
Macrophage-Derived Adenosine Deaminase 2 Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
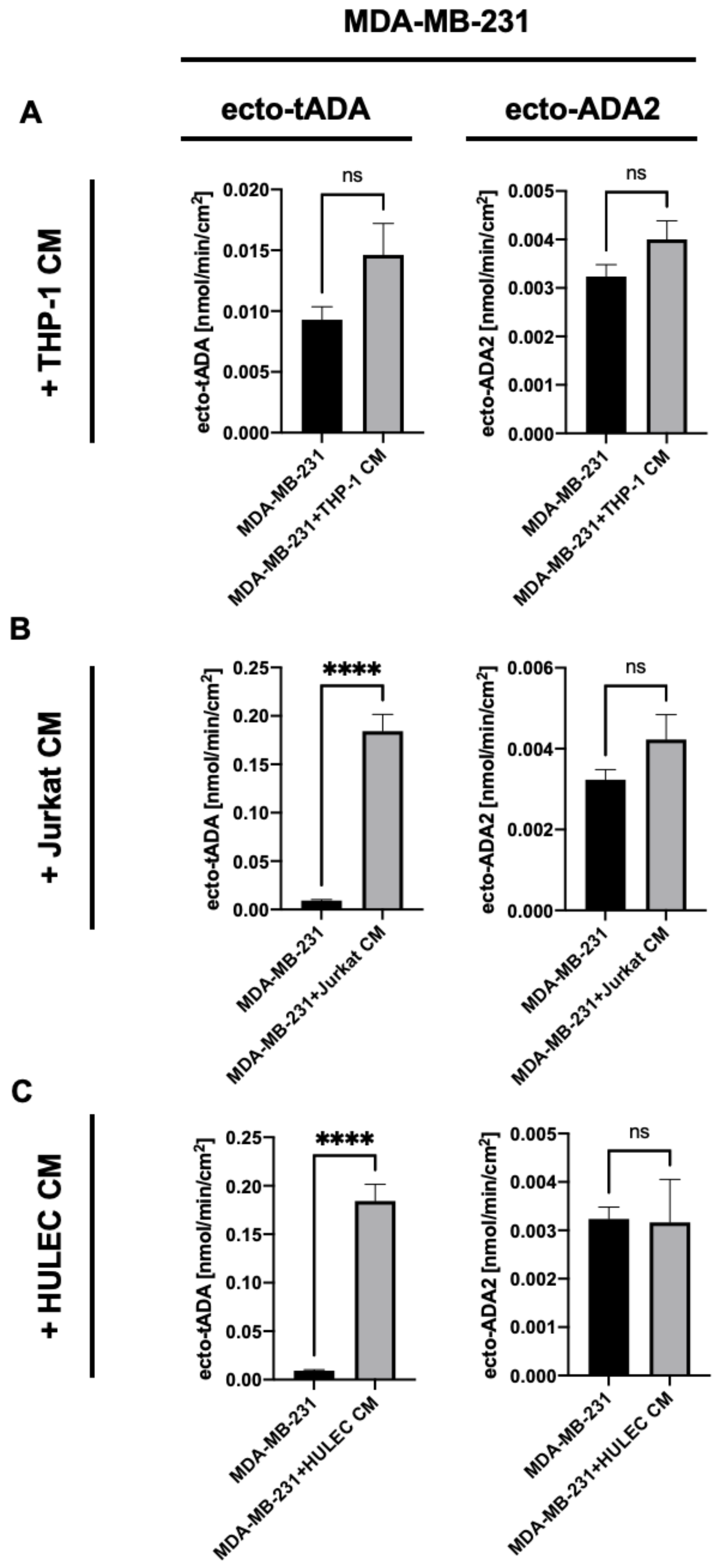
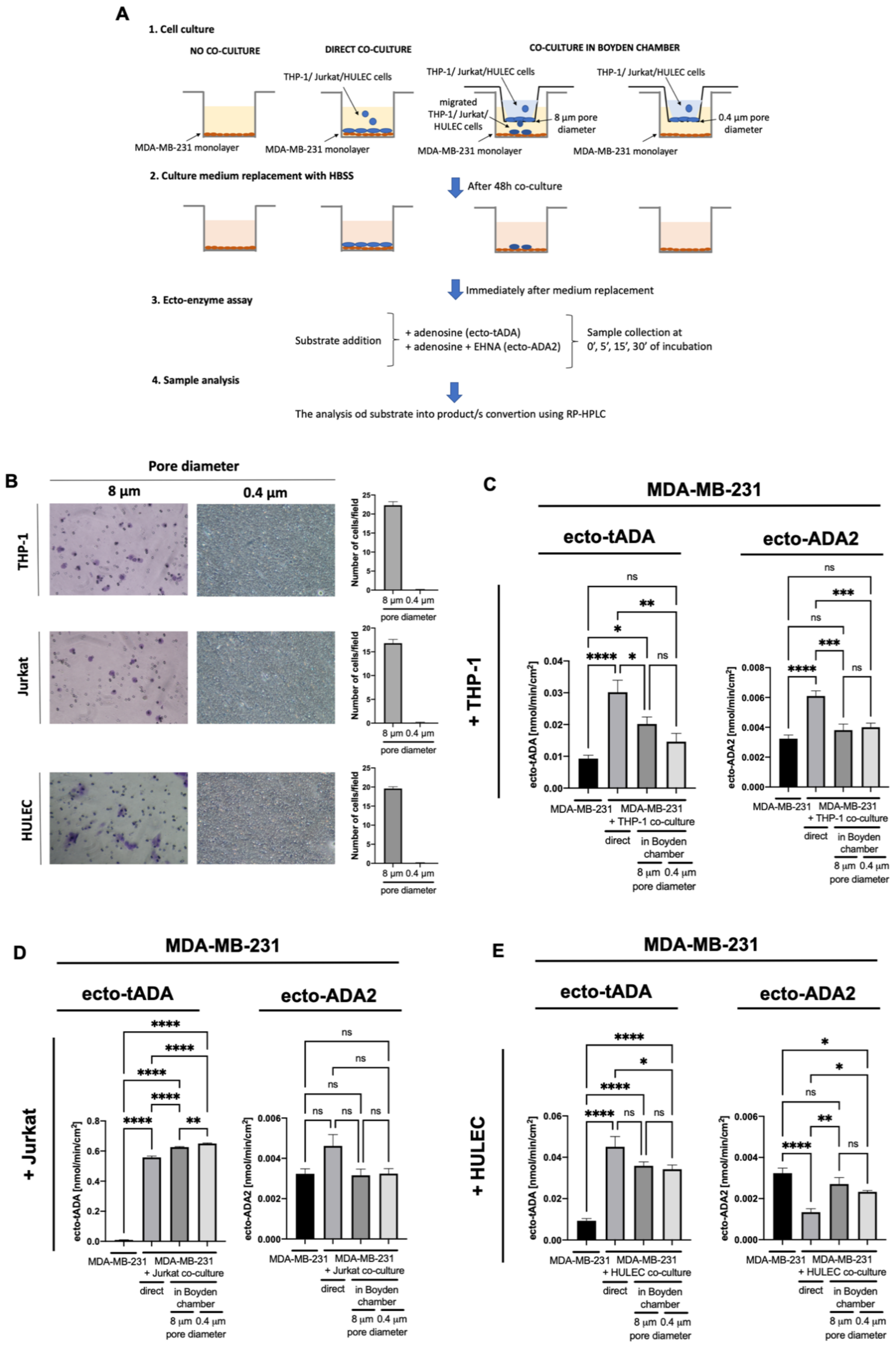
2.1. Immune and Endothelial Cells Modulate ADA1 Activity on the Surface of MDA-MB-231 Triple Negative Breast Cells
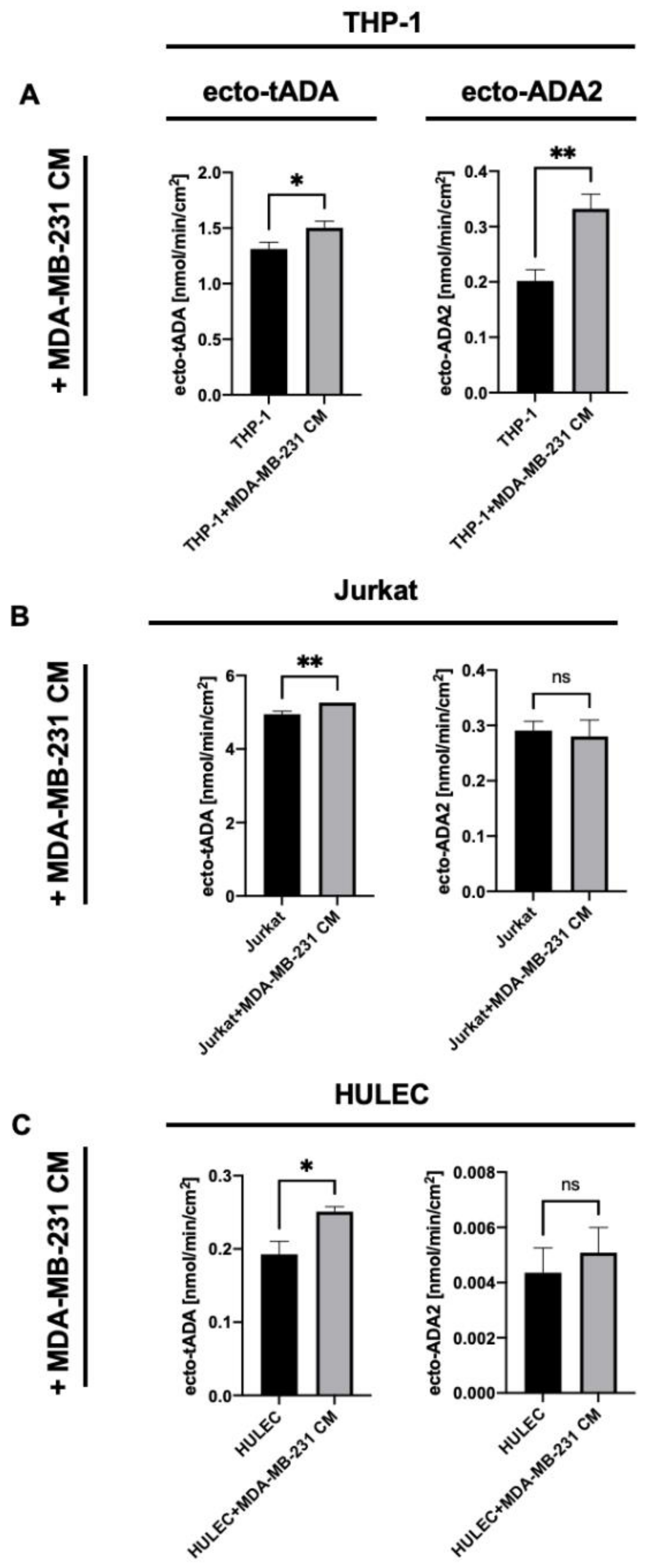
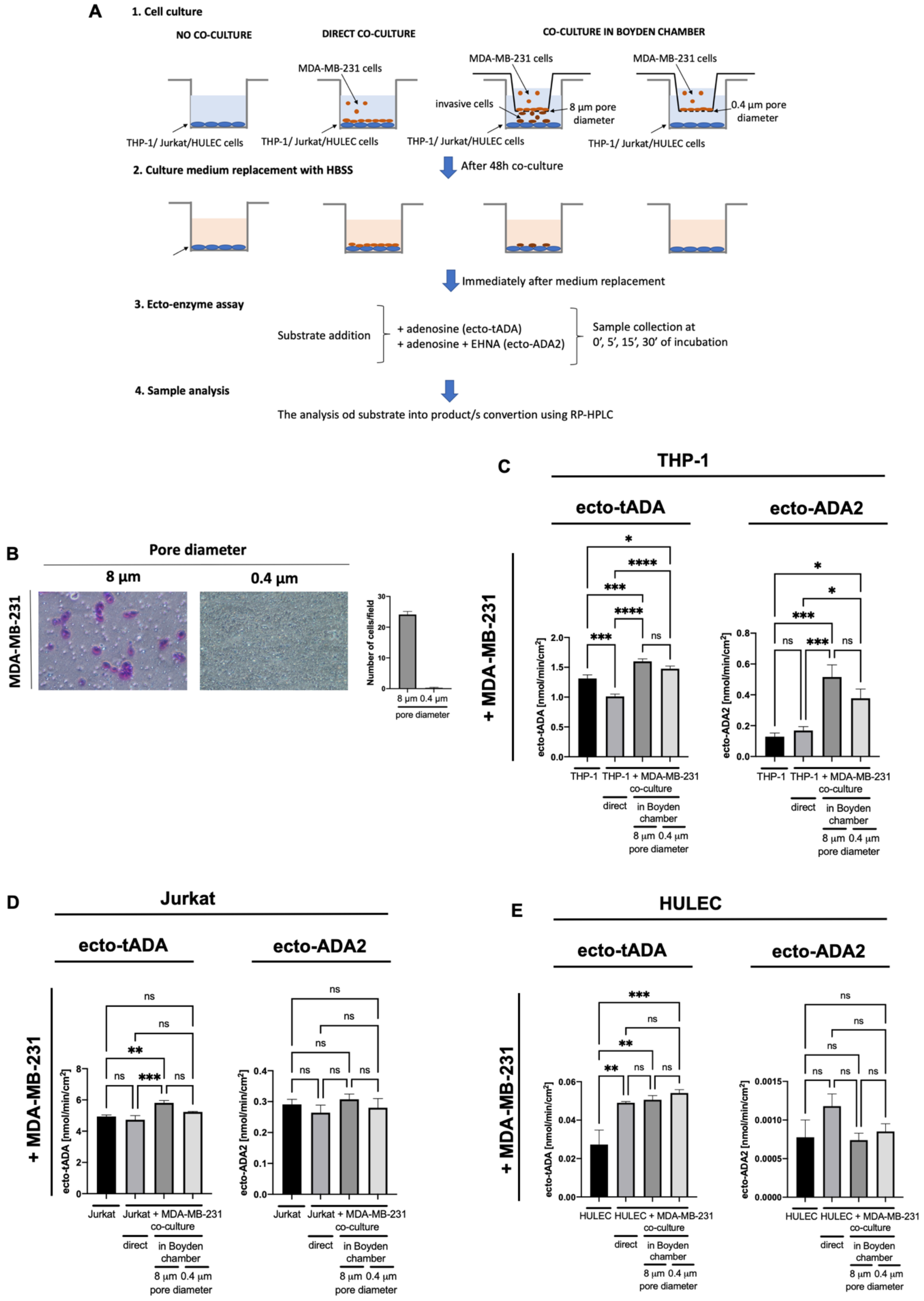
2.2. MDA-MB-231 Triple Negative Breast Cancer Cells Stimulate ADA2 Activity on the Surface of Monocytes/Macrophages and ADA1 Activity on Lymphocytes and Endothelial Cells
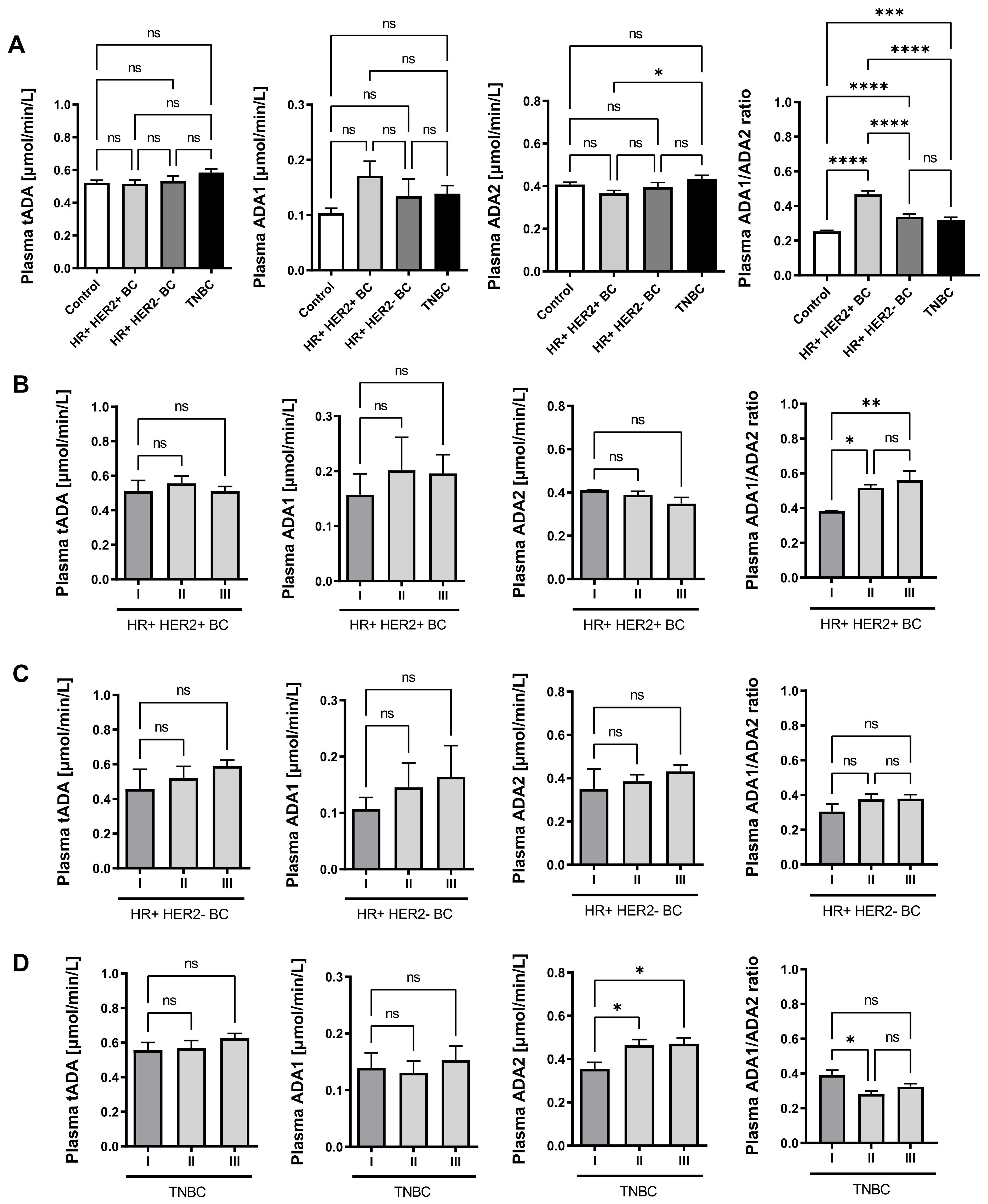
2.3. Plasma ADA Iso-Enzyme Pattern Is Adversely Deregulated in Different Subtypes Breast Cancers
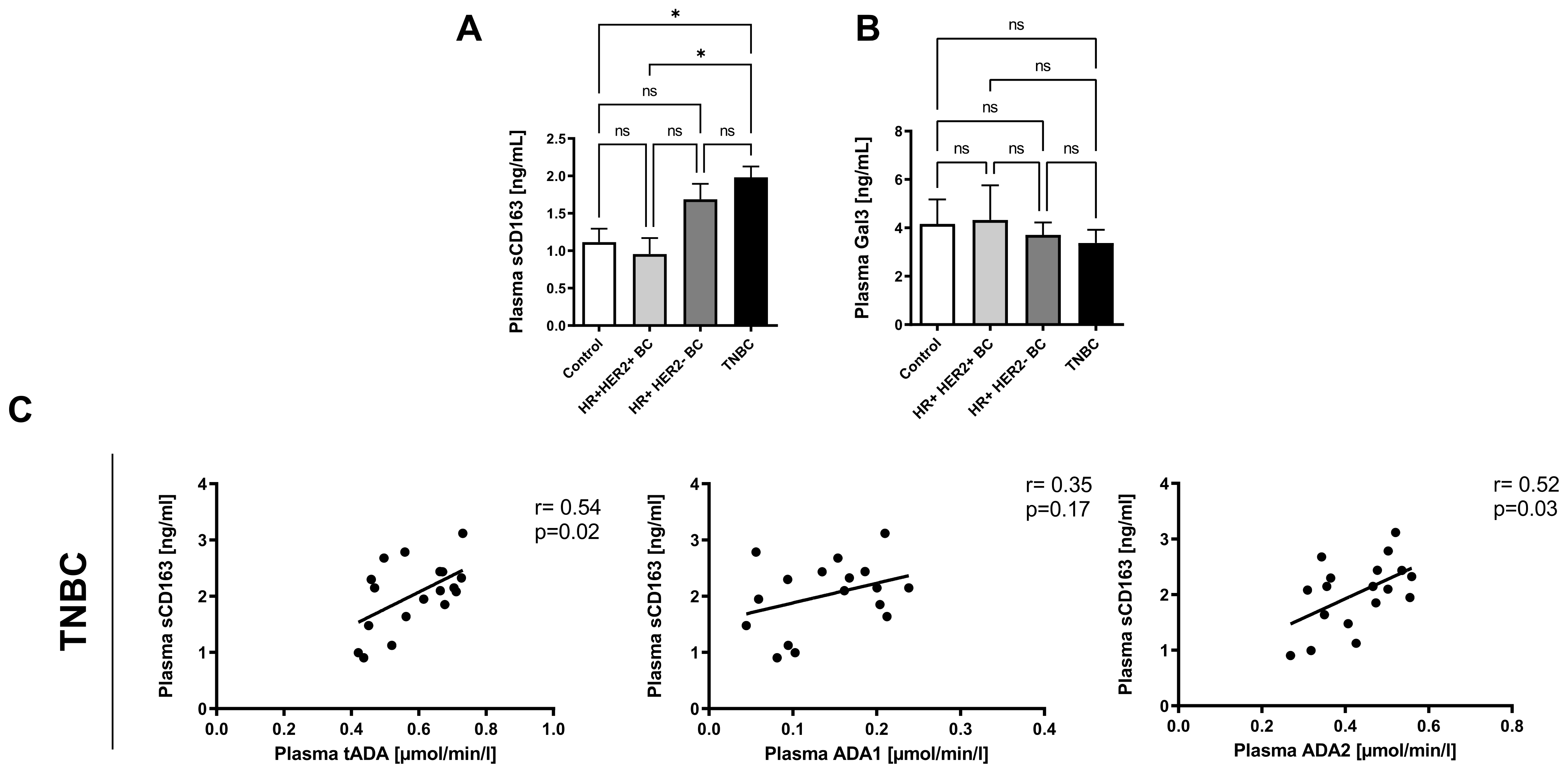
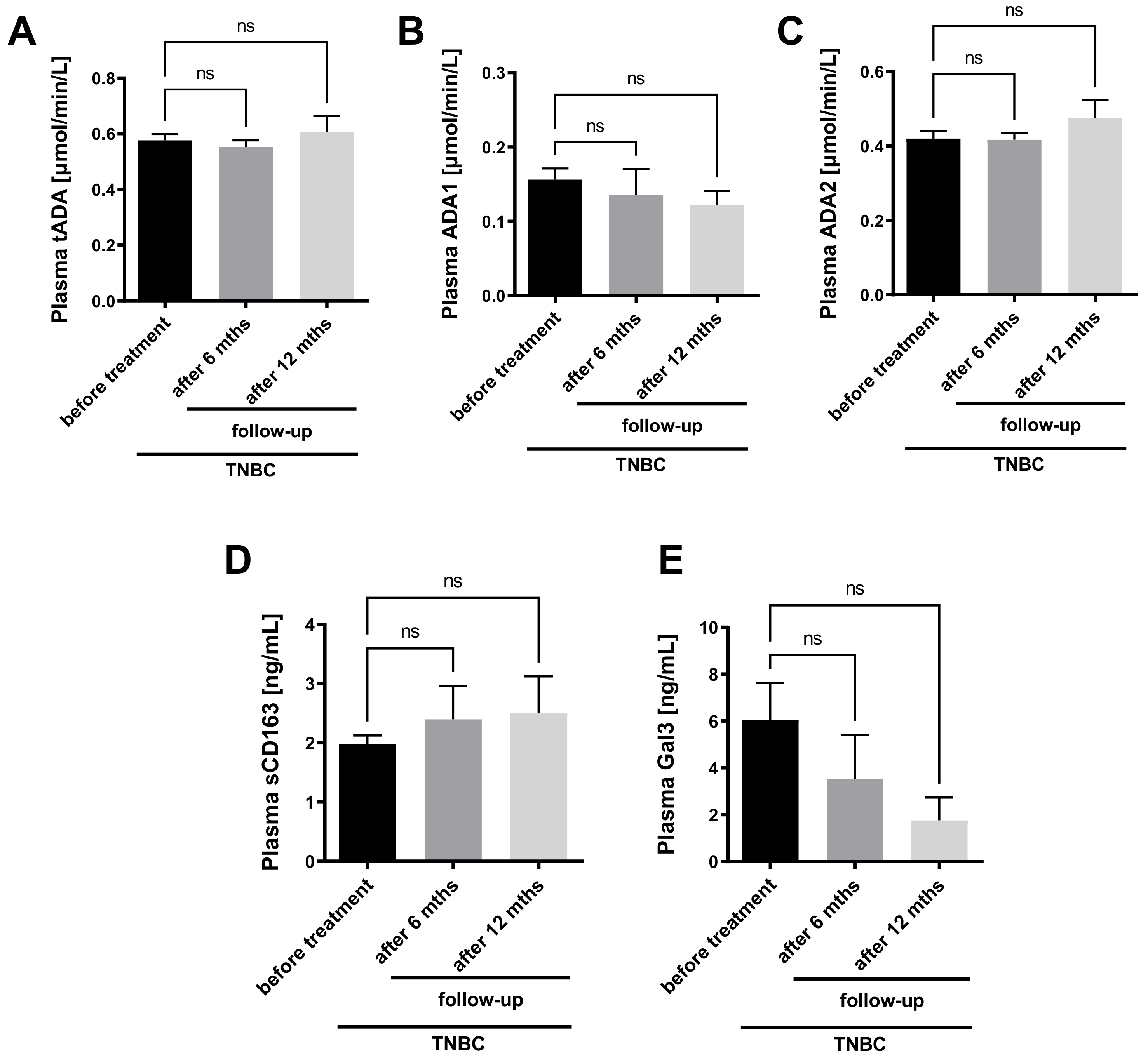
2.4. Plasma ADA2 Activity Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients
5.2. Determination of the Plasma Activities of ADA Iso-Enzymes
5.3. Detemination of Plasma Markers of Macrophage Polarization, Clinical Biochemistry Panel, Blood Morphology, Electrolites and Coagulation Parameters in Patients
5.4. Determination of Serum Amino Acid Metabolites
5.5. Cell Culture
5.6. Media Transfer
5.7. Co-Culture Conditions
5.8. The Analysis of Cell Migration Abilities
5.9. Determination of the Activities of ADA Iso-Enzymes in Human Cell Lines
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADA | Adenosine deaminase |
| ADA1 | Adenosine deaminase 1 |
| ADA2 | Adenosine deaminase 2 |
| ADMA | Asymmetric dimethylarginine |
| Ado | Adenosine |
| ALB | Albumin |
| ALP | Alkaline phosphatase |
| ALT | Alanine transaminase |
| APTT | Activated partial thromboplastin time |
| AR | Adenosine receptor |
| AST | Aspartate transaminase |
| ATP | Adenosine triphosphate |
| BC | Breast cancer |
| BUN | Blood urea nitrogen |
| Ca | Calcium |
| CD39 | Ecto-nucleoside triphosphate diphosphohydrolase |
| CD73 | Ecto-5’nucleotidase |
| CHOL | Total cholesterol |
| CM | Cell medium |
| Creat | Creatinine |
| CRPhs | High sensitive C reactive protein |
| dAdo | Deoxyadenosine |
| dCF | Deoxycoformycin |
| eADA1 | Ecto-adenosine deaminase 1 |
| eADA2 | Ecto-adenosine deaminase 2 |
