
Disease Modeling and Disease Gene Discovery in Cardiomyopathies: A Molecular Study of Induced Pluripotent Stem Cell Generated Cardiomyocytes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
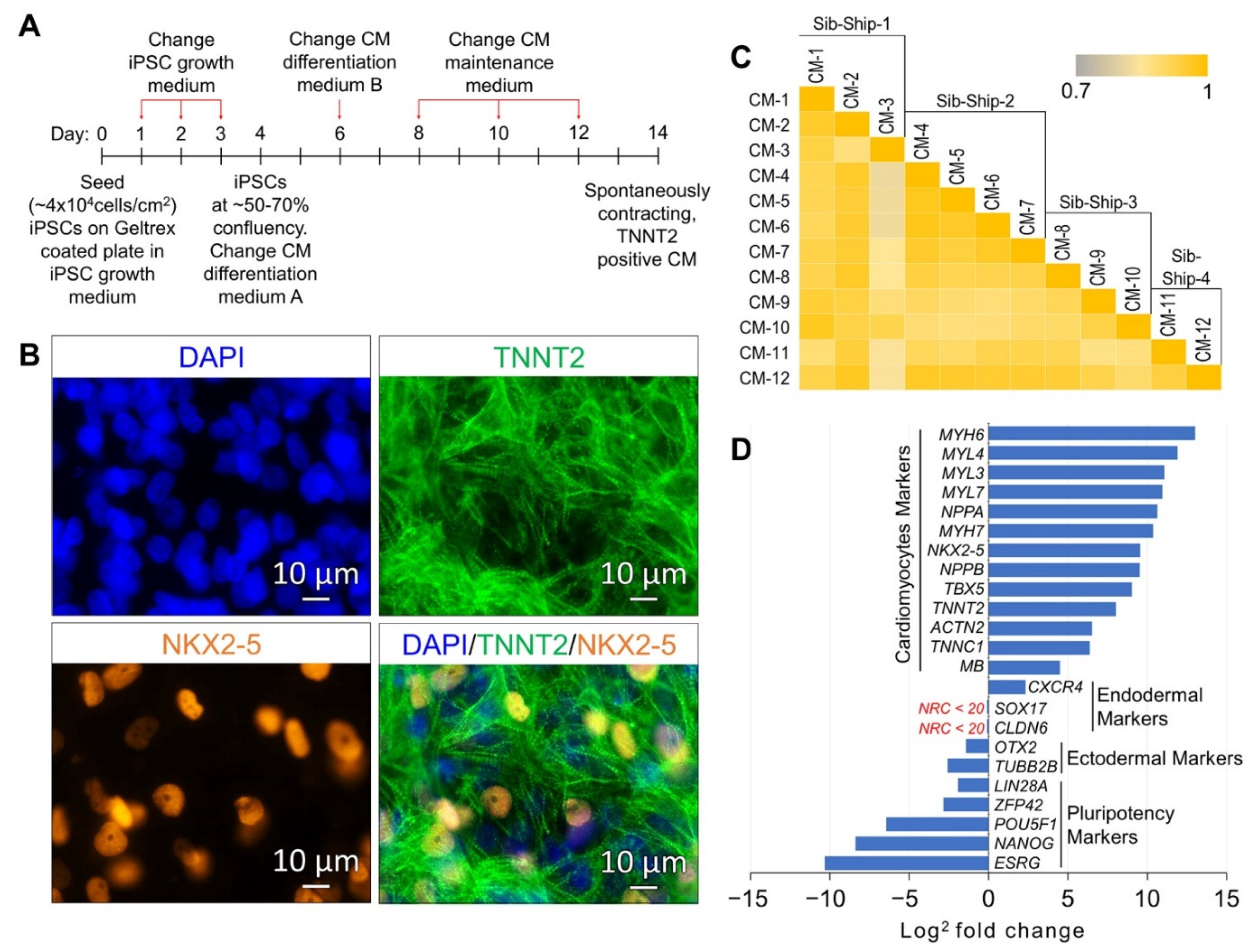
2.1. Differentiation and Lineage-Specific Functional Characteristics of Generated CMs
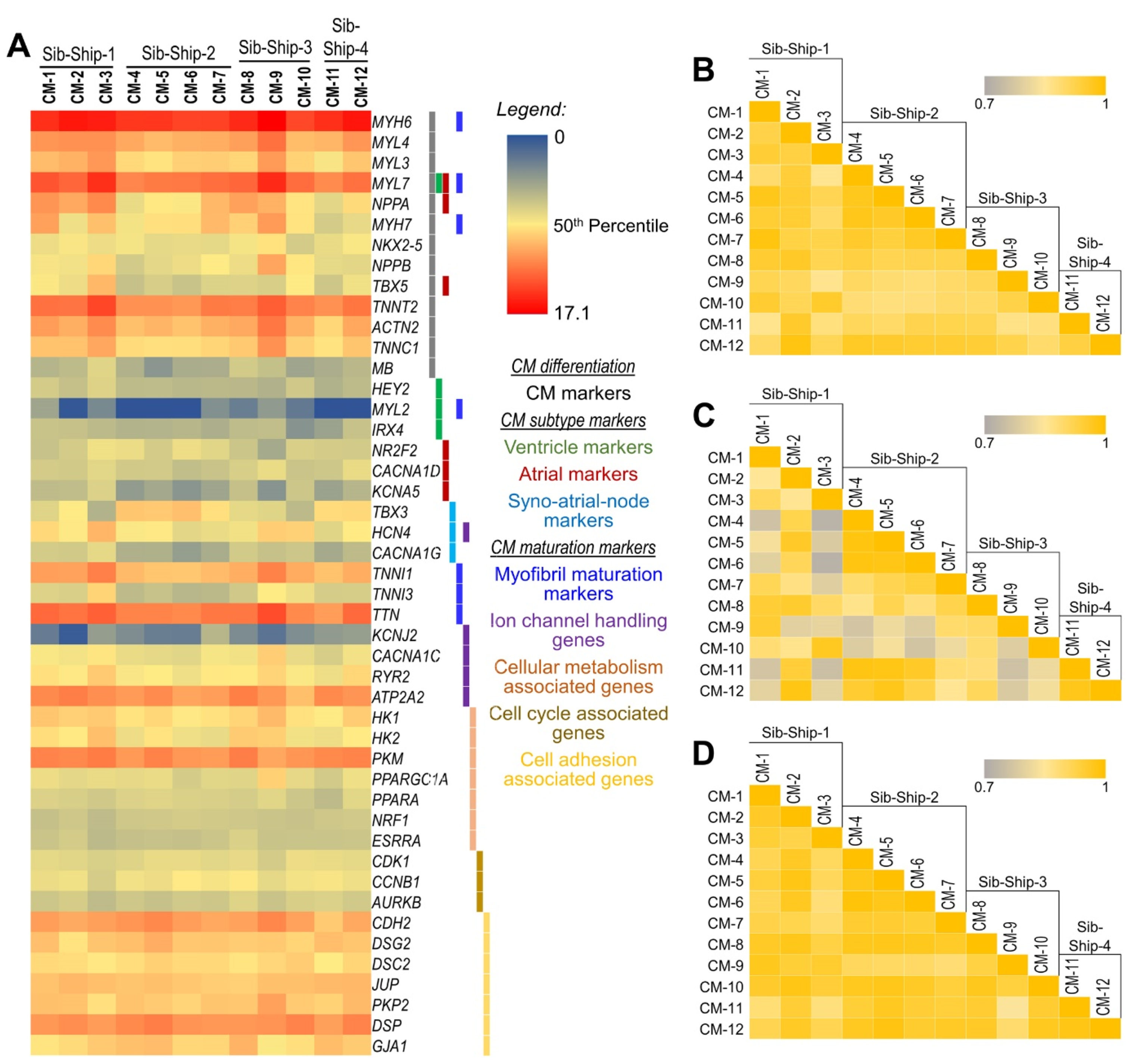
2.2. CM Subtypes and Maturation State
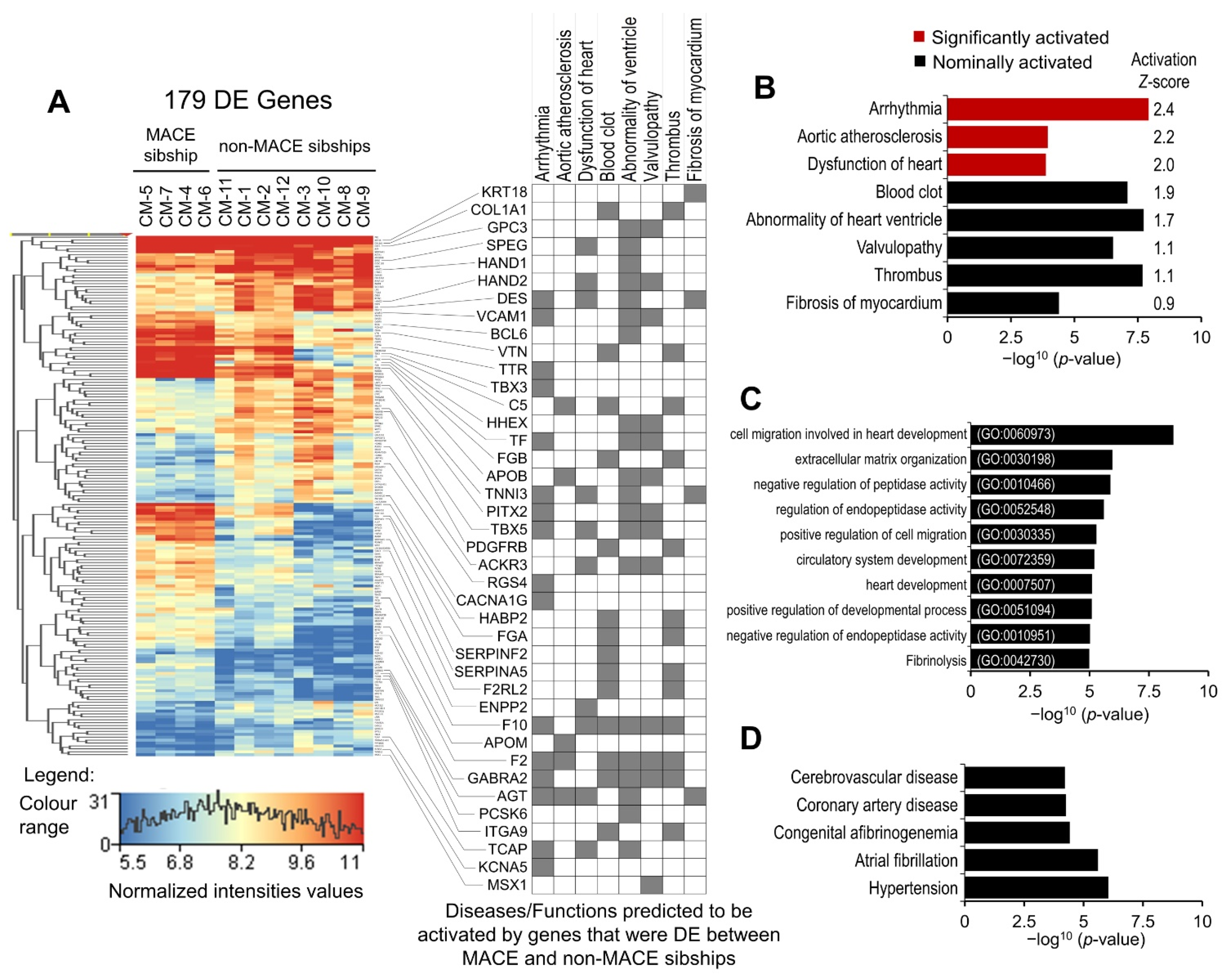
2.3. Differentially Expressed (DE) Genes
2.4. Functional Annotation Analysis of DE Genes
2.5. Modeling the Effect of Major Adverse Cardiac Event (MACE) in Familial Sample
3. Discussion
3.1. Characterization and Disease Modeling Potential of Generated CMs
3.2. MACE-Associated Transcriptomic Changes in Familial CM Samples
4. Materials and Methods
4.1. CMs Differentiation
4.2. Characterization of Generated CMs
4.3. RNA Extraction and Sequencing
4.4. Differential Gene Expression Analyses
