
Rare Variants in Autophagy and Non-Autophagy Genes in Late-Onset Pompe Disease: Suggestions of Their Disease-Modifying Role in Two Italian Families

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:1. Introduction
2. Results
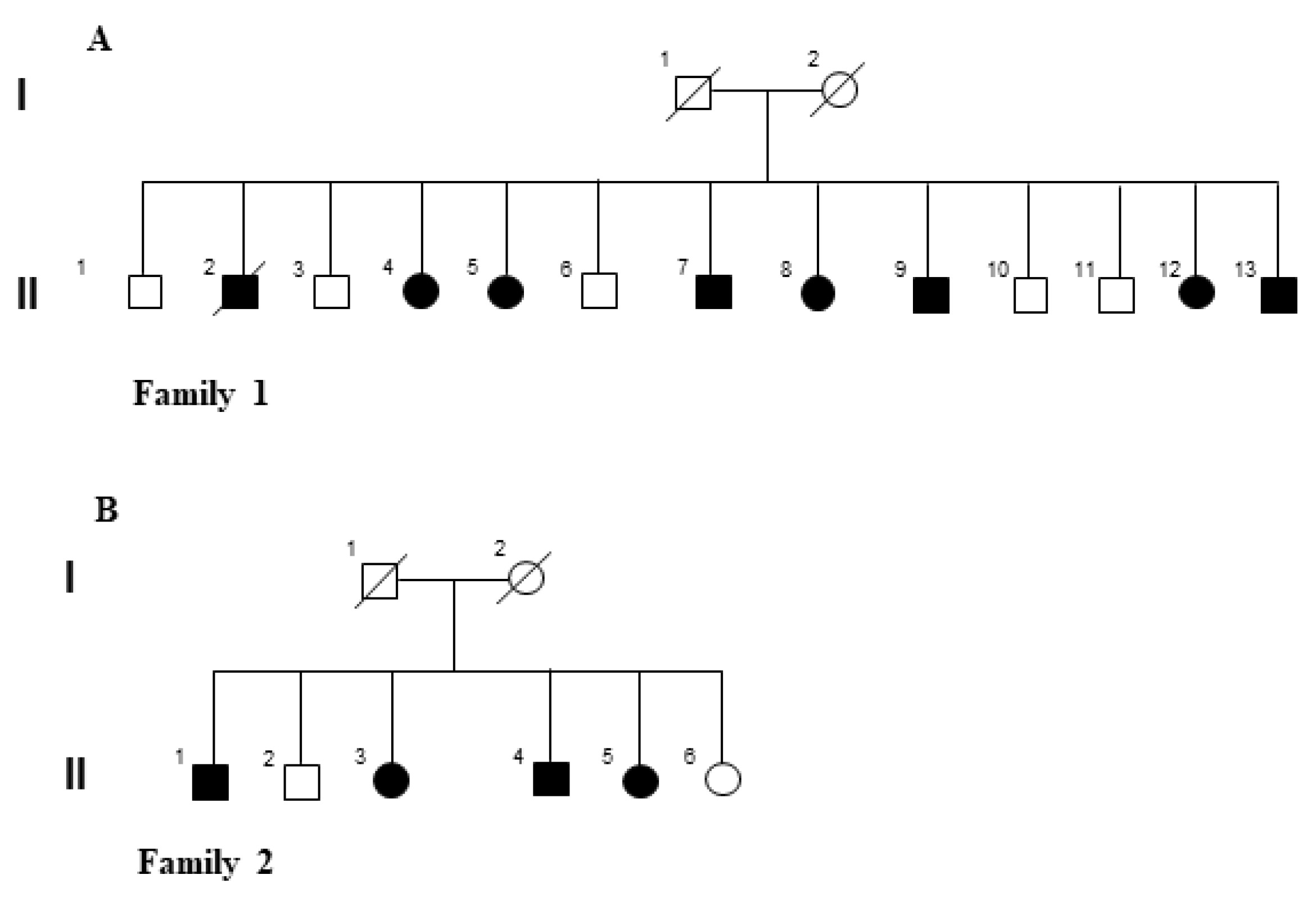
2.1. Clinical Evaluation of the LOPD Patients
2.2. Generation of a Phenotype Severity Index (SI) for LOPD
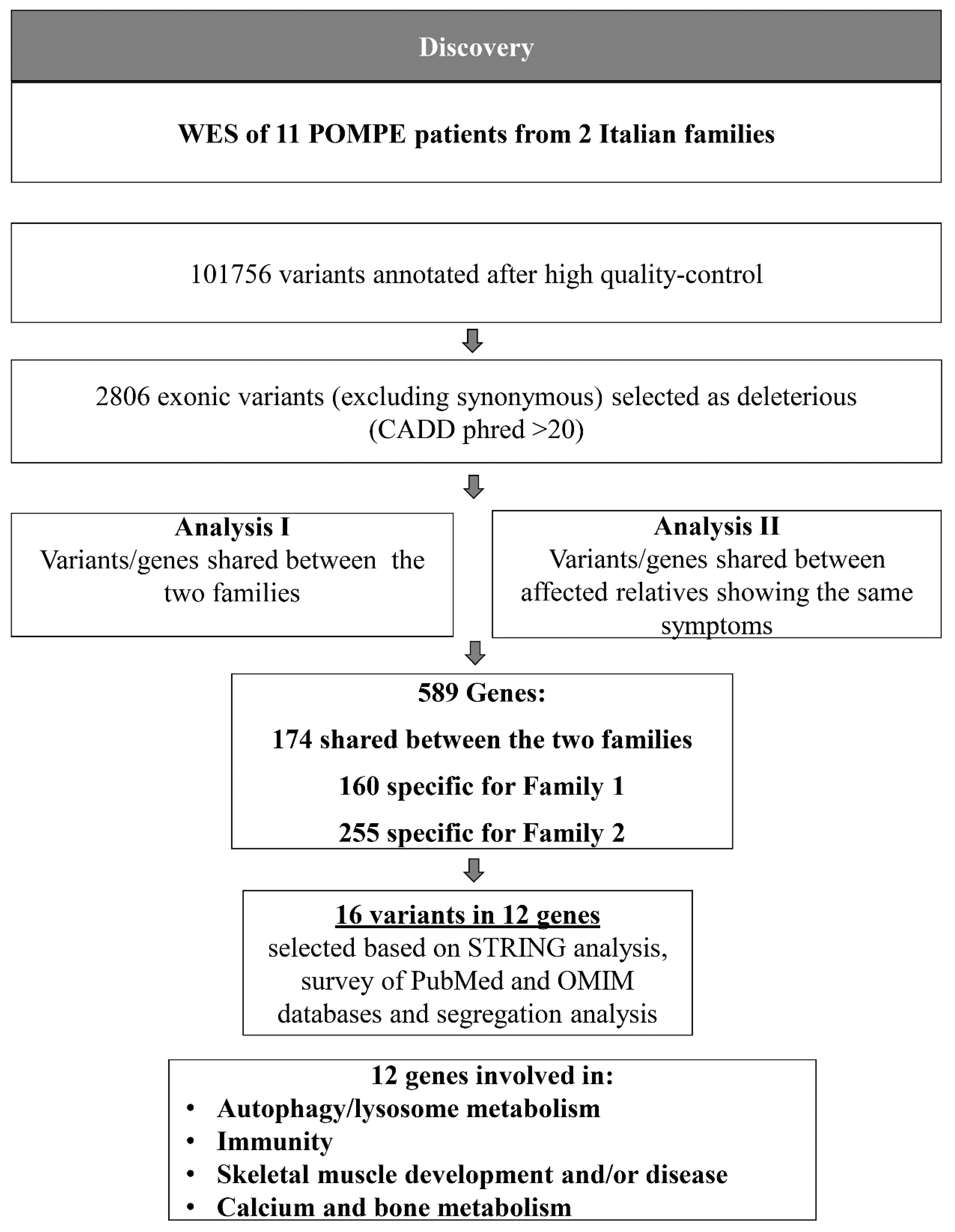
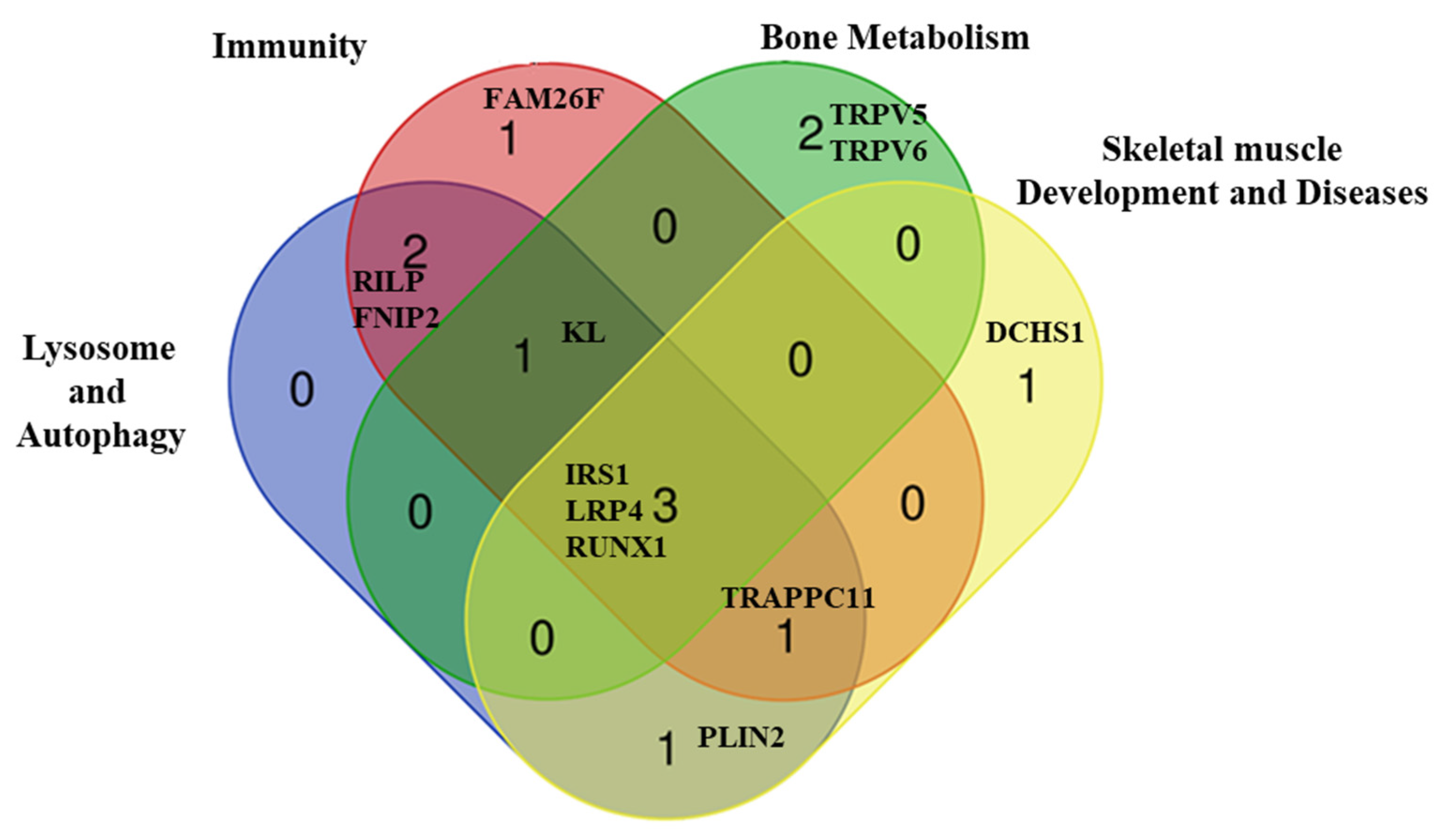
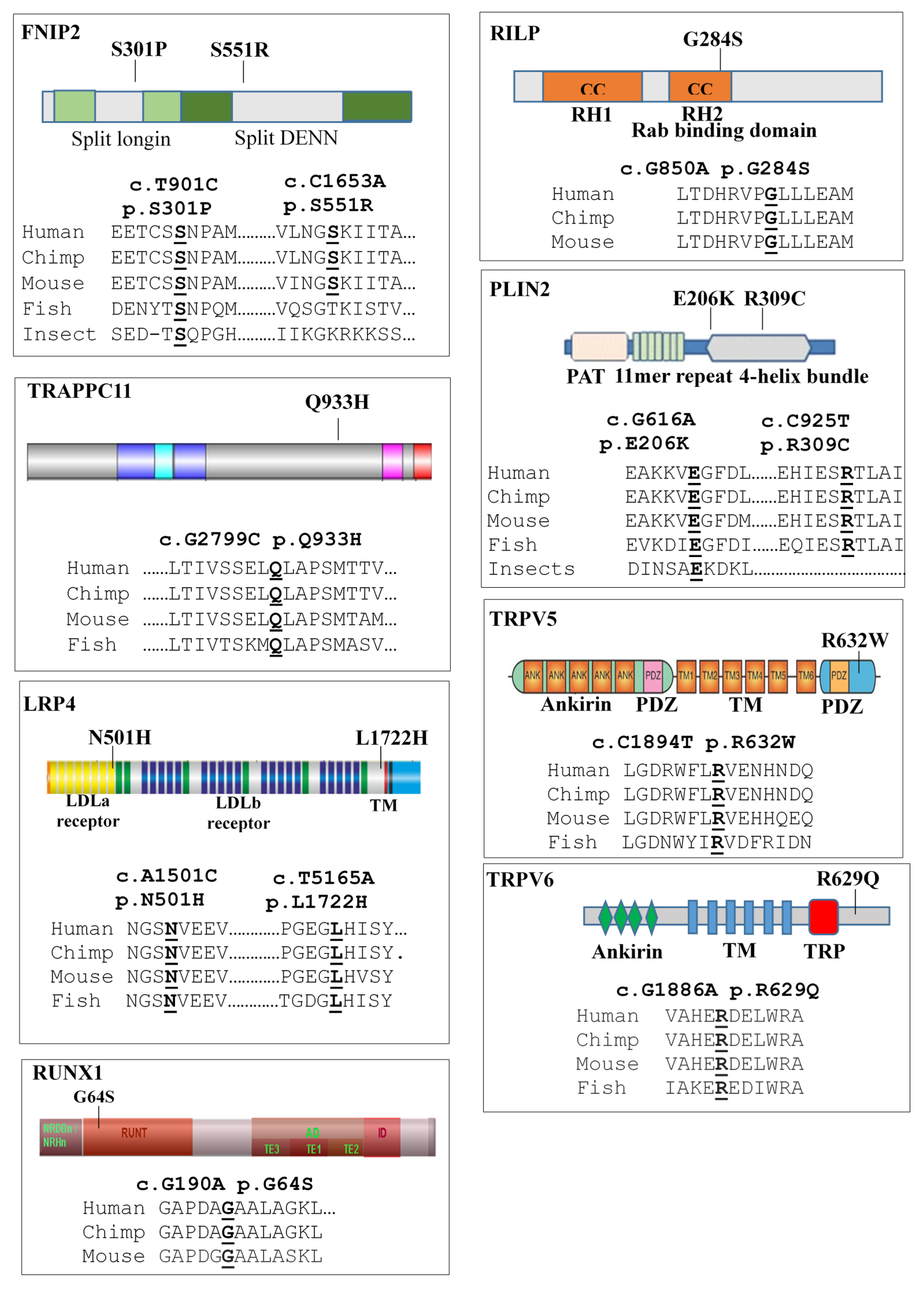
2.3. Comprehensive Genetic Analysis of the LOPD Patients
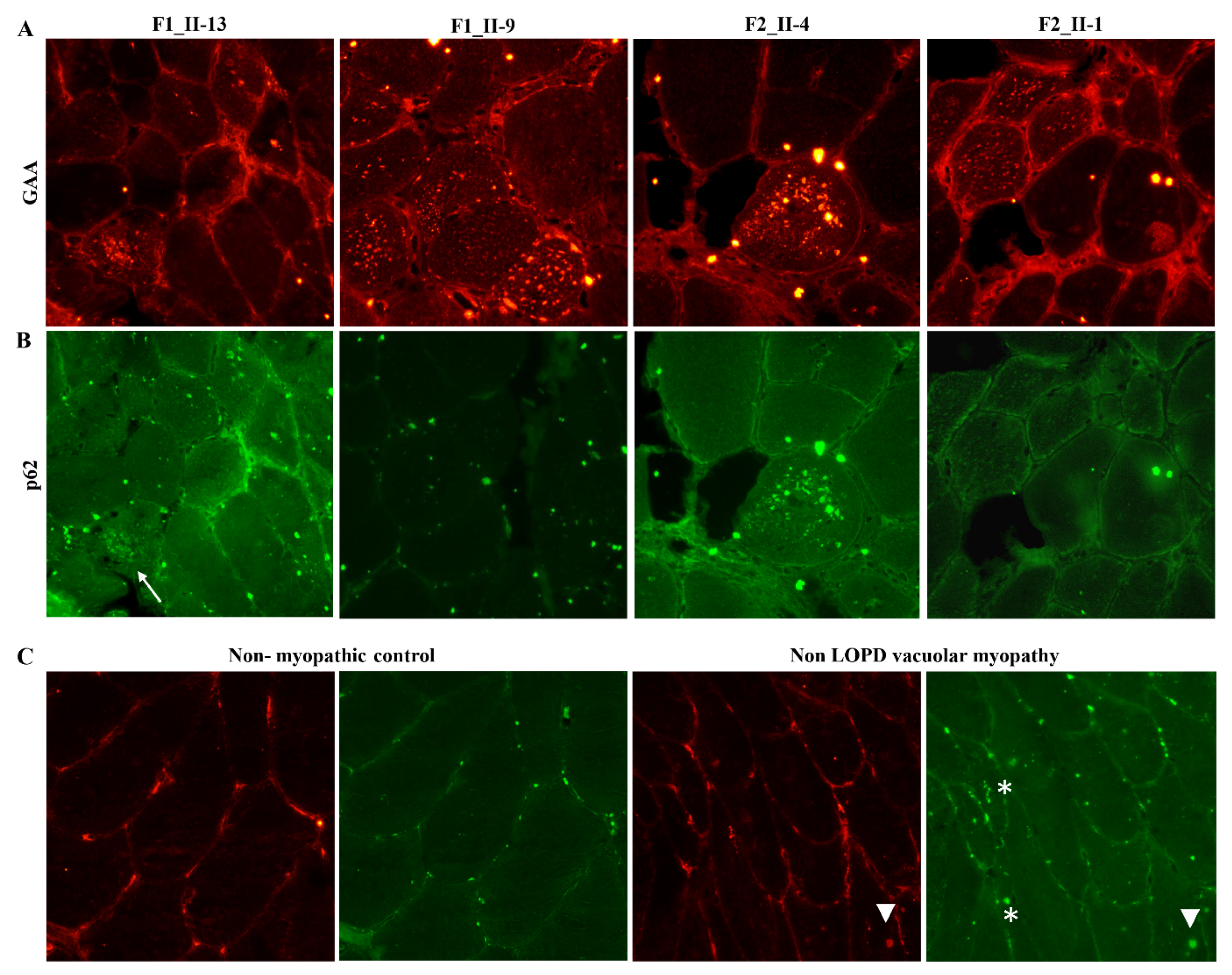
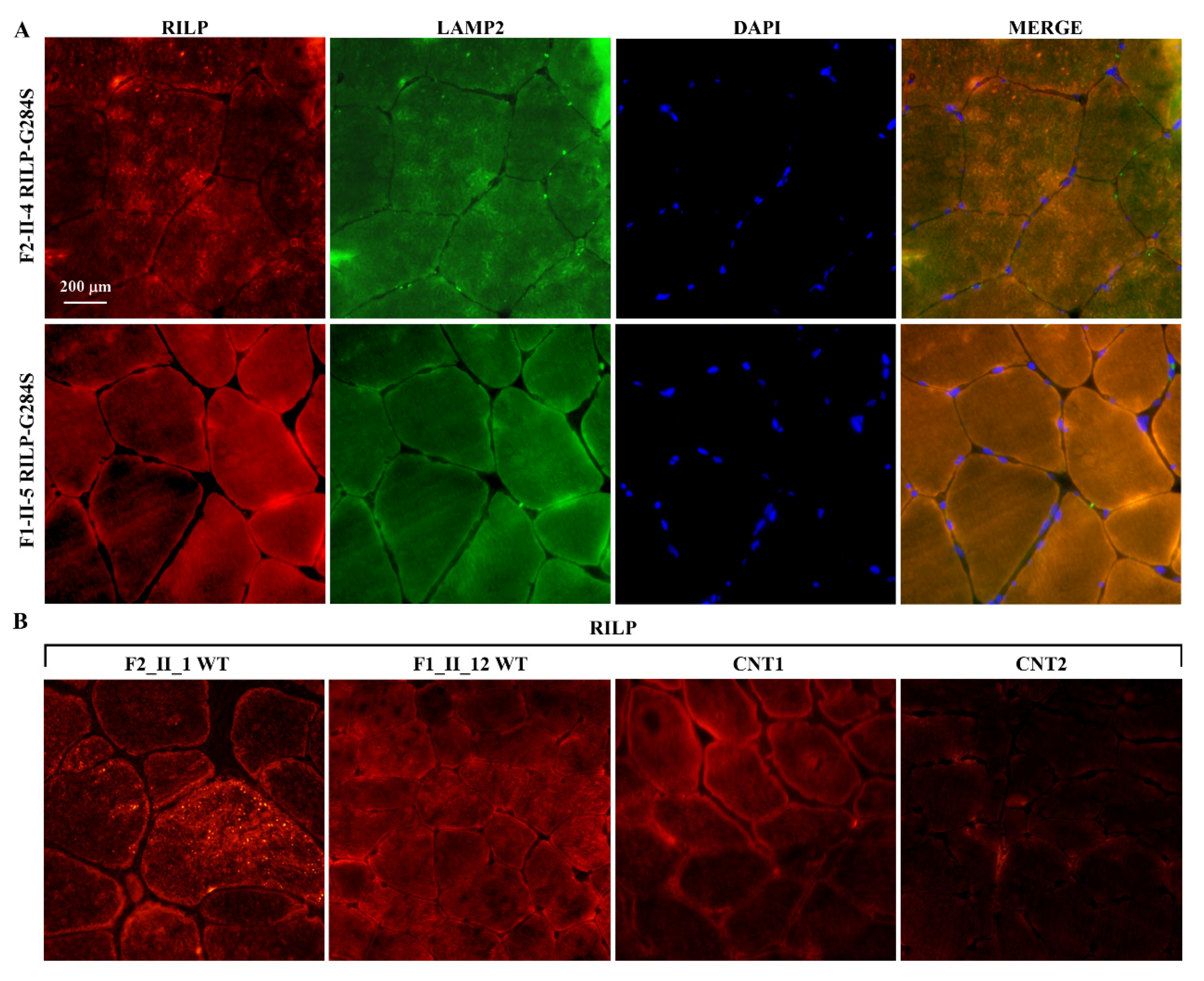
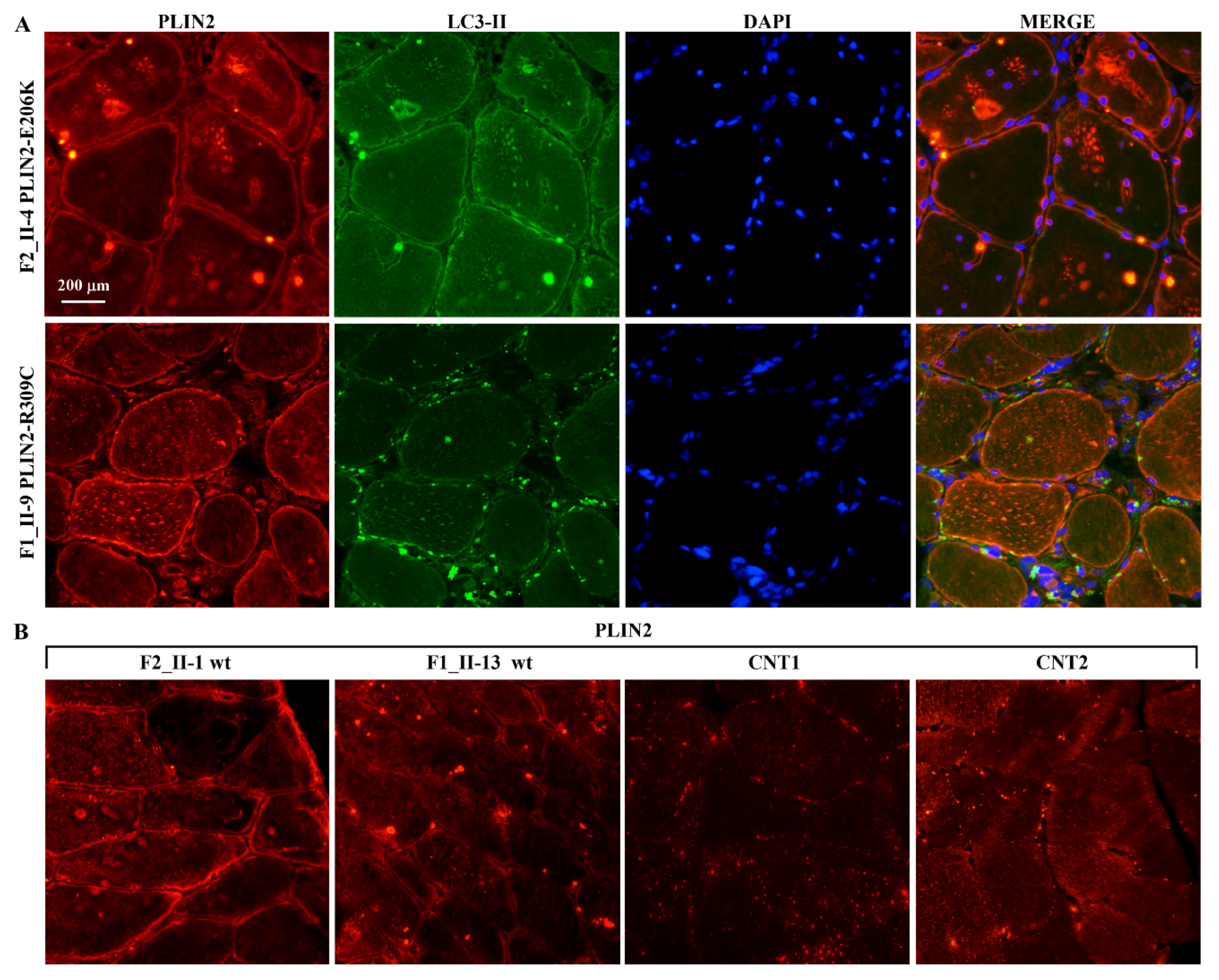
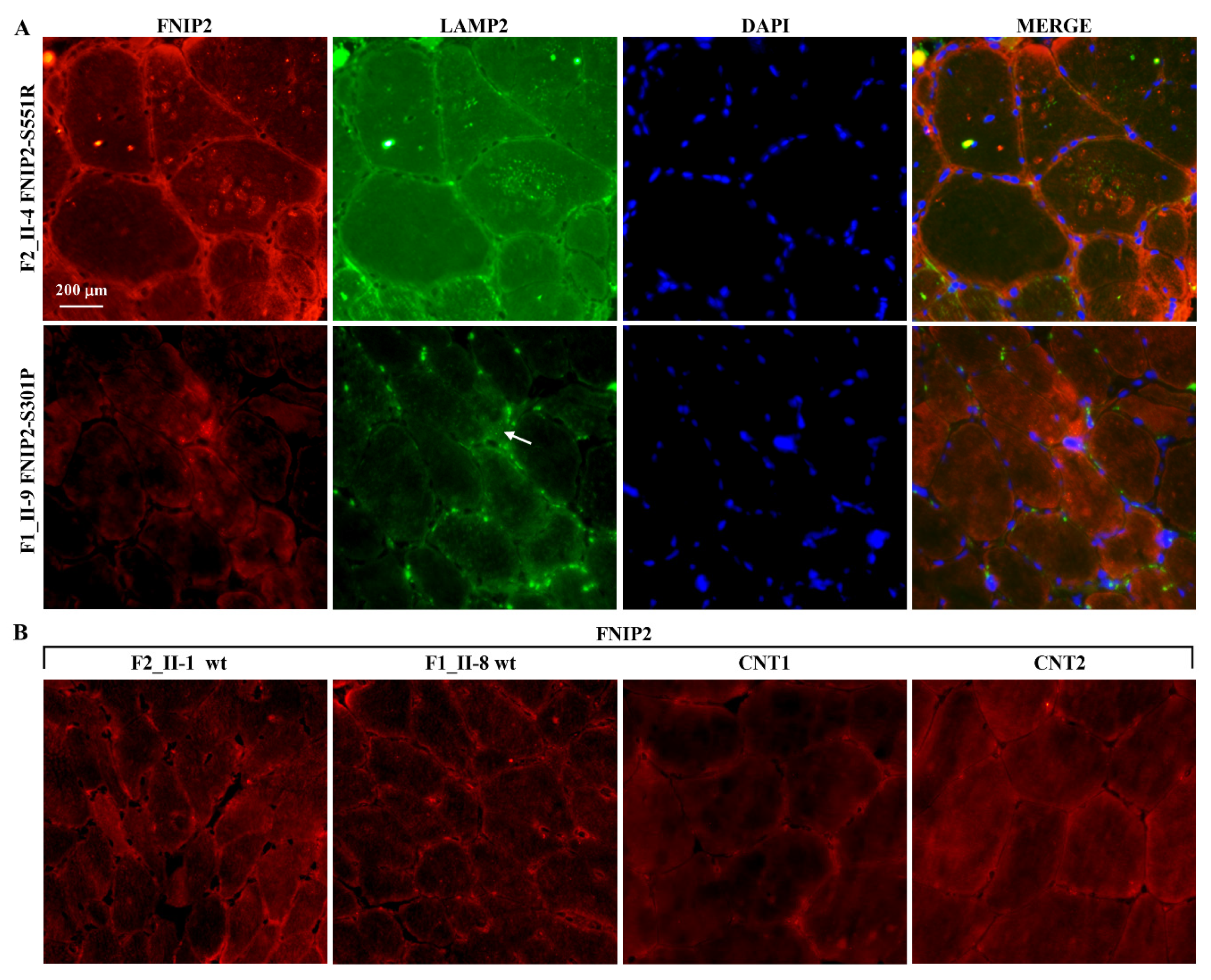
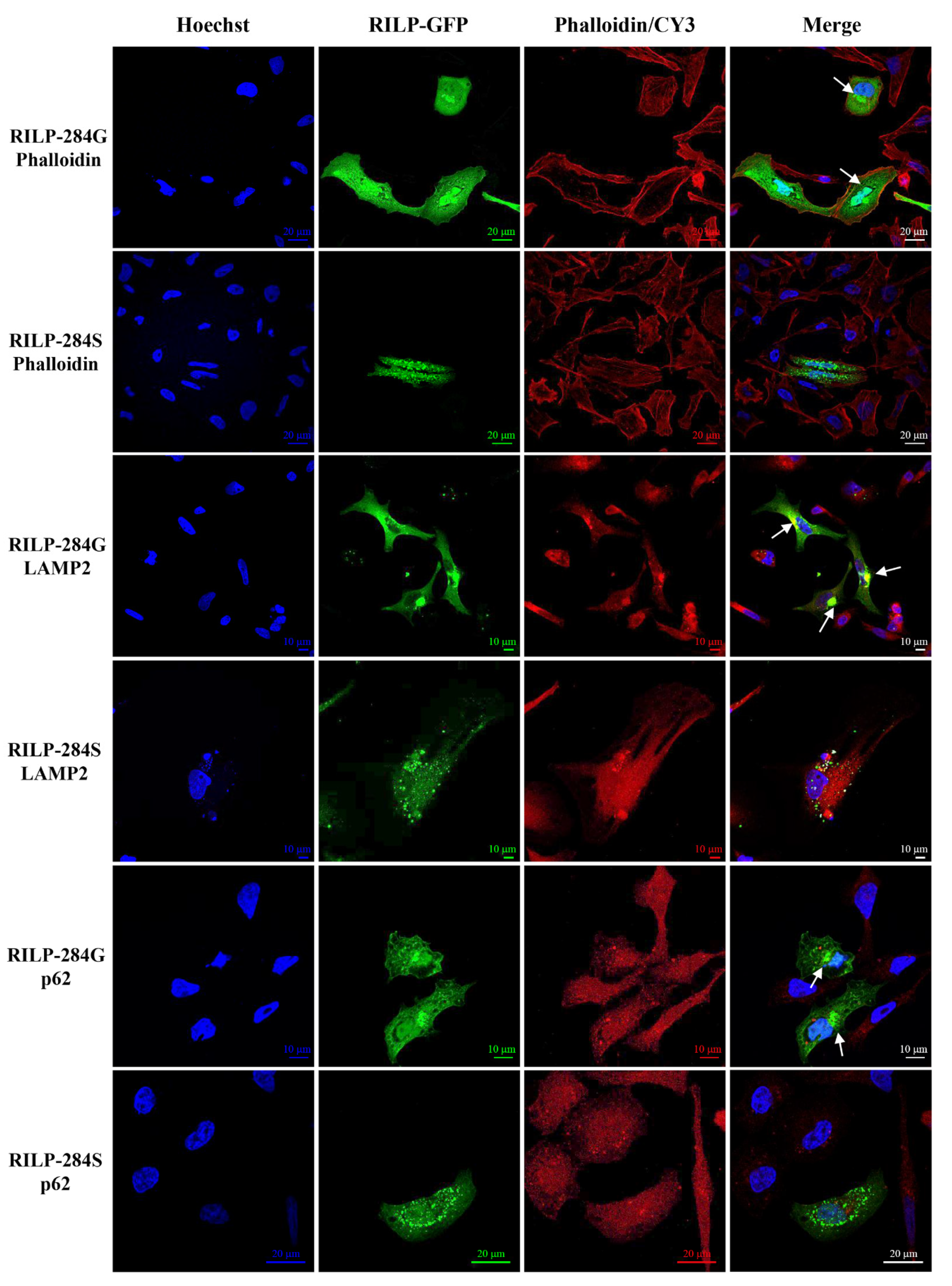
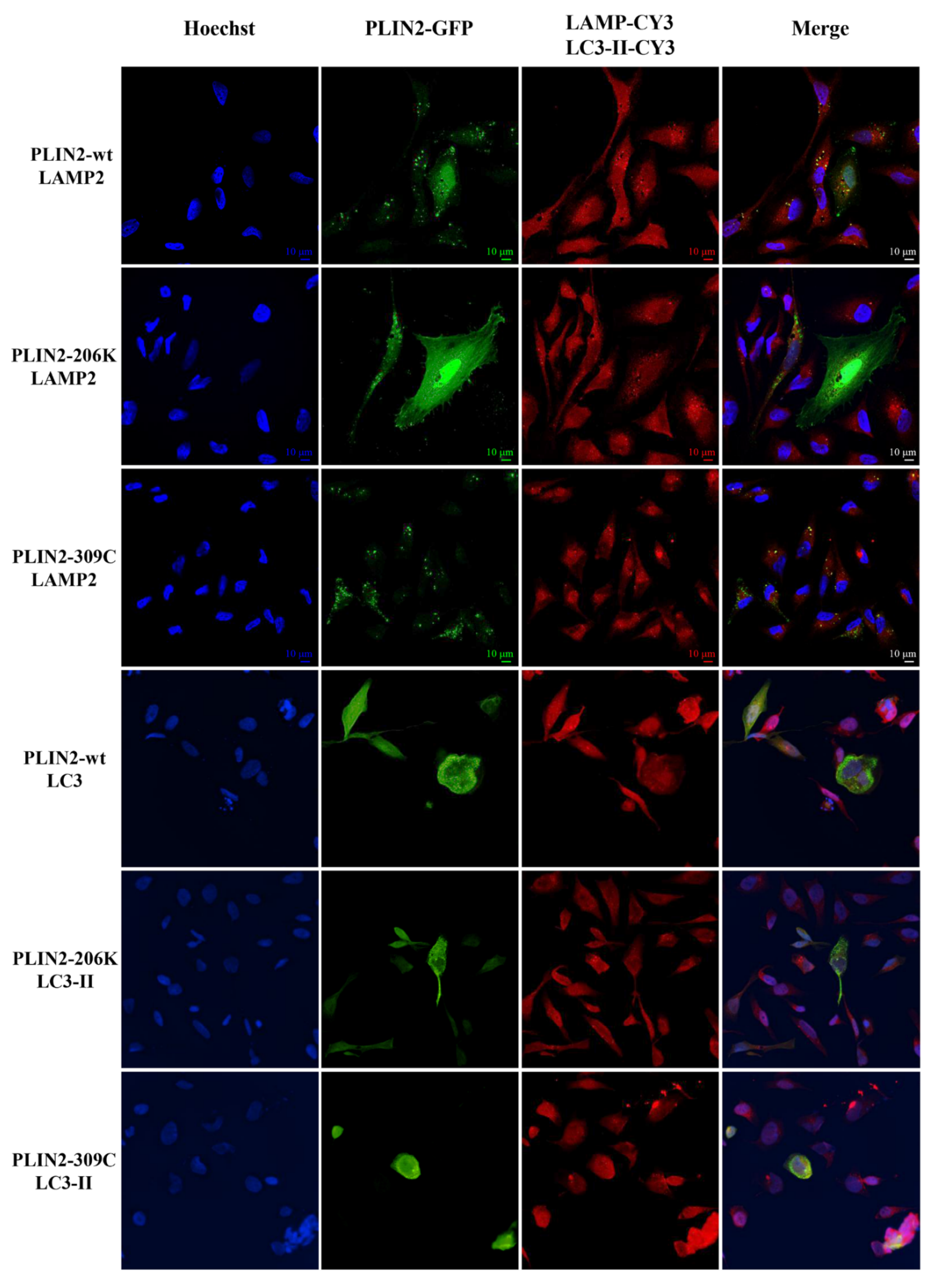
2.4. Expression Studies
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. WES Analysis
4.3. Immunofluorescence Staining of Skeletal Muscle
4.4. Plasmids Preparation, Cell Cultures and Transfection
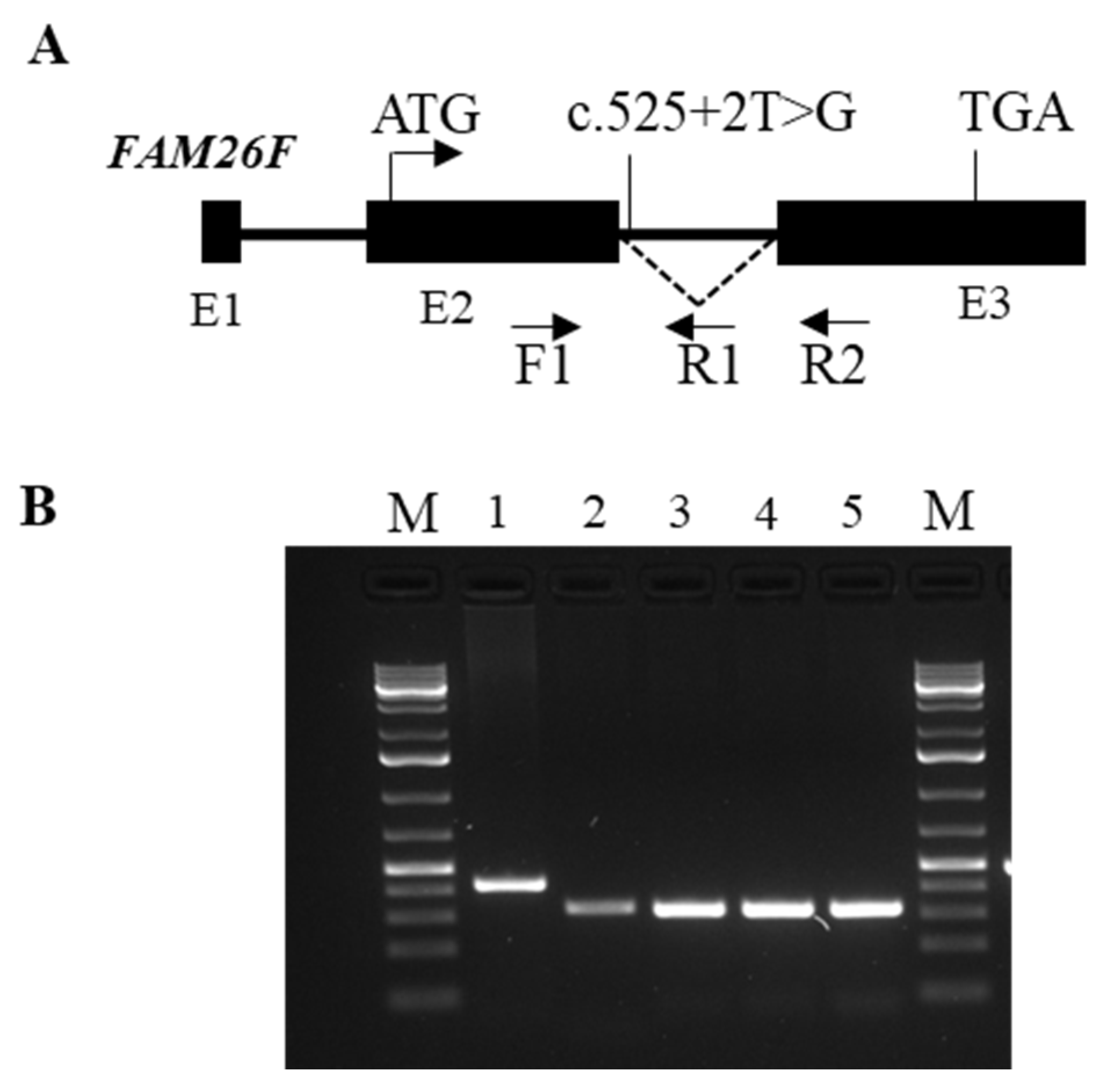
4.5. Expression Analysis of the Mutated FAM26Fc.525 + 2T > G Transcript
4.6. Statistical Analysis for Clinical Variables
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirschhorn, R.; Reuser, A.J. Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3389–3420. [Google Scholar]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- Raben, N.; Nichols, R.C.; Martiniuk, F.; Plotz, P.H. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum. Mol. Genet. 1996, 5, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Montalvo, A.L.; Bembi, B.; Donnarumma, M.; Filocamo, M.; Parenti, G.; Rossi, M.; Merlini, L.; Buratti, E.; De Filippi, P.; Dardis, A.; et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum. Mutat. 2006, 27, 999–1006. [Google Scholar] [CrossRef]
- Spada, M.; Porta, F.; Vercelli, L.; Pagliardini, V.; Chiadò-Piat, L.; Boffi, P.; Pagliardini, S.; Remiche, G.; Ronchi, D.; Comi, G.; et al. Screening for later-onset Pompe’s disease in patients with paucisymptomatic hyperCKemia. Mol. Genet. Metab. 2013, 109, 171–173. [Google Scholar] [CrossRef]
- Mehler, M.; Di Mauro, S. Residual acid maltase activity in late-onset acid maltase deficiency. Neurology 1977, 27, 178–184. [Google Scholar] [CrossRef]
- Umapathysivam, K.; Hopwood, J.J.; Meikle, P.J. Correlation of acid alpha-glucosidase and glycogen content in skin fibroblasts with age of onset in Pompe disease. Clin. Chim. Acta 2005, 36, 191–198. [Google Scholar] [CrossRef]
- Reuser, A.J.; Kroos, M.A.; Hermans, M.M.; Bijvoet, A.G.; Verbeet, M.P.; Van Diggelen, O.P.; Kleijer, W.J.; Van der Ploeg, A.T. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995, 3, S61–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.L.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.A.; Van der Kraan, M.; Van Diggelen, O.P.; Kleijer, W.J.; Reuser, A.J. Two extremes of the clinical spectrum of glycogen storage disease type II in one family: A matter of genotype. Hum. Mut. 1997, 9, 17–22. [Google Scholar] [CrossRef]