| EHNA | Erythro-9-(2-hydroxy-3-nonyl)adenine |
| ER | Estrogen receptors |
| FIB | Fibrinogen |
| Gal3 | Galectin 3 |
| Glu | Glucose |
| HDL | High density lipoproteins |
| HER2 | Human epidermal growth factor 2 |
| Hgb | Hemoglobin |
| HR | Hormone receptors |
| Ht | Hematocrit |
| HULEC | Human lung endothelial cell line |
| K | Potassium |
| Ki-67/Ki | Marker of proliferation Ki-67 |
| LDH | Lactate dehydrogenase |
| LDL | Low density lipoproteins |
| M2 | Macrophages M2 |
| MDA-MB-231 | Human triple negative breast cancer cell line |
| Mg | Magnesium |
| N.A. | Not available |
| Na | Sodium |
| Pho | Phosphorus |
| PLT | Platelets |
| PR | Progesterone receptors |
| PT | Prothrombin time |
| RBC | Red blood cells |
| RP-HPLC | Reverse phase high performance liquid chromatography |
| sCD163 | Soluble CD163 protein |
| tADA | Total adenosine deaminase |
| TAMs | Tumor associated macrophages |
| TG | Triglycerydes |
| THP-1 | Human monocytes/macrophages line |
| TILs | Tumor infiltrated lymphocytes |
| TNBC | Triple negative breast cancer |
| VEGF | Vascular endothelial growth factor |
| W APTT | Activated partial thromboplastin time factor |
| WBC | White blood cells |
| WPT | Prothrombin time factor |
References
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-Negative Breast Cancer: Treatment Challenges and Solutions. Breast Cancer. Targets Ther. 2016, 8, 93–107. [Google Scholar]
- Antonioli, L.; Colucci, R.; la Motta, C.; Tuccori, M.; Awwad, O.; da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine Deaminase in the Modulation of Immune System and Its Potential as a Novel Target for Treatment of Inflammatory Disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Gakis, C. Adenosine Deaminase (ADA) Isoenzymes ADA1 and ADA2: Diagnostic and Biological Role. Eur. Respir. J. 1996, 9, 632–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasyan, N.A.; Hairapetyan, H.L.; Sargisova, Y.G.; Mardanyan, S.S. ADA2 Isoform of Adenosine Deaminase from Pleural Fluid. FEBS Lett. 2005, 579, 643–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavialov, A.V.; Engström, A. Human ADA2 Belongs to a New Family of Growth Factors with Adenosine Deaminase Activity. Biochem. J. 2005, 391, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Casadó, V.; Ciruela, F.; Saura, C.; Mallol, J.; Canela, E.I.; Lluis, C. Cell Surface Adenosine Deaminase: Much More than an Ectoenzyme. Prog. Neurobiol. 1997, 52, 283–294. [Google Scholar] [CrossRef]
