shRNAmiR (a technique known as RNAi, RNA interference) that contributed to improvement in neurological and motor function in a cerebral ischaemia model in the first week following ischaemia. A month after ischaemia, deterioration of learning, memory and reversal learning was prevented by CDK5 RNAi [208]. Moreover, CDK5 RNAi induced astrocyte stellation and the release of BDNF in a Rac1- dependent manner that provided neuroprotection in astrocyte and neuron co-cultures [208]. Following up on this work, Becerra-Calixto and Cardona-Gómez [206] showed that one month after transplantation of CDK5 knock-down astrocytes into ischaemic rats, motor function recovery and endogenous astrocyte branching around blood vessels were increased, accompanied by increased expression of endothelial PECAM1 and cell proliferation marker, Ki67 in the subventricula zone. These effects were sustained for four months and helped prevent neuronal and astrocyte loss, which supported BBB recovery through secretion of BDNF by endogenous astrocytes in ischaemic rats [209]. Taken together these studies suggest that knockdown of CDK5 in astrocytes could be a potential therapeutic target to protect the BBB and enhance the recovery of the NVU and improve brain function following ischaemic stroke.
It has recently been reported that genetic manipulation can convert astrocytes into functional neurons [210]. Differentiation of stem/precursor cells towards neuronal fate is driven by miR-124 inhibition of REST (a transcriptional repressor of neuron-associated genes, including miR-124), an induction loop that is suppressed by polypyrimidine tract-binding protein (PTB). Downregulation of PTB removes this suppression (including in differentiated cells, e.g., fibroblasts), and contributes to specification of a neuronal cell fate. Depletion of PTB in astrocytes, even transient suppression by lentiviral delivery of shRNA, generated neurons [210]. In a model of Parkinson’s disease, midbrain astrocytes were converted into dopaminergic neurons [210]. There may be therapeutic opportunities in exploiting the prevalence of astrocytes in lesions following ischaemic stroke (due to greater survival and proliferative responses), by converting some astrocytes into neurons, although conversion efficiency reportedly varied across brain regions [210].
Neural precursor cell (NPC) transplantation is another potential option for improving stroke recovery. NPC transplantation in mice led to enhanced functional and structural plasticity up to 60 days post ischaemia. This relied on the ability of transplanted NPCs to localise in the penumbra to help promote GLT-1 upregulation in astrocytes and reduction of peri-ischaemic extracellular glutamate [211]. Secretion of VEGF by NPCs was required for the upregulation of GLT-1, as blocking VEGF during the first week after stroke reduced this upregulation and long-term behavioural recovery of these mice [211].
Furthermore, Luo et al. [212] has shown that co-transplantation of astrocytes with neural stem cells (NSCs) leads to increased survival and neuronal differentiation of NSCs following ischaemic stroke in rats compared with NSC transplantation alone. Therefore, astrocyte co-transplantation with NSC could be a novel strategy for repairing the brain following stroke injury.
Given the important role played by astrocytes in ischaemic stroke, models which can accurately mimic the NVU are essential for furthering our understanding of the mechanisms of astrocyte activation and BBB dysfunction to help develop new therapeutics. While traditional BBB models, which usually comprise of co-cultures of brain endothelial cells with astrocytes in static conditions, can give some important mechanistic information on astrocyte-endothelial interactions, advanced 3D or perfusion-based models are much more suited for investigating the complex molecular and cellular interactions of the NVU during ischaemic conditions [174,213]. Furthermore, these BBB models can give real-time information on permeability changes during ischaemia and release of inflammatory mediators as well as live imaging of the NVU/BBB. For example, perfused NVU/BBB-on-a-chip [214,215,216], dynamic in vitro BBB models [217,218] and 3D multi-cultures/organoids [219,220] can simulate the physiological characteristics of the NVU/BBB, and therefore can be used to model hypoxic/ischaemic conditions to understand the underlying pathophysiological mechanisms. Furthermore, these models can also assess/screen the potential of novel drug candidates and BBB penetration before confirmatory studies in animals, thereby reducing the use of animals, time and resources during the early stages of drug discovery.
9. Conclusions
Astrocytes are critical for BBB reconstruction and neuroprotection in the acute stages of ischaemic stroke, as they increase the uptake of extracellular glutamate and sodium/potassium-ATPase activity. Neurotrophic factors released by astrocytes facilitate neurological recovery in the chronic stages. Nevertheless, astrocytes form glial scars, which hinder axon regeneration. In addition, reactive astrocytes produce and release proinflammatory mediators (e.g., IL-6, TNF-α, IL-1α, IL-1β and IFN-γ), free radicals and neurotoxic molecules. Since a large number of astrocytes survive after stroke, and neurons cannot survive if neighbouring astrocytes are lost, astrocytes must be considered as an important treatment target for stroke. Understanding the signalling pathways and molecules secreted by astrocytes in cerebral ischaemia can help guide the development of novel neuroprotective treatments for ischaemic stroke. Indeed, preclinical studies have shown the potential of several target molecules to induce the survival of astrocytes, and reduce cerebral oedema, thereby improving stroke outcomes. Furthermore, genetically modified astrocytes, NPC and NSC have shown promise in experimental strokes by promoting neurorestoration. Furthermore, advanced in vitro models such as perfusion-based NVU/BBB models or organoids can give valuable information on the roles of astrocytes during hypoxic/ischaemic conditions, and screen potential therapeutic candidates for ischaemic stroke.
Author Contributions
Conceptualisation, R.C. and A.P.; writing—original draft preparation, A.P. and R.C.; writing—review and editing, A.P., A.S., S.J., J.S. and R.C. All authors have read and agreed to the published version of the manuscript.
Funding
A.P. is supported by the NSW Ministry of Health, Australia under the NSW Health Early-Mid Career Fellowships Scheme. All content is solely the responsibility of the authors and do not reflect the views of the NSW Health Entity. R.C. acknowledges funding from the Wellcome Trust (200633/z/16/z).
Conflicts of Interest
The authors declare no conflict of interest.