- Ausems, M.G.; ten Berg, K.; Beemer, F.A.; Wokke, J.H. Phenotypic expression of late-onset glycogen storage disease type II: Identification of asymptomatic adults through family studies and review of reported families. Neuromusc. Disord. 2000, 10, 467–471. [Google Scholar] [CrossRef]
- Semplicini, C.; Letard, P.; De Antonio, M.; Taouagh, N.; Perniconi, B.; Bouhour, F.; Echaniz-Laguna, A.; Orlikowski, D.; Sacconi, S.; Salort-Campana, E.; et al. Late onset Pompe disease in France: Molecular features and epidemiology from a nation-wide study. J. Inherit. Metab. Dis. 2018, 41, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Herzog, A.; Hartung, R.; Reuser, A.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A cross sectional single-centre study on the spectrum of Pompe disease, German patients: Molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet. J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforet, P.; Laloui, K.; Granger, B.; Hamroun, D.; Taouagh, N.; Hogrel, J.Y.; Orlikowski, D.; Bouhour, F.; Lacour, A.; Salort-Campana, E.; et al. The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease. Rev. Neurol. 2013, 169, 595–602. [Google Scholar] [CrossRef]
- Slonim, A.E.; Bulone, L.; Goldberg, T.; Minikes, J.; Slonim, E.; Galanko, J.; Martiniuk, F. Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy. Muscle Nerve 2007, 35, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Slonim, A.E.; Bulone, L.; Minikes, J.; Hays, A.P.; Shanske, S.; Tsujino, S.; DiMauro, S. Benign course of glycogen storage disease type IIb in two brothers: Nature or nurture? Muscle Nerve 2006, 33, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Sampaolo, S.; Esposito, T.; Farina, O.; Formicola, D.; Diodato, D.; Gianfrancesco, F.; Cipullo, F.; Cremone, G.; Cirillo, M.; Del Viscovo, L.; et al. Distinct disease phenotypes linked to different combinations of GAA mutations in a large late-onset GSDII sibship. Orphanet. J. Rare Dis. 2013, 8, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemans, M.L.C.; van Schie, S.P.M.; Janssens, A.C.J.W.; van Doorn, P.A.; Reuser, A.J.J.; van der Ploeg, A.T. Fatigue: An important feature of late-onset Pompe disease. J. Neurol. 2007, 254, 941–945. [Google Scholar] [CrossRef] [Green Version]
- American Thoracic Society/European Respiratory Society. ATS/ERS: Statement on respiratory muscle testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [Google Scholar] [CrossRef]
- Van den Berg, L.E.; Zandbergen, A.A.; van Capelle, C.I.; de Vries, J.M.; Hop, W.C.; van denHout, J.M.; Reuser, A.J.; Zillikens, M.C.; van der Ploeg, A.T. Low bone mass in Pompe disease: Muscular strength as a predictor of bone mineral density. Bone 2010, 47, 643–649. [Google Scholar] [CrossRef]
- Laforêt, P.; Petiot, P.; Nicolino, M.; Orlikowski, D.; Caillaud, C.; Pellegrini, N.; Froissart, R.; Petitjean, T.; Maire, I.; Chabriat, H.; et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology 2008, 70, 2063–2066. [Google Scholar] [CrossRef]
- Morris, D.A.; Blaschke, D.; Krebs, A.; Canaan-Kühl, S.; Plöckinger, U.; Knobloch, G.; Walter, T.C.; Kühnle, Y.; Boldt, L.H.; Kraigher-Krainer, E.; et al. Structural and functional cardiac analyses using modern and sensitive myocardial techniques in adult Pompe disease. Int. J. Cardiovasc. Imaging 2015, 31, 947–956. [Google Scholar] [CrossRef]