- Franco, R.; Pacheco, R.; Gatell, J.M.; Gallart, T.; Lluis, C. Enzymatic and Extraenzymatic Role of Adenosine Deaminase 1 in T-Cell-Dendritic Cell Contacts and in Alterations of the Immune Function. Crit. Rev. Immunol. 2007, 27, 495–509. [Google Scholar] [CrossRef]
- Hershfield, M.S. New Insights into Adenosine-Receptor-Mediated Immunosuppression and the Role of Adenosine in Causing the Immunodeficiency Associated with Adenosine Deaminase Deficiency. Eur. J. Immunol. 2005, 35, 25–30. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Mateuszuk, L.; Zukowska, P.; Jasztal, A.; Zabielska, M.A.; Toczek, M.; Zakrzewska, A.; Sitek, B.; Rogowski, J.; Lango, R.; et al. Increased Activity of Vascular Adenosine Deaminase in Atherosclerosis and Therapeutic Potential of Its Inhibition. Cardiovasc. Res. 2016, 112, 590–605. [Google Scholar] [CrossRef]
- Kutryb-Zajac, B.; Mierzejewska, P.; Sucajtys-Szulc, E.; Bulinska, A.; Zabielska, M.A.; Jablonska, P.; Serocki, M.; Koszalka, P.; Milczarek, R.; Jasztal, A.; et al. Inhibition of LPS-Stimulated Ecto-Adenosine Deaminase Attenuates Endothelial Cell Activation. J. Mol. Cell. Cardiol. 2019, 128, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Zavialov, A.V.; Lauvau, G. Human Adenosine Deaminase 2 Induces Differentiation of Monocytes into Macrophages and Stimulates Proliferation of T Helper Cells and Macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Gao, S.; Kajigaya, S.; Diamond, C.; Alemu, L.; Ombrello, A.; Young, N.S. Analysis of Deficiency of Adenosine Deaminase 2 Pathogenesis Based on Single Cell RNA Sequencing of Monocytes. Blood 2019, 134, 2317. [Google Scholar] [CrossRef]