References
- GBD 2016 Stroke Collaborators. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Gribkoff, V.K.; Starrett, J.E., Jr.; Dworetzky, S.I.; Hewawasam, P.; Boissard, C.G.; Cook, D.A.; Frantz, S.W.; Heman, K.; Hibbard, J.R.; Huston, K.; et al. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat. Med. 2001, 7, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cabrer, P.; Campos, F.; Sobrino, T.; Castillo, J. Targeting the ischemic penumbra. Stroke 2011, 42, S7–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Ma, H.; Ringleb, P.A.; Parsons, M.W.; Churilov, L.; Bendszus, M.; Levi, C.R.; Hsu, C.; Kleinig, T.J.; Fatar, M.; et al. Extending thrombolysis to 4·5-9 h and wake-up stroke using perfusion imaging: A systematic review and meta-analysis of individual patient data. Lancet 2019, 394, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.C.V.; Donnan, G.A.; Lees, K.R.; Hacke, W.; Khatri, P.; Hill, M.D.; Goyal, M.; Mitchell, P.J.; Saver, J.L.; Diener, H.C.; et al. Endovascular stent thrombectomy: The new standard of care for large vessel ischaemic Stroke. Lancet Neurol. 2015, 14, 846–854. [Google Scholar] [CrossRef]
- Campbell, B.C.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Sardar, P.; Chatterjee, S.; Giri, J.; Kundu, A.; Tandar, A.; Sen, P.; Nairooz, R.; Huston, J.; Ryan, J.J.; Bashir, R.; et al. Endovascular therapy for acute ischaemic stroke: A systematic review and meta-analysis of randomized trials. Eur. Heart J. 2015, 36, 2373–2380. [Google Scholar] [CrossRef] [Green Version]
- Zerna, C.; Thomalla, G.; Campbell, B.C.V.; Rha, J.H.; Hill, M.D. Current practice and future directions in the diagnosis and acute treatment of ischaemic Stroke. Lancet 2018, 392, 1247–1256. [Google Scholar] [CrossRef]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Neuhaus, A.A.; Couch, Y.; Hadley, G.; Buchan, A.M. Neuroprotection in stroke: The importance of collaboration and reproducibility. Brain 2017, 140, 2079–2092. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.; Goyal, M.; Menon, B.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- García-Cabezas, M.A.; John, Y.J.; Barbas, H.; Zikopoulos, B. Distinction of Neurons, Glia and Endothelial Cells in the Cerebral Cortex: An Algorithm Based on Cytological Features. Front. Neuroanat. 2016, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Lent, R.; Azevedo, F.; Andrade-Moraes, C.H.; Pinto, A.V.O. How many neurons do you have? Some dogmas of quantitative neuroscience under revision. Eur. J. Neurosci. 2012, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The Search for True Numbers of Neurons and Glial Cells in the Human Brain: A Review of 150 Years of Cell Counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, C.C.; Stimpson, C.D.; Raghanti, M.A.; Wildman, D.E.; Uddin, M.; Grossman, L.I.; Goodman, M.; Redmond, J.C.; Bonar, C.J.; Erwin, J.M.; et al. Evolution of increased glia-neuron ratios in the human frontal cortex. Proc. Natl. Acad. Sci. USA 2006, 103, 13606–13611. [Google Scholar] [CrossRef] [Green Version]
- Pakkenberg, B.; Gundersen, H. Total number of neurons and glial cells in human brain nuclei estimated by the disector and the fractionator. J. Microsc. 1988, 150, 1–20. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Anderson, M.F.; Blomstrand, F.; Blomstrand, C.; Eriksson, P.S.; Nilsson, M. Astrocytes and stroke: Networking for survival? Neurochem. Res. 2003, 28, 293–305. [Google Scholar] [CrossRef]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic Stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.H.; Cho, J.; Kwon, D.H.; Park, J.Y.; Woo, J.; Lee, J.M.; Lee, S.; Ko, H.Y.; Won, W.; Kim, R.G.; et al. Excessive Astrocytic GABA Causes Cortical Hypometabolism and Impedes Functional Recovery after Subcortical Stroke. Cell Rep. 2020, 32, 107861. [Google Scholar] [CrossRef]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair After Ischemic Stroke. Trans. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özen, I.; Deierborg, T.; Miharada, K.; Padel, T.; Englund, E.; Genové, G.; Paul, G. Brain pericytes acquire a microglial phenotype after Stroke. Acta Neuropathol. 2014, 128, 381–396. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Poskanzer, K.E.; Molofsky, A.V. Dynamism of an Astrocyte In Vivo: Perspectives on Identity and Function. Annu. Rev. Physiol. 2018, 80, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, S.A.; Schnell, O.; Pöschl, J.; Eigenbrod, S.; Kretzschmar, H.A.; Tonn, J.C.; Schüller, U. ALDH1A1 is a marker of astrocytic differentiation during brain development and correlates with better survival in glioblastoma patients. Brain Pathol. 2012, 22, 788–797. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Dallérac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef]
- Rosenberg, P.A.; Amin, S.; Leitner, M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J. Neurosci. 1992, 12, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Contributions of Glycogen to Astrocytic Energetics during Brain Activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, R.A. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can. J. Physiol. Pharmacol. 1992, 70, S138–S144. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Bergher, J.P.; Anderson, C.M.; Treadway, J.L.; Fosgerau, K.; Swanson, R.A. Astrocyte glycogen sustains neuronal activity during hypoglycemia: Studies with the glycogen phosphorylase inhibitor CP-316, 819 ([R-R*, S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther. 2007, 321, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.B.; Heimbürger, C.M.; Bouman, S.D.; Schousboe, A.; Waagepetersen, H.S. Robust glycogen shunt activity in astrocytes: Effects of glutamatergic and adrenergic agents. Neuroscience 2009, 158, 284–292. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Heales, S.J.; Peuchen, S.; Barker, J.E.; Land, J.M.; Clark, J.B. Nitric oxide-mediated mitochondrial damage: A potential neuroprotective role for glutathione. Free Radic Biol Med. 1996, 21, 995–1001. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Martinez, A.D.; Retamal, M.A. Gap junction channels and hemichannels in the CNS: Regulation by signaling molecules. Neuropharmacology 2013, 75, 567–582. [Google Scholar] [CrossRef]