- Kulessa, M.; Weyer-Menkhoff, I.; Viergutz, L.; Kornblum, C.; Claeys, K.G.; Schneider, I.; Plöckinger, U.; Young, P.; Boentert, M.; Vielhaber, S.; et al. An integrative correlation of myopathology, phenotype and genotype in late onset Pompe disease. Neuropathol. Appl. Neurobiol. 2020, 46, 359–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierzba-Bobrowicz, T.; Lewandowska, E.; Lugowska, A.; Rola, R.; Stepień, T.; Ryglewicz, D.; Pasennik, E. Adult glycogenosis type II (Pompe’s disease): Morphological abnormalities in muscle and skin biopsies compared with acid alpha-glucosidase activity. Folia Neuropathol. 2007, 45, 179–186. [Google Scholar] [PubMed]
- Durst, R.; Sauls, K.; Peal, D.S.; deVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Sampaolo, S.; Napolitano, F.; Tirozzi, A.; Reccia, M.G.; Lombardi, L.; Farina, O.; Barra, A.; Cirillo, F.; Melone, M.A.B.; Gianfrancesco, F.; et al. Identification of the first dominant mutation of LAMA5 gene causing a complex multisystem syndrome due to dysfunction of the extracellular matrix. J. Med. Genet. 2017, 54, 710–720. [Google Scholar] [CrossRef]
- Napolitano, F.; Terracciano, C.; Bruno, G.; De Blasiis, P.; Lombardi, L.; Gialluisi, A.; Gianfrancesco, F.; De Giovanni, D.; Tummolo, A.; Di Iorio, G.; et al. Novel autophagic vacuolar myopathies: Phenotype and genotype features. Neuropathol. Appl. Neurobiol. 2021, 2021. [Google Scholar] [CrossRef]
- Alathari, B.E.; Sabta, A.A.; Kalpana, C.A.; Vimaleswaran, K.S. Vitamin D pathway-related gene polymorphisms and their association with metabolic diseases: A literature review. J. Diabetes Metab. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hagemans, M.L.; Winkel, L.P.; Van Doorn, P.A.; Hop, W.J.; Loonen, M.C.; Reuser, A.J.; Van der Ploeg, A.T. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain 2005, 128, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Domingues-Faria, C.; Boirie, Y.; Walrand, S. Vitamin D and muscle trophicity. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 169–174. [Google Scholar] [CrossRef]
- Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019, 20, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Arribas, M.; Pedro, J.M.; Gómez-Sánchez, R.; Yakhine-Diop, S.M.; Martínez-Chacón, G.; Uribe-Carretero, E.; De Castro, D.C.; Casado-Naranjo, I.; López de Munaín, A.; Niso-Santano, M.; et al. Pompe disease and autophagy: Partners in crime, or cause and consequence? Curr. Med. Chem. 2016, 23, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Fanin, M.; Masiero, E.; Angelini, C.; Sandri, M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy 2012, 8, 1697–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimbeni, A.C.; Fanin, M.; Angelini, C.; Sandri, M. Autophagy dysregulation in Danon disease. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef] [Green Version]
- De Martín Garrido, N.; Aylett, C.H.S. Nutrient signaling and lysosome positioning crosstalk through a multifunctional protein, Folliculin. Front. Cell Dev. Biol. 2020, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef]