- Caorsi, R.; Penco, F.; Schena, F.; Gattorno, M. Monogenic Polyarteritis: The Lesson of ADA2 Deficiency. Pediatr. Rheumatol. 2016, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghaei, M.; Karami-Tehrani, F.; Salami, S.; Atri, M. Diagnostic Value of Adenosine Deaminase Activity in Benign and Malignant Breast Tumors. Arch. Med. Res. 2010, 41, 14–18. [Google Scholar] [CrossRef]
- Aghaei, M.; Karami-Tehrani, F.; Salami, S.; Atri, M. Adenosine Deaminase Activity in the Serum and Malignant Tumors of Breast Cancer: The Assessment of Isoenzyme ADA1 and ADA2 Activities. Clin. Biochem. 2005, 38, 887–891. [Google Scholar] [CrossRef]
- Mahajan, M.; Tiwari, N.; Sharma, R.; Kaur, S.; Singh, N. Oxidative Stress and Its Relationship With Adenosine Deaminase Activity in Various Stages of Breast Cancer. Indian J. Clin. Biochem. 2013, 28, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple Negative Breast Cancer: Key Role of Tumor-Associated Macrophages in Regulating the Activity of Anti-PD-1/PD-L1 Agents. Biochim. Biophys. Acta. Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Purines, Purinergic Receptors, and Cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Gao, Z.-G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, Inflammation and Cancer: A Leading Role for Adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Koszalka, P.; Mierzejewska, P.; Bulinska, A.; Zabielska, M.A.; Brodzik, K.; Skrzypkowska, A.; Zelazek, L.; Pelikant-Malecka, I.; Slominska, E.M.; et al. Adenosine Deaminase Inhibition Suppresses Progression of 4T1 Murine Breast Cancer by Adenosine Receptor-Dependent Mechanisms. J. Cell. Mol. Med. 2018, 22, 5939–5954. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Smyth, M.J. Extracellular Adenosine Triphosphate and Adenosine in Cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [Green Version]
- Podgorska, M.; Kocbuch, K.; Pawelczyk, T. Recent Advances in Studies on Biochemical and Structural Properties of Equilibrative and Concentrative Nucleoside Transporters. Acta Biochim. Pol. 2005, 52, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Casado, F.J.; Pastor-Anglada, M. Nucleoside Transporter Proteins. Curr. Vasc. Pharmacol. 2009, 7, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Smolenski, R.T.; Kochan, Z.; McDouall, R.; Seymour, A.M.L.; Yacoub, M.H. Adenosine uptake and metabolism in human endothelial cells. Adv. Exp. Med. Biol. 1994, 370, 435–438. [Google Scholar] [CrossRef]