- Li, X.; Zhao, H.; Tan, X.; Kostrzewa, R.M.; Du, G.; Chen, Y.; Zhu, J.; Miao, Z.; Yu, H.; Kong, J.; et al. Inhibition of connexin43 improves functional recovery after ischemic brain injury in neonatal rats. Glia 2015, 63, 1553–1567. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Bennet, L.; Nicholson, L.F.; Danesh-Meyer, H.; O’Carroll, S.J.; Gunn, A.J. A key role for connexin hemichannels in spreading ischemic brain injury. Curr. Drug Targets 2013, 14, 36–46. [Google Scholar] [CrossRef]
- Kim, Y.; Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; O’Carroll, S.J.; Zhang, J. Connexins and Pannexins in cerebral ischemia. Biochim. Biophys. Acta Biomembr. 2018, 1860, 224–236. [Google Scholar] [CrossRef]
- Zündorf, G.; Kahlert, S.; Reiser, G. Gap-junction blocker carbenoxolone differentially enhances NMDA-induced cell death in hippocampal neurons and astrocytes in co-culture. J. Neurochem. 2007, 102, 508–521. [Google Scholar] [CrossRef]
- Takano, K.; Ogawa, M.; Kawabe, K.; Moriyama, M.; Nakamura, Y. Inhibition of gap junction elevates glutamate uptake in cultured astrocytes. Neurochem. Res. 2018, 43, 59–65. [Google Scholar] [CrossRef]
- Xu, L.J.; Emery, J.F.; Ouyang, Y.B.; Voloboueva, L.A.; Giffard, R.G. Astrocyte Targeted Overexpression of Hsp72 or SOD2 Reduces Neuronal Vulnerability to Forebrain Ischemia. Glia 2010, 58, 1042–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated micro. Glia Nat. 2017, 541, 481–487. [Google Scholar]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, L.C.; Kasner, S.E.; Gorman, M.; Atkinson, R.P.; Funakoshi, Y.; Ishibashi, H. Arundic Acid (ONO-2506) Stroke Study Group. Effect of arundic acid on serum S-100beta in ischemic Stroke. J. Neurol. Sci. 2006, 251, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Barreto, G.E.; Gonzalez, J.; Torres, Y.; Morales, L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci. Res. 2011, 71, 107–113. [Google Scholar] [CrossRef]
- Jean-Claude, P.; Bordey, A. The multifaceted subventricular zone astrocyte: From a metabolic and pro-neurogenic role to acting as a neural stem cell. Neuroscience 2016, 323, 20–28. [Google Scholar]
- Acaz-Fonseca, E.; Ortiz-Rodriguez, A.; Azcoitia, I.; Garcia-Segura, L.M.; Arevalo, M.A. Notch signaling in astrocytes mediates their morphological response to an inflammatory challenge. Cell Death Discov. 2019, 5, 85. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome analysis of gene expression provides new insights into the effect of mild therapeutic hypothermia on primary human cortical astrocytes cultured under hypoxia. Front. Cell Neurol. 2017, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D.; et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Fernandez, E.; Staursky, D.; Lucas, K.; Nguyen, B.V.; Li, M.; Liu, Y.; Washington, C.; Coolen, L.M.; Fan, F.; Roman, R.J. 20-HETE Enzymes and Receptors in the Neurovascular Unit: Implications in Cerebrovascular Disease. Front. Neurol. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Mechanisms of Astrocyte-Mediated Cerebral Edema. Neurochem. Res. 2015, 40, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Sykova, E. Glial diffusion barriers during aging and pathological states. Prog. Brain Res. 2001, 132, 339–363. [Google Scholar]
- Chen, R.L.; Lai, U.H.; Zhu, L.L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species (ROS) formation in the brain at different oxygen Levels: Role of hypoxia inducible factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.A.; Papadakis, M.; Chen, R.L.; Buchan, A.M. Cerebral blood flow alteration in neuroprotection following cerebral ischaemia. J. Physiol. 2011, 589, 4105–4114. [Google Scholar] [CrossRef]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas-Rivera, A.; Campero-Romero, A.N.; Heras-Romero, Y.; Penagos-Puig, A.; Rincón-Heredia, R.; Tovar-Y.-Romo, L.B. Early Post-stroke Activation of Vascular Endothelial Growth Factor Receptor 2 Hinders the Receptor 1-Dependent Neuroprotection Afforded by the Endogenous Ligand. Front. Cell Neurosci. 2019, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Takahashi, T.; Ishikawa, M.; Onodera, O.; Shimohata, T.; Del Zoppo, G.J. Angiogenesis in the ischemic core: A potential treatment target? J. Cereb. Blood Flow Metab. 2019, 39, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood–brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davson, H.; Oldendorf, W.H. Symposium on membrane transport. Transport in the central nervous system. Proc. R. Soc. Med. 1967, 60, 326–329. [Google Scholar]
- Stewart, P.A.; Wiley, M.J. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: A study using quail-chick transplantation chimeras. Dev. Biol. 1981, 84, 183–192. [Google Scholar] [CrossRef]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood–brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Wilkinson, M.; Hume, R.; Strange, R.; Bell, J.E. Glial and neuronal differentiation in the human fetal brain 9–23 weeks of gestation. Neuropathol. Appl. Neurobiol. 1990, 16, 193–204. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The perivascular astroglial sheath provides a complete covering of the brain microvessels: An electron microscopic 3D reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Lee, S.-W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Tao-Cheng, J.H.; Nagy, Z.; Brightman, M.W. Tight junctions of brain endothelium in vitro are enhanced by astro. Glia J. Neurosci. Off. J. Soc. Neurosci. 1987, 7, 3293–3299. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar] [CrossRef]