4.5. Disease and Function Annotation Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary Definitions and Classification of the Cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Júnior, A.L.; Ferrari, F.; Max, R.; Ritt, L.E.F.; Stein, R. Importance of Genetic Testing in Dilated Cardiomyopathy: Applications and Challenges in Clinical Practice. Arq. Bras. Cardiol. 2019, 113, 274–281. [Google Scholar] [CrossRef]
- Cahill, T.J.; Ashrafian, H.; Watkins, H. Genetic Cardiomyopathies Causing Heart Failure. Circ. Res. 2013, 113, 660–675. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited Cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Monserrat, L.; Hermida, M.; Bouzas, B.; Mosquera, I.; Peteiro, J.; Álvarez, N.; Penas-Lado, M.; Crespo, M.; Castro-Beiras, A.; Mahon, N. Miocardiopatía dilatada familiar en pacientes trasplantados por miocardiopatía dilatada idiopática. Rev. Española Cardiol. 2002, 55, 725–732. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lowe, A.M.; Colan, S.D.; Sleeper, L.A.; Orav, E.J.; Clunie, S.; Messere, J.; Cox, G.F.; Lurie, P.R.; Hsu, D.; et al. Incidence, Causes, and Outcomes of Dilated Cardiomyopathy in Children. JAMA 2006, 296, 1867–1876. [Google Scholar] [CrossRef]