- Canbolat, O.; Durak, I.; Çetin, R.; Kavutcu, M.; Demirci, S.; Öztürk, S. Activities of Adenosine Deaminase, 5′-Nucleotidase, Guanase, and Cytidine Deaminase Enzymes in Cancerous and Non-Cancerous Human Breast Tissues. Breast Cancer Res. Treat. 1996, 37, 189–193. [Google Scholar] [CrossRef]
- Sharma, S.; Desai, P.B.; Metgudmath, R.B. Evaluation of Serum Adenosine Deaminase and Retinol in Patients with Laryngeal Cancer. Indian J. Pharm. Biol. Res. 2013, 1, 4. [Google Scholar]
- Eroĝlu, A.; Canbolat, O.; Demirci, S.; Kocaoĝlu, H.; Eryavuz, Y.; Akgül, H. Activities of Adenosine Deaminase and 5′-Nucleotidase in Cancerous and Noncancerous Human Colorectal Tissues. Med. Oncol. 2000, 17, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Nishizaki, T. Anticancer Effect of Adenosine on Gastric Cancer via Diverse Signaling Pathways. World J. Gastroenterol. 2015, 21, 10931–10935. [Google Scholar] [CrossRef]
- Xia, B.; Wang, J. Effects of Adenosine on Apoptosis of Ovarian Cancer A2780 Cells via ROS and Caspase Pathways. OncoTargets Ther. 2019, 12, 9473–9480. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediat. Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage Polarization: Anti-Cancer Strategies to Target Tumor-Associated Macrophage in Breast Cancer. J. Cell. Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Schaar, B.; Tallapragada, S.; Dorigo, O. Tumor Associated Macrophages in Gynecologic Cancers. Gynecol. Oncol. 2018, 149, 205–213. [Google Scholar] [CrossRef]
- García-Teijido, P.; Cabal, M.L.; Fernández, I.P.; Pérez, Y.F. Tumor-Infiltrating Lymphocytes in Triple Negative Breast Cancer: The Future of Immune Targeting. Clin. Med. Insights Oncol. 2016, 10, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Speiser, D.E.; Verdeil, G. More T Cells versus Better T Cells in Patients with Breast Cancer. Cancer Discov. 2017, 7, 1062–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Mustafa, D.; Zheng, P.