- Patabendige, A.; Skinner, R.A.; Morgan, L.; Abbott, N.J. A detailed method for preparation of a functional and flexible blood-brain barrier model using porcine brain endothelial cells. Brain Res. 2013, 1521, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, M.S.; Krizanac-Bengez, L.; Macchia, F.; Naftalin, R.J.; Pedley, K.C.; Mayberg, M.R.; Marroni, M.; Leaman, S.; Stanness, K.A.; Janigro, D.; et al. Mechanisms of glucose transport at the blood-brain barrier: An in vitro study. Brain Res. 2001, 904, 20–30. [Google Scholar] [CrossRef]
- Helms, H.C.; Madelung, R.; Waagepetersen, H.S.; Nielsen, C.U.; Brodin, B. In Vitro evidence for the brain glutamate efflux hypothesis: Brain endothelial cells cocultured with astrocytes display a polarized brain-to-blood transport of glutamate. Glia 2012, 60, 882–893. [Google Scholar] [CrossRef]
- Baello, S.; Iqbal, M.; Gibb, W.; Matthews, S.G. Astrocyte-mediated regulation of multidrug resistance p-glycoprotein in fetal and neonatal brain endothelial cells: Age-dependent effects. Physiol. Rep. 2016, 4, e12853. [Google Scholar] [CrossRef] [Green Version]
- Demeuse, P.; Kerkhofs, A.; Struys-Ponsar, C.; Knoops, B.; Remacle, C.; van den Bosch de Aguilar, P. Compartmentalized coculture of rat brain endothelial cells and astrocytes: A syngenic model to study the blood-brain barrier. J. Neurosci. Methods 2002, 121, 21–31. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T.; Ting, P.; Martinez, H.; Klatzo, I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol. 1985, 68, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.A.; Mesquita, M.; Ashioti, M.; Beech, J.S.; Williams, S.C.R.; Irving, E.; Cash, D. Late changes in blood-brain barrier permeability in a rat tMCAO model of stroke detected by gadolinium-enhanced MRI. Neurol. Res. 2020, 42, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.G.; Xue, D.; Preston, E.; Karbalai, H.; Buchan, A.M. Biphasic opening of the blood–brain barrier following transient focal ischemia: Effects of hypothermia. Can. J. Neurol. Sci. 1999, 26, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D.; et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in Stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Flores, S.; Berge, E.; Whittle, I.R. Surgical decompression for cerebral oedema in acute ischaemic Stroke. Cochr. Datab. Syst. Rev. 2012, 1, Cd003435. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, L.; Kardos, L.; Szász, J.; Szatmári, S.; Bereczki, D.; Hegedüs, K.; Csiba, L. Factors influencing hemorrhagic transformation in ischemic stroke: A clinicopathological comparison. Eur. J. Neurol. 2006, 13, 1251–1255. [Google Scholar] [CrossRef]
- Szepesi, R.; Csokonay, Á.; Murnyák, B.; Kouhsari, M.C.; Hofgárt, G.; Csiba, L.; Hortobágyi, T. Haemorrhagic transformation in ischaemic stroke is more frequent than clinically suspected—A neuropathological study. J. Neurol. Sci. 2016, 368, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Balami, J.S.; Chen, R.L.; Grunwald, I.Q.; Buchan, A.M. Neurological complications of acute ischaemic Stroke. Lancet Neurol. 2011, 10, 357–371. [Google Scholar] [CrossRef]
- Simard, J.M.; Kent, T.A.; Chen, M.; Tarasov, K.V.; Gerzanich, V. Brain oedema in focal ischaemia: Molecular pathophysiology and theoretical implications. Lancet Neurol. 2007, 6, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S. Aquaporins. Curr. Biol. 2013, 23, R52–R55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelhus, E.A.; Ottersen, O.P. Physiological roles of aquaporin-4 in brain. Physiol. Rev. 2013, 93, 1543–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara-Chikuma, M.; Verkman, A.S. Physiological roles of glycerol-transporting aquaporins: The aquaglyceroporins. Cell Mol. Life Sci. 2006, 63, 1386–1392. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Salman, M.M.; Pickel, S.U.; Jennings, J.; Törnroth-Horsefield, S.; Conner, M.T.; Bill, R.M.; Conner, A.C. Water channel pore size determines exclusion properties but not solute selectivity. Sci. Rep. 2019, 9, 20369. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Smith, A.J.; Phuan, P.W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Exp. Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus 2007, 22, E2. [Google Scholar] [CrossRef] [Green Version]
- Michinaga, S.; Koyama, Y. Pathogenesis of brain edema and investigation into anti-edema drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef] [Green Version]
- Frydenlund, D.S.; Bhardwaj, A.; Otsuka, T.; Mylonakou, M.N.; Yasumura, T.; Davidson, K.G.; Zeynalov, E.; Skare, O.; Laake, P.; Haug, F.M.; et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13532–13536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic Stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Fukuda, A.M.; Ambadipudi, K.; Rashid, F.; Binder, D.; Verkman, A.; Ashwal, S.; Obenaus, A.; Badaut, J. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. Blood Flow Metab. 2017, 37, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [Green Version]
- Abir-Awan, M.; Kitchen, P.; Salman, M.M.; Conner, M.T.; Conner, A.C.; Bill, R.M. Inhibitors of Mammalian Aquaporin Water Channels. Int. J. Mol. Sci. 2019, 20, 1589. [Google Scholar] [CrossRef] [Green Version]
- Kourghi, M.; Pei, J.V.; De Ieso, M.L.; Flynn, G.; Yool, A.J. Bumetanide Derivatives AqB007 and AqB011 Selectively Block the Aquaporin-1 Ion Channel Conductance and Slow Cancer Cell Migration. Mol. Pharmacol. 2016, 89, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Migliati, E.R.; Amiry-Moghaddam, M.; Froehner, S.C.; Adams, M.E.; Ottersen, O.P.; Bhardwaj, A. Na(+)-K (+)-2Cl (-) cotransport inhibitor attenuates cerebral edema following experimental stroke via the perivascular pool of aquaporin. Neurocrit. Care 2010, 13, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Detmers, F.J.; de Groot, B.L.; Müller, E.M.; Hinton, A.; Konings, I.B.; Sze, M.; Flitsch, S.L.; Grubmüller, H.; Deen, P.M. Quaternary ammonium compounds as water channel blockers. Specificity, potency, and site of action. J. Biol. Chem. 2006, 281, 14207–14214. [Google Scholar] [CrossRef] [Green Version]