- Harrison, R.E.; Bucci, C.; Vieira, O.V.; Schroer, T.A.; Grinstein, S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: Role of Rab7 and RILP. Mol. Cell Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Seifan, S.; Claessens, T.; Behrends, C.; Kamps, M.A.F.; Rozycka, E.; Kemp, A.J.; Nookala, R.K.; Blenis, J.; Coull, B.J.; et al. FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy 2014, 10, 1749–1760. [Google Scholar] [CrossRef]
- Shen, K.; Rogala, K.B.; Chou, H.-T.; Huang, R.K.; Yu, Z.; Sabatini, D.M. Cryo-EM structure of the human FLCN-FNIP2-Rag-ragulator complex. Cell 2019, 179, 1319. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P. Folliculin–A tumor suppressor at the intersection of metabolic signaling and membrane traffic. Small GTPases 2017, 8, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, L.S.; and Linehan, W.M. FLCN: The causative gene for birt-hogg dubé syndrome. Gene 2018, 640, 28–42. [Google Scholar] [CrossRef]
- Rudolf, R.; Deschenes, M.R.; Sandri, M. Neuromuscular junction degeneration in muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Barik, A.; Lu, Y.; Sathyamurthy, A.; Bowman, A.; Shen, C.; Li, L.; Xiong, W.C.; Mei, L. LRP4 is critical for neuromuscular junction maintenance. J. Neurosci. 2014, 34, 13892–13905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzella, F.; Codeluppi, L.; Lusuardi, M.; Galeone, C.; Valzania, F.; Facciolongo, N. Acute respiratory failure as presentation of late-onset Pompe disease complicating the diagnostic process as a labyrinth: A case report. Multidiscip. Respir. Med. 2018, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Stanga, D.; Zhao, Q.; Milev, M.P.; Saint-Dic, D.; Jimenez-Mallebrera, C.; Sacher, M. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to pre-autophagosomal membranes. Traffic 2019, 20, 325–345. [Google Scholar] [CrossRef] [PubMed]
- Sacher, M.; Shahrzad, N.; Kamel, H.; Milev, M.P. TRAPPopathies: An emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic 2019, 20, 5–26. [Google Scholar] [CrossRef] [Green Version]
- Magné, J.; Aminoff, A.; Perman Sundelin, J.; Mannila, M.N.; Gustafsson, P.; Hultenby, K.; Wernerson, A.; Bauer, G.; Listenberger, L.; Neville, M.J.; et al. The minor allele of the missense polymorphism Ser251Pro in perilipin 2 (PLIN2) disrupts an α-helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans. FASEB J. 2013, 27, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Najt, C.P.; Lwande, J.S.; McIntosh, A.L.; Senthivinayagam, S.; Gupta, S.; Kuhn, L.A.; Atshaves, B.P. Structural and functional assessment of perilipin 2 lipid binding domain(s). Biochemistry 2014, 53, 7051–7066. [Google Scholar] [CrossRef]
- Sentinelli, F.; Capoccia, D.; Incani, M.; Bertoccini, L.; Severino, A.; Pani, M.G.; Manconi, E.; Cossu, E.; Leonetti, F.; Baroni, M.G. The perilipin 2 (PLIN2) gene Ser251Pro missense mutation is associated with reduced insulin secretion and increased insulin sensitivity in Italian obese subjects. Diabetes Metab Res. Rev. 2016, 32, 550–556. [Google Scholar] [CrossRef]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [Green Version]