-P.; van der Weiden, M.; Sacchetti, A.; Brandt, M.; Chrifi, I.; Tempel, D.; Leenen, P.J.M.; Duncker, D.J.; et al. Activation of CECR1 in M2-like TAMs Promotes Paracrine Stimulation-Mediated Glial Tumor Progression. Neuro-Oncology 2017, 19, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist Antibodies to Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) as Anti-Angiogenic Agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef] [Green Version]
- Bousquet, G.; el Bouchtaoui, M.; Sophie, T.; Leboeuf, C.; de Bazelaire, C.; Ratajczak, P.; Giacchetti, S.; de Roquancourt, A.; Bertheau, P.; Verneuil, L.; et al. Targeting Autophagic Cancer Stem-Cells to Reverse Chemoresistance in Human Triple Negative Breast Cancer. Oncotarget 2017, 8, 35205–35221. [Google Scholar] [CrossRef] [Green Version]
- Bender, R.J.; mac Gabhann, F. Expression of VEGF and Semaphorin Genes Define Subgroups of Triple Negative Breast Cancer. PLoS ONE 2013, 8, e61788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-H.; Avraham, H.K.; Jiang, S.; Avraham, S. Vascular Endothelial Growth Factor Modulates the Transendothelial Migration of MDA-MB-231 Breast Cancer Cells through Regulation of Brain Microvascular Endothelial Cell Permeability. J. Biol. Chem. 2003, 278, 5277–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernatchez, P.N.; Soker, S.; Sirois, M.G. Vascular Endothelial Growth Factor Effect on Endothelial Cell Proliferation, Migration, and Platelet-Activating Factor Synthesis Is Flk-1-Dependent. J. Biol. Chem. 1999, 274, 31047–31054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolenski, R.T.; Lachno, D.R.; Ledingham, S.J.M.; Yacoub, M.H. Determination of Sixteen Nucleotides, Nucleosides and Bases Using High-Performance Liquid Chromatography and Its Application to the Study of Purine Metabolism in Hearts for Transplantation. J. Chromatogr. B. Biomed. Sci. Appl. 1990, 527, 414–420. [Google Scholar] [CrossRef]
- Mierzejewska, P.; Zabielska, M.A.; Kutryb-Zajac, B.; Tomczyk, M.; Koszalka, P.; Smolenski, R.T.; Slominska, E.M. Impaired L-Arginine Metabolism Marks Endothelial Dysfunction in CD73-Deficient Mice. Mol. Cell. Biochem. 