- Popescu, E.S.; Pirici, I.; Ciurea, R.N.; Bălşeanu, T.A.; Cătălin, B.; Mărgăritescu, C.; Mogoantă, L.; Hostiuc, S.; Pirici, D. Three-dimensional organ scanning reveals brain edema reduction in a rat model of strok e treated with an aquaporin 4 inhibitor. Rom. J. Morphol. Embryol. 2017, 58, 59–66. [Google Scholar] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Brown, J.E.; Bill, R.M.; Conner, A.C.; Conner, M.T. Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur. J. Neurosci. 2017, 46, 2542–2547. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, P.; Day, R.E.; Taylor, L.H.; Salman, M.M.; Bill, R.M.; Conner, M.T.; Conner, A.C. Identification and Molecular Mechanisms of the Rapid Tonicity-induced Relocalization of the Aquaporin 4 Channel. J. Biol. Chem. 2015, 290, 16873–16881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, E.; Enzmann, G.U.; Lin, S.; Ghavampour, S.; Hannocks, M.J.; Zuber, B.; Rüegg, M.A.; Sorokin, L.; Engelhardt, B. Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia 2012, 60, 1646–1659. [Google Scholar] [CrossRef]
- Ciappelloni, S.; Bouchet, D.; Dubourdieu, N.; Boué-Grabot, E.; Kellermayer, B.; Manso, C.; Marignier, R.; Oliet, S.H.R.; Tourdias, T.; Groc, L. Aquaporin-4 Surface Trafficking Regulates Astrocytic Process Motility and Synaptic Activity in Health and Autoimmune Disease. Cell Rep. 2019, 27, 3860–3872.e4. [Google Scholar] [CrossRef] [Green Version]
- Neely, J.D.; Amiry-Moghaddam, M.; Ottersen, O.P.; Froehner, S.C.; Agre, P.; Adams, M.E. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl. Acad. Sci. USA 2001, 98, 14108–14113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Shinjyo, N.; de Pablo, Y.; Pekny, M.; Pekna, M. Complement peptide C3a promotes astrocyte survival in response to ischemic stress. Mol. Neurobiol. 2016, 53, 3076–3087. [Google Scholar] [CrossRef]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019. [Google Scholar] [CrossRef]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017, 2, 28. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef]
- Rawlinson, C.; Jenkins, S.; Thei, L.; Dallas, M.; Chen, R.L. Post-ischaemic immunological response in the brain: Targeting microglial activation in ischaemic stroke therapy. Brain Sci. Neurogl. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Xu, J.; Zheng, Y.; Wei, Y.; Zhu, X.; Lo, E.H.; Moskowitz, M.A.; Sims, J.R. HMGB1 promotes MMP-9 upregulation through TLR4 after cerebral ischemia. Stroke 2010, 41, 2077–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, H.; Hong, J.; Cho, I.; Jang, Y.H.; Lee, H.; Kim, D.; Yu, S.; Lee, S. TLR2-induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Mol. Brain 2015, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Signaling to NF-kB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Dvoriantchikova, G.; Barakat, D.; Brambilla, R.; Agudelo, C.; Hernandez, E.; Bethea, J.R.; Shestopalov, V.I.; Ivanov, D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 2009, 30, 175–185. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.; Weinstein, J.R. Neuroimmune Response in Ischemic Preconditioning. Neurotherapeutics 2016, 13, 748–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, C.; Deng, J.; Wang, B.; Cheng, D.; Yang, Q.; Dong, H.; Xiong, L. Reactive Oxygen Species Scavenger Inhibits STAT3 Activation After Transient Focal Cerebral Ischemia–Reperfusion Injury in Rats. Anest. Analg. 2011, 113, 153–159. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Deng, Y.; Shangguan, S.; Zhou, F.; Zhou, W.; Li, X.; Li, Y.; Chen, G. Notch signaling in cerebrovascular diseases. Mol. Med. Rep. 2016, 14, 2883–2898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. J. Cerebr. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villringer, K.; Sanz Cuesta, B.E.; Ostwaldt, A.-C.; Grittner, U.; Brunecker, P.; Khalil, A.A.; Schindler, K.; Eisenblätter, O.; Audebert, H.; Fiebach, J.B.; et al. DCE-MRI blood–brain barrier assessment in acute ischemic Stroke. Neurology 2017, 88, 433–440. [Google Scholar] [CrossRef]
- Arba, F.; Leigh, R.; Inzitari, D.; Warach, S.J.; Luby, M.; Lees, K.R. Blood–Brain barrier leakage increases with small vessel disease in acute ischemic Stroke. Neurology 2017, 89, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Gauberti, M.; Montagne, A.; Marcos-Contreras, O.A.; Béhot, A.L.; Maubert, E.; Vivien, D. Ultra-Sensitive Molecular MRI of Vascular Cell Adhesion Molecule-1 Reveals a Dynamic Inflammatory Penumbra After Strokes. Stroke 2013, 44, 1988–1996. [Google Scholar] [CrossRef] [Green Version]
- Quenault, A.; Martinez de Lizarrondo, S.; Etard, O.; Gauberti, M.; Orset, C.; Haelewyn, B.; Segal, H.C.; Rothwell, P.M.; Vivien, D.; Touzé, E.; et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain 2016, 140, 146–157. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic Analysis of Reactive Astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raker, C.; Schleif, M.; Blank, N.; Matušková, H.; Ulas, T.; Händler, K.; Torres, S.V.; Schumacher, T.; Tai, K.; Schultze, J.L.; et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia 2018, 67, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, K.P.; Cekanaviciute, E.; Mamer, L.E.; Buckwalter, M.S. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after Stroke. J. Neuroinflamm. 2010, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cua, R.C.; Lau, L.W.; Keough, M.B.; Midha, R.; Apte, S.S.; Yong, V.W. Overcoming Neurite-Inhibitory Chondroitin Sulfate Proteoglycans in the Astrocyte Matrix. Glia 2013, 61, 972–984. [Google Scholar] [CrossRef]
- Yang, T.; Dai, Y.J.; Chen, G.; Cui, S.S. Dissecting the Dual Role of the Glial Scar and Scar-Forming Astrocytes in Spinal Cord Injury. Front. Cell Neurosci. 2020, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, M.; Tian, L.; Chen, J.; Liu, R.; Ning, B. Reactive Astrogliosis: Implications in Spinal Cord Injury Progression and Therapy. Oxid. Med. Cell Longev. 2020, 2020, 9494352. [Google Scholar] [CrossRef]