- Papadimas, G.K.; Terzis, G.; Methenitis, S.; Spengos, K.; Papadopoulos, C.; Vassilopoulou, S.; Kavouras, S.; Michelakakis, H.; Manta, P. Body composition analysis in late-onset Pompe disease. Mol. Genet. Metab. 2011, 102, 41–43. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.; Stähelin, H.B.; Walter, P. Vitamin D effects on bone and muscle. Int. J. Vitam. Nutr. Res. 2011, 81, 264–272. [Google Scholar] [CrossRef]
- Peng, J.B.; Suzuki, Y.; Gyimesi, G.; Hediger, M.A. TRPV5 and TRPV6 calcium-selective channels. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018. [Google Scholar]
- Van Abel, M.; Hoenderop, J.G.; Bindels, R.J. The epithelial calcium channels TRPV5 and TRPV6: Regulation and implications for disease. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 371, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Earley, S. Vanilloid and melastatin transient receptor potential channels in vascular smooth muscle. Microcirculation 2010, 17, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenderop, J.G.; Voets, T.; Hoefs, S.; Weidema, F.; Prenen, J.; Nilius, B.; Bindels, R.J. Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 andTRPV6. EMBO J. 2003, 22, 776–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanoevelen, J.; Janssens, A.; Huitema, L.F.; Hammond, C.L.; Metz, J.R.; Flik, G.; Voets, T.; Schulte-Merker, S. Trpv5/6 is vital for epithelial calcium uptake and bone formation. FASEB J. 2011, 25, 3197–3207. [Google Scholar] [CrossRef] [Green Version]
- van der Eerden, B.C.; Hoenderop, J.G.; de Vries, T.J.; Schoenmaker, T.; Buurman, C.J.; Uitterlinden, A.G.; Pols, H.A.; Bindels, R.J.; van Leeuwen, J.P. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc. Natl. Acad. Sci. USA 2005, 102, 17507–17512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Ni, B.; Yang, Y.O.; Ye, T.; Chen, A. Knockout of TRPV6 causes osteopenia in mice by increasing osteoclastic differentiation and activity. Cell Physiol. Biochem. 2014, 33, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.; Bivi, N.; Farrow, E.; Lezcano, V.; Plotkin, L.I.; White, K.E.; Bellido, T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011, 49, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Komaba, H.; Kaludjerovic, J.; Hu, D.Z.; Nagano, K.; Amano, K.; Ide, N.; Sato, T.; Densmore, M.J.; Hanai, J.I.; Olauson, H.; et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 2017, 92, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Erben, R.G. Update on FGF23 and Klotho signaling. Mol. Cell Endocrinol. 2016, 432, 56–65. [Google Scholar] [CrossRef]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Clin. Investig. 2007, 117, 2684–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc. Natl. Acad. Sci. USA 2008, 105, 3455–3460. [Google Scholar] [CrossRef] [Green Version]
- Ogata, N.; Chikazu, D.; Kubota, N.; Terauchi, Y.; Tobe, K.; Azuma, Y.; Ohta, T.; Kadowaki, T.; Nakamura, K.; Kawaguchi, H. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J. Clin. Investig. 2000, 105, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and longterm outcomes in late-onset Pompe disease following glucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef]