2019, 458, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olkowicz, M.; Debski, J.; Jablonska, P.; Dadlez, M.; Smolenski, R.T. Application of a New Procedure for Liquid Chromatography/Mass Spectrometry Profiling of Plasma Amino Acid-Related Metabolites and Untargeted Shotgun Proteomics to Identify Mechanisms and Biomarkers of Calcific Aortic Stenosis. J. Chromatogr. A 2017, 1517, 66–78. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control (n = 18) | HR+ HER2+ (n = 12) | HR+ HER2- (n = 16) | TNBC (n = 19) |
|---|---|---|---|---|
| CRPhs, mg/dL | 0.37 ± 0.05 | 0.34 ± 0.02 | 0.44 ± 0.04 | 0.38 ± 0.08 |
| LDL, mg/dL | 109 ± 10.2 | 120 ± 21.8 | 105 ± 6.57 | 130 ± 17 |
| HDL, mg/dL | 65.4 ± 5.82 | 54.6 ± 5.82 | 55.4 ± 4.77 | 41.2 ± 5.82 * |
| CHOL, mg/dL | 199 ± 13.8 | 201 ± 17.6 | 195.3 ± 9.80 | 204.3 ± 20.5 |
| TG, mg/dL | 123 ± 18.9 | 116 ± 18.4 | 173 ± 32.8 | 164.3 ± 15.44 |
| ALB, g/dL | 4.75 ± 0.12 | 4.09 ± 0.50 | 4.80 ± 0.32 | 4.20 ± 0.19 |
| Total protein, g/dL | 8.23 ± 0.34 | 6.44 ± 1.25 | 6.60 ± 0.5 | 7.36 ± 0.30 |
| ALP, U/L | 59.7 ± 10.1 | 43.7 ± 4.24 | 63.6 ± 11.2 | 64.6 ± 12.08 |
| Ca, mg/dL | 13.6 ± 4.00 | 9.92 ± 1.27 | 9.69 ± 0.87 | 11.3 ± 1.57 |
| LDH, U/L | 353 ± 21.3 | 358 ± 29.9 | 425 ± 23.5 | 406 ± 21.2 |
| Mg, mg/dL | 2.90 ± 0.55 | 2.14 ± 0.54 | 2.17 ± 0.33 | 2.45 ± 0.40 |
| Pho, mg/dL | 4.06 ± 0.20 | 3.9 ± 0.23 | 4.47 ± 0.40 | 3.95 ± 0.10 |
| AST, U/L | 22.3 ± 1.88 | 18.34 ± 2.37 | 23.49 ± 1.90 | 24.5 ± 3.57 |
| ALT, U/L | 21.6 ± 2.85 | 18.0 ± 4.00 | 19.29 ± 2.24 | 18.55 ± 2.17 |
| Urea, mg/dL | 30.4 ± 1.52 | 28.3 ± 2.58 | 38.2 ± 3.02 | 27.23 ± 3.29 |
| ADMA, μmol/L | 0.73 ± 0.08 | 0.93 ± 0.1 | 0.97 ± 0.09 | 1.23 ± 0.14 ** |
| ADMA/L-arginine ratio | 0.017 ± 0.002 | 0.021 ± 0.005 | 0.022 ± 0.002 | 0.026 ± 0.003 |
| Parameter | HR+ HER2+ (n = 12) | HR+ HER2- (n = 16) | TNBC (n = 19) |
|---|---|---|---|
| Histological type | |||
| Ductal | 8 | 13 | 16 |
| Lobular | 2 | 1 | 1 |
| N.A. | 2 | 2 | 2 |
| Involved lymph nodes | 2.00 ± 1.19 | 3.21 ± 1.53 | 2.69 ± 0.95 |
| Ki, % | 26.6 ± 3.39 | 14.1 ± 2.00 $ | 42.4 ± 5.35 *** |
| Hgb, g/dL | 13.4 ± 0.46 | 13.6 ± 0.40 | 12.9 ± 0.32 |
| Ht, % | 39.1 ± 1.19 | 40.2 ± 1.09 | 38.1 ± 0.91 |
| RBC, 106/μL | 4.64 ± 0.17 | 4.62 ± 0.15 | 4.20 ± 0.12 |