- Rocamonde, B.; Paradells, S.; Barcia, C.; Garcia Esparza, A.; Soria, J.M. Lipoic acid treatment after brain injury: Study of the glial reaction. Clin. Dev. Immunol. 2013, 2013, 521939. [Google Scholar] [CrossRef]
- Abeysinghe, H.C.S.; Bokhari, L.; Dusting, G.J.; Roulston, C.L. Cyclosporine A Reduces Glial Scarring and Facilitates Functional Recovery Following Transient Focal Ischemia. J. Neurol. Neurophysiol. 2015, 6, 2. [Google Scholar]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids CNS axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Raposo, C.; Schwartz, M. Glial scar and immune cell involvement in tissue remodeling and repair following acute CNS injuries. Glia 2014, 62, 1895–1904. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.P.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 135, a020370. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.A.; Johnson, J.A.; Li, L. Identification of Astrocyte Secreted Proteins with a Combination of Shotgun Proteomics and Bioinformatics. J. Proteome Res. 2009, 8, 4135–4143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Elshourbagy, N.A.; Liao, W.S.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA 1985, 82, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Bortolotto, V.; Canonico, P.L.; Sortino, M.A.; Grilli, M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood–Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deli, M.A.; Abrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell. Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A. The value of in vitro models of the blood-brain barrier and their uses. Altern. Lab. Anim. 2012, 40, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Mun-Bryce, S.; Rosenberg, G.A. Matrix metalloproteinases in cerebrovascular disease. J. Cereb. Blood Flow Metab. 1998, 18, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Hadman, M.; De Sevilla, L.; Wade, M.F.; Mahesh, V.B.; Brann, D.W. Astrocyte protection of neurons: Role of transforming growth factor-beta signaling via a c-Jun-AP-1 protective pathway. J. Biol. Chem. 2003, 278, 43329–43339. [Google Scholar] [CrossRef] [Green Version]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. The Role of Astrocytes in neuroprotection after Brain Stroke: Potential in Cell Therapy. Front. Mol. Neurosci. 2017, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Wu, Y.; Yang, Z.; Kalueff, A.; Tsao, Y.; Dong, Y.; Su, K. Astrocyte-Conditioned Medium Protects Prefrontal Cortical Neurons from Glutamate-Induced Cell Death by Inhibiting TNF-α Expression. Neuroimmunomodulation 2019, 26, 33–42. [Google Scholar] [CrossRef]
- Lu, X.; Al-Aref, R.; Zhao, D.; Shen, J.; Yan, Y.; Gao, Y. Astrocyte-conditioned medium attenuates glutamate-induced apoptotic cell death in primary cultured spinal cord neurons of rats. Neurol. Res. 2015, 37, 803–808. [Google Scholar] [CrossRef]
- Bicknell, B.A.; Pujic, Z.; Dayan, P.; Goodhill, G.J. Control of neurite growth and guidance by an inhibitory cell-body signal. PLoS Comput. Biol. 2018, 14, e1006218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trendelenburg, G.; Dirnagl, U. Neuroprotective role of astrocytes in cerebral ischemia: Focus on ischemic preconditioning. Glia 2005, 50, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Helena, S.; Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Yang, H.; Manaenko, A.; Lu, J.; Mei, Q.; Hu, Q. Potential of Exosomes for the Treatment of Stroke; SAGE Publications: Los Angeles, CA, USA, 2019. [Google Scholar]
- Venturini, A.; Passalacqua, M.; Pelassa, S.; Pastorino, F.; Tedesco, M.; Cortese, K.; Gagliani, M.C.; Leo, G.; Maura, G.; Guidolin, D.; et al. Exosomes from Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 2019, 10, 1452. [Google Scholar] [CrossRef] [Green Version]
- Pei, X.; Li, Y.; Zhu, L.; Zhou, Z. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic Stroke. Exp. Cell Res. 2019, 382, 111474. [Google Scholar] [CrossRef] [PubMed]
- Hira, K.; Ueno, Y.; Tanaka, R.; Miyamoto, N.; Yamashiro, K.; Inaba, T.; Urabe, T.; Okano, H.; Hattori, N. Astrocyte-Derived Exosomes Treated with a Semaphorin 3A Inhibitor Enhance Stroke Recovery via Prostaglandin D2 Synthase. Stroke 2018, 49, 2483–2494. [Google Scholar] [CrossRef]
- Taylor, A.R.; Robinson, M.B.; Gifondorwa, D.J.; Tytell, M.; Milligan, C.E. Regulation of heat shock protein 70 release in astrocytes: Role of signaling kinases. Dev. Neurobiol. 2007, 67, 1815–1829. [Google Scholar] [CrossRef]
- Xu, L.; Cao, H.; Xie, Y.; Zhang, Y.; Du, M.; Xu, X.; Ye, R.; Liu, X. Exosome-shuttled miR-92b-3p from ischemic preconditioned astrocytes protects neurons against oxygen and glucose deprivation. Brain Res. 2019, 17, 66–73. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol. Ther. Nucl. Acids 2017, 7, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Lafourcade, C.; Ramírez, J.P.; Luarte, A.; Fernández, A.; Wyneken, U. MiRNAs in Astrocyte-Derived Exosomes as Possible Mediators of Neuronal Plasticity. J. Exp. Neurosci. 2016, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zagrean, A.M.; Hermann, D.M.; Opris, I.; Zagrean, L.; Popa-Wagner, A. Multicellular Crosstalk Between Exosomes and the Neurovascular Unit After Cerebral Ischemia. Therapeutic Implications. Front. Neurosci. 2018, 12, 811. [Google Scholar] [CrossRef]
- Paterno, R.; Chillon, J. Potentially Common therapeutic targets for multiple sclerosis and ischemic Stroke. Front. Physiol. 2018, 9, 855. [Google Scholar] [CrossRef]
- Verma, R.; Mishra, V.; Sasmal, D.; Raghubir, R. Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur. J. Pharmacol. 2010, 638, 65–71. [Google Scholar] [CrossRef]