- Nayak, S.; Doerfler, P.A.; Porvasnik, S.L.; Cloutier, D.D.; Khanna, R.; Valenzano, K.J.; Herzog, R.W.; Byrne, B.J. Immune Responses and Hypercoagulation in ERT for Pompe Disease Are Mutation and rhGAA Dose Dependent. PLoS ONE 2014, 9, e98336. [Google Scholar] [CrossRef] [PubMed]
- Malik, U.; Javed, A. FAM26F: An enigmatic protein having a complex role in the immune system. Int. Rev. Immunol. 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Malik, U.; Javed, A.; Ali, A.; Asghar, K. Structural and functional annotation of human FAM26F: A multifaceted protein having a critical role in the immune system. Gene 2017, 597, 66–75. [Google Scholar] [CrossRef]
- World Health Organization. Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ. Tech. Rep. Ser. 2000, 894, 1–253. [Google Scholar]
- Pascarella, A.; Terracciano, C.; Farina, O.; Lombardi, L.; Esposito, T.; Napolitano, F.; Franzese, G.; Panella, G.; Tuccillo, F.; la Marca, G.; et al. Vacuolated PAS-positive lymphocytes as a hallmark of Pompe disease and other myopathies related to impaired autophagy. J. Cell Physiol. 2018, 233, 5829–5837. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | GAA Mut | Sex | Age | AAO | MMTMRC | 6MWT | GSCG | FVC | Δ-FVC | BMI | VDD | BF | BMD T-Score TB/FN/LSV | BAD | MVP | IgG–rhGAA | DBS GAA | PPL | VMF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 II-4 | p.R40X p.N882fs | F | 67 | 50 | 79 | 297 | 12 | 79 | 35 | 22.20 | 6.64 | YES | −2.9/−3/−3 | YES | YES | 1200 | 2.70 | 8 | 0.53 |
| F1 II-5 | p.R40X p.N882fs | F | 65 | 53 | 82 | 225 | 12 | 62 | 46 | 28.00 | 11.6 | NO | −1.3/−1.5/−2.6 | YES | YES | 6400 | 0.12 | 69 | 0.01 |
| F1 II-7 | p.R40X p.N882fs | M | 61 | 42 | 76 | 324 | 7 | 98 | 17 | 28.10 | 28.4 | NO | −1.1/−1.5/−0.5 | YES | YES | 1600 | 0.11 | 56 | 4.11 |
| F1 II-8 | p.R40X p.N882fs | F | 59 | 47 | 86 | 325 | 9 | 90 | 18 | 33.10 | 18.5 | YES | 0.5/−0.7/−0.5 | np | YES | 1000 | 0.04 | 80 | 0.45 |
| F1 II-9 | p.R40X p.N882fs | M | 58 | 38 | 74 | 254 | 9 | 83 | 20 | 21.40 | 12.7 | NO | −0.8/−1.4/0.2 | YES | YES | 3200 | 0.09 | 48 | 11.18 |
| F1 II-12 | p.R40X p.N882fs | F | 51 | 41 | 78 | 268 | 8 | 82 | 21 | 28.70 | 40.2 | NO | 1.1/2.2/0.6 | YES | YES | 1350 | 0.12 | 16 | 3.59 |
| F1 II-13 | p.R40X p.N882fs | M | 50 | 36 | 75 | 254 | 6 | 105 | 18 | 20.10 | 41.3 | NO | 1.0/−0.8/−0.5 | YES | NO | 600 | 0.31 | 59 | 1.30 |
| F2 II-1 | c.-32-13T > G p.R375L | M | 51 | 44 | 99 | 450 | 5 | 89 | 17 | 31.80 | 8.90 | NO | −0.3/−0.5/0.9 | NO | NO | 1000 | 0.79 | 52 | 0.05 |
| F2 II-3 | c.-32-13T > G p.R375L | F | 48 | 41 | 97 | 325 | 7 | 79 | 21 | 26.50 | 30.1 | NO | 0.4/0.1/1.3 | NO | NO | 1010 | 0.13 | 45 | 0.12 |
| F2 II-4 | c.-32-13T > G p.R375L | M | 46 | 38 | 89 | 114 | 21 | 42 | n.p. | 39.18 | 8.4 | NO | −0.8/−1.2/−0.5 | NO | NO | 850 | 0.78 | 58 | 5.28 |
| F2 II-5 | c.-32-13T > G p.R375L | F | 41 | 29 | 83 | 10 | 26 | 36 | n.p. | 26.10 | 7.11 | YES | −0.7/−2.1/1.9 | NO | NO | 900 | 0.98 | 63 | 15.31 |
| F1 II:4 | F1 II:5 | F1 II:7 | F1 II:8 | F1 II:9 | F1 II:12 | F1 II:13 | F2 II:1 | F2 II:3 | F2 II:4 | F2 II:5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AAO | 1 | 1 | 2 | 2 | 3 | 2 | 3 | 2 | 2 | 3 | 3 |
| MMT-MRC | 3 | 2 | 3 | 2 | 3 | 3 | 3 | 1 | 1 | 2 | 2 |
| 6MWT | 3 | 4 | 3 | 2 | 4 | 3 | 4 | 2 | 2 | 6 | 6 |