| WBC, 103/μL | 7.25 ± 0.36 | 7.19 ± 0.35 | 6.86 ± 0.68 |
| PLT, 103/μL | 245 ± 15.7 | 266 ± 22.6 | 291 ± 19.3 |
| BUN mg/dL | 12.3 ± 1.32 | 19.0 ± 3.93 | 15.7 ± 1.32 |
| Creat, mg/dL | 0.71 ± 0.04 | 0.94 ± 0.17 | 0.74 ± 0.04 |
| Glu, mg/dL | 103 ± 3.47 | 96.6 ± 3.17 | 106 ± 5.54 |
| K, mmol/L | 4.29 ± 0.13 | 4.52 ± 0.11 | 4.30 ± 0.09 |
| Na mmol/L | 139.3 ± 1.15 | 140 ± 0.71 | 139 ± 0.40 |
| PT, s | 11.3 ± 0.17 | 11.6 ± 0.20 | 11.4 ± 0.12 |
| WPT | 105.3 ± 1.81 | 102 ± 1.67 | 104 ± 1.14 |
| INR | 0.95 ± 0.02 | 0.98 ± 0.02 | 0.95 ± 0.01 |
| APTT | 30.3 ± 0.82 | 28.3 ± 0.91 | 28.3 ± 0.56 |
| WAPPT | 0.91 ±0.02 | 0.85 ± 0.03 | 0.85 ± 0.02 |
| FIB, g/L | 3.83 ± 0.46 | 3.32 ± 0.23 | 3.78 ± 0.70 |
| tADA | ADA1 | ADA2 | ||||
|---|---|---|---|---|---|---|
| Parameter | Correlation Coefficient | p-Value | Correlation Coefficient | p-Value | Correlation Coefficient | p-Value |
| Galectin-3, ng/mL | −0.08 | NS | −0.28 | NS | −0.37 | NS |
| ADMA/L-arginine ratio | 0.40 | NS | 0.54 | 0.03 | 0.42 | NS |
| ADMA, μmol/L | 0.51 | 0.03 | 0.73 | 0.0007 | 0.48 | 0.04 |
| hsCRP, mg/dL | 0.35 | NS | 0.63 | 0.008 | 0.05 | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutryb-Zajac, B.; Harasim, G.; Jedrzejewska, A.; Krol, O.; Braczko, A.; Jablonska, P.; Mierzejewska, P.; Zielinski, J.; Slominska, E.M.; Smolenski, R.T. Macrophage-Derived Adenosine Deaminase 2 Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3764. https://doi.org/10.3390/ijms22073764
Kutryb-Zajac B, Harasim G, Jedrzejewska A, Krol O, Braczko A, Jablonska P, Mierzejewska P, Zielinski J, Slominska EM, Smolenski RT. Macrophage-Derived Adenosine Deaminase 2 Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2021; 22(7):3764. https://doi.org/10.3390/ijms22073764
Chicago/Turabian StyleKutryb-Zajac, Barbara, Gabriela Harasim, Agata Jedrzejewska, Oliwia Krol, Alicja Braczko, Patrycja Jablonska, Paulina Mierzejewska, Jacek Zielinski, Ewa M. Slominska, and Ryszard. T. Smolenski. 2021. "Macrophage-Derived Adenosine Deaminase 2 Correlates with M2 Macrophage Phenotype in Triple Negative Breast Cancer" International Journal of Molecular Sciences 22, no. 7: 3764. https://doi.org/10.3390/ijms22073764