- Ouyang, Y.; Voloboueva, L.A.; Xu, L.; Giffard, R.G. Selective Dysfunction of Hippocampal CA1 Astrocytes Contributes to Delayed Neuronal Damage after Transient Forebrain Ischemia. J. Neurosci. 2017, 27, 4253–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Chen, D.; Gao, F.; Lv, H.; Zhang, G.; Sun, X.; Liu, L.; Mo, D.; Ma, N.; Song, L.; et al. Exosomes derived from microRNA-138-5p-overexpressing bone marrow derived mesenchymal stem cells confer neuroprotection to astrocytes following ischemic stroke via inhibition of LCN. J. Biol. Eng. 2019, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Kastrisianaki, E.; Giambanco, I.; Donato, R. S100B protein stimulates microglia migration via rage-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 2011, 286, 7214–7226. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Mori, T.; Tateishi, N.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Ikuta, F.; Asano, T. Astrocytic Activation and Delayed Infarct Expansion after Permanent Focal Ischemia in Rats. Part I: Enhanced Astrocytic Synthesis of S-100β in the Periinfarct Area Precedes Delayed Infarct Expansion. J. Cerebr. Blood Flow Metab. 2002, 22, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Rempe, D. Targeting astrocytes for stroke therapy. Neurotherapeutics 2010, 7, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Asano, T.; Mori, T.; Shimoda, T.; Shinagawa, R.; Satoh, S.; Yada, N.; Katsumata, S.; Matsuda, S.; Kagamiishi, Y.; Tateishi, N. Arundic acid (ONO-2506) ameliorates delayed ischemic brain damage by preventing astrocytic overproduction of S100B. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 127–142. [Google Scholar] [CrossRef]
- Tateishi, N.; Mori, T.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Matsui, T.; Asano, T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)-()-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J. Cereb. Blood Flow Metab. 2002, 22, 723–734. [Google Scholar] [PubMed] [Green Version]
- Zlokovic, B.V.; Gottesman, R.F.; Bernstein, K.E.; Seshadri, S.; McKee, A.; Snyder, H.; Greenberg, S.M.; Yaffe, K.; Schaffer, C.B.; Yuan, C.; et al. Vascular contributions to cognitive impairment and dementia (VCID): A report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 2020, 16, 1714–1733. [Google Scholar] [CrossRef]
- Becerra-Calixto, A.; Cardona-Gómez, G.P. Neuroprotection Induced by Transplanted CDK5 Knockdown Astrocytes in Global Cerebral Ischemic Rats. Mol. Neurobiol. 2017, 54, 6681–6696. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vargas, J.A.; Múnera, A.; Cardona-Gómez, G.P. CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1937–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada-Duque, R.A.; Palacio-Castañeda, V.; Cardona-Gómez, G.P. CDK5 knockdown in astrocytes provide neuroprotection as a trophic source via RacMol. Cell. Neurosci. 2015, 68, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Posada-Duque, R.; Cardona-Gómez, G.P. Recovery of Neurovascular Unit Integrity by CDK5-KD Astrocyte Transplantation in a Global Cerebral Ischemia Model. Mol. Neurobiol. 2018, 55, 8563–8585. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Bacigaluppi, M.; Russo, G.L.; Peruzzotti-Jametti, L.; Rossi, S.; Sandrone, S.; Butti, E.; De Ceglia, R.; Bergamaschi, A.; Motta, C.; Gallizioli, M.; et al. Neural Stem Cell Transplantation Induces Stroke Recovery by Upregulating Glutamate Transporter GLT-1 in Astrocytes. J. Neurosci. 2016, 36, 10529–10544. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Guo, K.; Fan, W.; Lu, Y.; Chen, L.; Wang, Y.; Shao, Y.; Wu, G.; Xu, J.; Lü, L. Niche astrocytes promote the survival, proliferation and neuronal differentiation of co-transplanted neural stem cells following ischemic stroke in rats. Exp. Ther. Med. 2017, 13, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Cucullo, L. In-Vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 38, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 573775. [Google Scholar] [CrossRef]
- Cucullo, L.; Hossain, M.; Tierney, W.; Janigro, D. A new dynamic in vitro modular capillaries-venules modular system: Cerebrovascular physiology in a box. BMC Neurosci. 2013, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Cucullo, L.; Couraud, P.O.; Weksler, B.; Romero, I.A.; Hossain, M.; Rapp, E.; Janigro, D. Immortalized human brain endothelial cells and flow-based vascular modeling: A marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2008, 28, 312–328. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [Green Version]
- Nzou, G.; Wicks, R.T.; VanOstrand, N.R.; Mekky, G.A.; Seale, S.A.; El-Taibany, A.; Wicks, E.E.; Nechtman, C.M.; Marrotte, E.J.; Makani, V.S.; et al. Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci. Rep. 2020, 10, 9766. [Google Scholar] [CrossRef]
Figure 1.
The diverse functions of astrocytes during physiological and pathophysiological conditions. Astrocytes have several important roles in the healthy and diseased brain. They are the main housekeeping cells of the brain, and can exert either protective or detrimental effects on neurons during pathophysiological conditions. Astrocytes are essential for the development and maintenance of the BBB, homeostasis of the brain microenvironment, cerebral blood flow regulation, neurotransmitter uptake, synaptogenesis, neurogenesis and release of neurotrophic factors and energy supply to neurons. Astrocytes also play a major role during pathophysiological conditions. Neuronal survival heavily depends on astrocytes. For example, neurons do not survive if neighbouring astrocytes are lost during an ischaemic stroke. Cerebral ischaemia leads astrocytes to change their morphology and function, causing astrocytes to become reactive, thereby producing several proinflammatory modulators and participating in glial scar formation. Their neuroprotective roles include clearing glutamate from synaptic regions, secretion of neurotrophic factors and promotion of angiogenesis, neurogenesis, synaptogenesis and axonal remodelling. Figure created with BioRender.com.