| GSCG | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 3 | 4 |
| FVC | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 4 |
| Δ-FVC | 4 | 4 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 4 | 4 |
| BMI | 0 | 1 | 1 | 2 | 1 | 1 | 0 | 2 | 1 | 3 | 1 |
| VDD | 2 | 2 | 1 | 2 | 2 | 0 | 0 | 2 | 0 | 2 | 2 |
| BMD | 2 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| BF | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| BAD | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| MVP | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| IgG-rhGAA | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| ERT-AE | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severity Index | 21 | 23 | 16 | 17 | 21 | 15 | 16 | 11 | 11 | 28 | 28 |
| CLINICAL OUTCOMES | SEVERITY | N | Mean (SD) | p |
|---|---|---|---|---|
| 6MWT (m) | VM/M | 6 | 324.3 (69.5) | 0.03 |
| M/S | 5 | 180.0 (116.6) | ||
| FVC (%) | VM/M | 6 | 90.5 (9.73) | 0.01 |
| M/S | 5 | 60.4 (21.1) | ||
| ΔFVC (%) | VM/M | 6 3 | 18.6 (13.0) | 0.02 |
| M/S | 33.6 (18.8) | |||
| VDD | VM/M | 6 5 | 27.9 (11.4) | 0.01 |
| M/S | 9.29 (2.4) | |||
| BMD femoral neck | VM/M | 6 5 | -0.20 (0.37) | 0.03 |
| M/S | -1.80 (0.77) | |||
| GSGC | VM/M | 6 5 | 7.0 (1.41) | 0.01 |
| M/S | 16.0 (7.17) |
| Type of Analysis | Patient ID Family 1 | Gene | Omim/RF | Inheritance | SKM Related Disease | A | I | M | CVM | O/BF | VD | Ob |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity score > 20 F1 and F2 | F1 and F2 | RILP | nr | nr | nr | yes | yes | nr | nr | nr | nr | nr |
| F1 and F2 | FNIP2 | nr | nr | nr | yes | yes | nr | nr | nr | nr | nr | |
| F2 | TRAPPC11 | LGMDR18 | AR | yes | yes | yes | yes | nr | nr | nr | nr | |
| BMI | F1 and F2 | PLIN2 | Obesity | nr | nr | yes | nr | yes | nr | nr | nr | yes |
| BMD/BF/VDD F1 | II-4, II-5 | TRPV6 | HRPTTN | AR | nr | nr | nr | nr | nr | yes | yes | nr |
| II-4, II-5, II-7, II-9, II-12, II-13 (II-8 only IRS1) | IRS1 | NIDDM | RF | nr | yes | yes | yes | nr | yes | yes | yes | |
| KL | HFTC3 | nr | nr | yes | yes | nr | nr | yes | yes | nr | ||
| TRPV5 | nr | nr | nr | nr | nr | nr | nr | yes | yes | nr | ||
| BMD/BF/VDD F2 | II-5 | LRP4 | CMS17 | AR | yes | yes | yes | yes | nr | yes | nr | yes |
| RUNX1 | FPDMM | AD | nr | yes | yes | yes | nr | yes | yes | nr | ||
| MVP F1 | II-4, II-5, II-7, II-8, II-9, II-12 | DCHS1* | MVP2 | AD | yes | nr | nr | yes | nr | nr | nr | nr |
| AR-ERT F1 | II-5 | FAM26F | nr | nr | nr | nr | yes | nr | nr | nr | nr | nr |
| Type of Analysis | Family | Gene | avsnp147 | Accession Number | Exonic Function | Nucleotide Change | AA Change | MAF Max | CADD Phred | SIFT | PolyPhen2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity score > 20 | F1 and F2 | RILP | rs61735419 | NM_031430 | NSV | c.G850A | p.G284S | 0.04 | 27.4 | T | D |
| F1 | FNIP2 | rs148251675 | NM_020840 | NSV | c.T901C | p.S301P | 0.02 | 23 | T | T | |
| F2 | FNIP2 | rs62001914 | NM_020840 | NSV | c.C1653A | p.S551R | 0.03 | 25 | D | T | |
| F2 | TRAPPC11 | rs62617790 | NM_021942 | NSV | c.G2799C | p.Q933H | 0.13 | 23 | D | D | |
| BMI | F1 | PLIN2 | rs759915698 | NM_001122 | NSV | c.C925T | p.R309C | 0.00003 | 24 | D | D |
| F2 | PLIN2 | nr | NM_001122 | NSV | c.G616A | p.E206K | na | 27 | D | D | |
| BMD/BF/VDD | F1 | TRPV6 | rs778016744 | NM_018646 | NSV | c.G1886A | p.R629Q | 0.000008 | 24 | T | P |
| F1 | TRPV5 | nr | NM_019841 | NSV | c.C1894T | p.R632W | na | 33 | D | D | |
| F1 | KL | rs9536314 | NM_004795 | NSV | c.T1054G | p.F352V | 0.19 | 27 | D | D | |
| F1 and F2 | IRS1 | rs1801278 | NM_005544 | NSV | c.G2911A | p.G971R | 0.08 | 24 | T | P | |
| F2 | LRP4 | rs72897663 | NM_002334 | NSV | c.A1501C | p.N501H | 0.07 | 21 | T | T | |
| F2 | LRP4 | rs117936904 | NM_002334 | NSV | c.T5165A | p.L1722H | 0.02 | 29 | D | D | |
| F2 | RUNX1 | nr | NM_001754 | NSV | c.G190A | p.G64S | na | 23 | T | P | |
| MVP | F1 | DCHS1* | rs35599968 | NM_003737 | NSV | c.G8480C | p.R2827P | 0.10 | 20 | T | T |
| F1 | DCHS1* | rs376287018 | NM_003737 | NFins | c.99_100insCTG | p.G34insLG | 0.10 | nr | nr | nr | |
| AR-ERT F1 | F1 | FAM26F | rs117361304 | NM_001010919 | splicing | c.525 + 2T > G | nr | 0.09 | 25.4 | nr | nr |
| VDR | GC/DBP | |||||||
|---|---|---|---|---|---|---|---|---|
| ID | Sex | Age | AAO | VDD (ng/mL) | rs2228570 c.T2C p.M1T FokI Variant | rs7975232 G/T ApaI | rs731236 c.T1056C p.I352I TaqI Variant | rs4588 c.C1307A p.T436K |
| F1 II-4 | F | 67 | 50 | 6.64 | CC | GT | TC | CC |
| F1 II-5 | F | 65 | 53 | 11.6 | CC | GG | TT | CC |
| F1 II-7 | M | 61 | 42 | 28.4 | CC | GG | TT | CC |
| F1 II-8 | F | 59 | 47 | 18.5 | CC | GT | TC | CC |
| F1 II-9 | M | 58 | 38 | 12.7 | TC | GT | TC | CC |
| F1 II-12 | F | 51 | 41 | 40.2 | TC | TT | CC | CC |
| F1 II-13 | M | 50 | 36 | 41.3 | CC | GT | TC | CC |
| F2 II-1 | M | 51 | 44 | 8.90 | CC | TT | TC | CC |
| F2 II-3 | F | 48 | 41 | 30.1 | CC | TT | TT | CA |
| F2 II-4 | M | 46 | 38 | 8.4 | CC | TT | CC | CC |
| F2 II-5 | F | 41 | 29 | 7.11 | CC | TT | TC | CC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napolitano, F.; Bruno, G.; Terracciano, C.; Franzese, G.; Palomba, N.P.; Scotto di Carlo, F.; Signoriello, E.; De Blasiis, P.; Navarro, S.; Gialluisi, A.; et al. Rare Variants in Autophagy and Non-Autophagy Genes in Late-Onset Pompe Disease: Suggestions of Their Disease-Modifying Role in Two Italian Families. Int. J. Mol. Sci. 2021, 22, 3625. https://doi.org/10.3390/ijms22073625
Napolitano F, Bruno G, Terracciano C, Franzese G, Palomba NP, Scotto di Carlo F, Signoriello E, De Blasiis P, Navarro S, Gialluisi A, et al. Rare Variants in Autophagy and Non-Autophagy Genes in Late-Onset Pompe Disease: Suggestions of Their Disease-Modifying Role in Two Italian Families. International Journal of Molecular Sciences. 2021; 22(7):3625. https://doi.org/10.3390/ijms22073625
Chicago/Turabian StyleNapolitano, Filomena, Giorgia Bruno, Chiara Terracciano, Giuseppina Franzese, Nicole Piera Palomba, Federica Scotto di Carlo, Elisabetta Signoriello, Paolo De Blasiis, Stefano Navarro, Alessandro Gialluisi, and et al. 2021. "Rare Variants in Autophagy and Non-Autophagy Genes in Late-Onset Pompe Disease: Suggestions of Their Disease-Modifying Role in Two Italian Families" International Journal of Molecular Sciences 22, no. 7: 3625. https://doi.org/10.3390/ijms22073625