Figure 1.
The diverse functions of astrocytes during physiological and pathophysiological conditions. Astrocytes have several important roles in the healthy and diseased brain. They are the main housekeeping cells of the brain, and can exert either protective or detrimental effects on neurons during pathophysiological conditions. Astrocytes are essential for the development and maintenance of the BBB, homeostasis of the brain microenvironment, cerebral blood flow regulation, neurotransmitter uptake, synaptogenesis, neurogenesis and release of neurotrophic factors and energy supply to neurons. Astrocytes also play a major role during pathophysiological conditions. Neuronal survival heavily depends on astrocytes. For example, neurons do not survive if neighbouring astrocytes are lost during an ischaemic stroke. Cerebral ischaemia leads astrocytes to change their morphology and function, causing astrocytes to become reactive, thereby producing several proinflammatory modulators and participating in glial scar formation. Their neuroprotective roles include clearing glutamate from synaptic regions, secretion of neurotrophic factors and promotion of angiogenesis, neurogenesis, synaptogenesis and axonal remodelling. Figure created with BioRender.com.

Figure 2.
Blood–brain barrier (BBB) dysfunction following ischaemic stroke. (1) Simplified structure of the neurovascular unit (NVU) comprising brain endothelial cells, astrocytes, pericytes and neurons. Brain endothelial cells form tight junctions between them that control the movement of molecules through the paracellular pathway by showing size and charge selectivity, thereby forming a selective barrier between the brain microenvironment and the systemic circulation. Pericytes partially cover the endothelial cells and are embedded in the basement membrane. They are spread discontinuously along the microvessel, and maintain the barrier properties of brain endothelial cells, regulate capillary diameter, cerebral blood flow and angiogenesis. Perivascular endfeet from multiple astrocytes ensheath endothelial cells, allowing intricate cell–cell communications to help maintain the BBB phenotype of brain endothelial cells. Astrocytes secrete soluble factors that are important for the development and maintenance of the BBB. In addition, astrocytes contribute to brain water homeostasis via aquaporin 4 (AQP4) water channels, which are expressed on the endfeet of astrocytes. (2) The schematic on the right demonstrates BBB dysfunction due to cerebral ischaemia. Loss of tissue perfusion due to ischaemic stroke leads to neuronal injury and death. This causes an increase in the release of proinflammatory mediators that activates endothelial cells, astrocytes and pericytes. The resulting neurovascular inflammation disrupts tight junctions, leading to paracellular leakage and cerebral oedema. During ischaemia, AQP4 lose their polarisation from astrocyte endfeet. A dual role is played by AQP4 in cytotoxic and vasogenic oedema, where AQP4 deficiency leads to a reduction in cytotoxic oedema and improved neurological outcomes, and vasogenic oedema is increased when AQP4 is knocked down. The activated brain endothelium attracts leukocytes, which results in increased transcytosis across the BBB and neuroinflammation. This proinflammatory environment leads to changes in astrocyte morphology: “reactive astrogliosis”. Reactive astrocytes can secrete proinflammatory mediators such as IL-1α, IL-6 and TNF-α that cause further BBB disruption and neuronal injury. In addition, ischaemic stroke causes pericyte detachment and death in rigor, which can lead to vessel constriction and the no-reflow phenomenon. Figure created with BioRender.com.
Figure 2.
Blood–brain barrier (BBB) dysfunction following ischaemic stroke. (1) Simplified structure of the neurovascular unit (NVU) comprising brain endothelial cells, astrocytes, pericytes and neurons. Brain endothelial cells form tight junctions between them that control the movement of molecules through the paracellular pathway by showing size and charge selectivity, thereby forming a selective barrier between the brain microenvironment and the systemic circulation. Pericytes partially cover the endothelial cells and are embedded in the basement membrane. They are spread discontinuously along the microvessel, and maintain the barrier properties of brain endothelial cells, regulate capillary diameter, cerebral blood flow and angiogenesis. Perivascular endfeet from multiple astrocytes ensheath endothelial cells, allowing intricate cell–cell communications to help maintain the BBB phenotype of brain endothelial cells. Astrocytes secrete soluble factors that are important for the development and maintenance of the BBB. In addition, astrocytes contribute to brain water homeostasis via aquaporin 4 (AQP4) water channels, which are expressed on the endfeet of astrocytes. (2) The schematic on the right demonstrates BBB dysfunction due to cerebral ischaemia. Loss of tissue perfusion due to ischaemic stroke leads to neuronal injury and death. This causes an increase in the release of proinflammatory mediators that activates endothelial cells, astrocytes and pericytes. The resulting neurovascular inflammation disrupts tight junctions, leading to paracellular leakage and cerebral oedema. During ischaemia, AQP4 lose their polarisation from astrocyte endfeet. A dual role is played by AQP4 in cytotoxic and vasogenic oedema, where AQP4 deficiency leads to a reduction in cytotoxic oedema and improved neurological outcomes, and vasogenic oedema is increased when AQP4 is knocked down. The activated brain endothelium attracts leukocytes, which results in increased transcytosis across the BBB and neuroinflammation. This proinflammatory environment leads to changes in astrocyte morphology: “reactive astrogliosis”. Reactive astrocytes can secrete proinflammatory mediators such as IL-1α, IL-6 and TNF-α that cause further BBB disruption and neuronal injury. In addition, ischaemic stroke causes pericyte detachment and death in rigor, which can lead to vessel constriction and the no-reflow phenomenon. Figure created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. https://doi.org/10.3390/ijms22084280
AMA Style
Patabendige A, Singh A, Jenkins S, Sen J, Chen R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. International Journal of Molecular Sciences. 2021; 22(8):4280. https://doi.org/10.3390/ijms22084280
Chicago/Turabian StylePatabendige, Adjanie, Ayesha Singh, Stuart Jenkins, Jon Sen, and Ruoli Chen. 2021. "Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke" International Journal of Molecular Sciences 22, no. 8: 4280. https://doi.org/10.3390/ijms22084280
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.