- Lee, D.S.; Pencina, M.J.; Benjamin, E.J.; Wang, T.J.; Levy, D.; O’Donnell, C.J.; Nam, B.-H.; Larson, M.G.; D’Agostino, R.B.; Vasan, R.S. Association of Parental Heart Failure with Risk of Heart Failure in Offspring. N. Engl. J. Med. 2006, 355, 138–147. [Google Scholar] [CrossRef]
- Brandão, K.O.; Tabel, V.A.; Atsma, D.E.; Mummery, C.L.; Davis, R.P. Human pluripotent stem cell models of cardiac disease: From mechanisms to therapies. Dis. Model. Mech. 2017, 10, 1039–1059. [Google Scholar] [CrossRef]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Insights into Molecular, Cellular, and Functional Phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- Pálóczi, J.; Szántai, Á.; Kobolák, J.; Bock, I.; Ruivo, E.; Kiss, B.; Gáspár, R.; Pipis, J.; Ocsovszki, I.; Táncos, Z.; et al. Systematic analysis of different pluripotent stem cell-derived cardiac myocytes as potential testing model for cardiocytoprotection. Vasc. Pharmacol. 2020, 133-134, 106781. [Google Scholar] [CrossRef]
- Kumar, S.; Blangero, J.; Curran, J.E. Induced Pluripotent Stem Cells in Disease Modeling and Gene Identification. Methods Mol. Biol. 2018, 1706, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Curran, J.E.; Espinosa, E.C.; Glahn, D.C.; Blangero, J. Highly efficient induced pluripotent stem cell reprogramming of cryopreserved lymphoblastoid cell lines. J. Biol. Methods 2020, 7, 124. [Google Scholar] [CrossRef]
- Kumar, S.; Curran, J.E.; Glahn, D.C.; Blangero, J. Utility of Lymphoblastoid Cell Lines for Induced Pluripotent Stem Cell Generation. Stem Cells Int. 2016, 2016, 2349261. [Google Scholar] [CrossRef]
- Cai, W.-F.; Kang, K.; Huang, W.; Liang, J.-L.; Feng, Y.-L.; Liu, G.-S.; Chang, D.-H.; Wen, Z.-L.; Paul, C.; Xu, M.; et al. CXCR4 attenuates cardiomyocytes mitochondrial dysfunction to resist ischaemia-reperfusion injury. J. Cell. Mol. Med. 2015, 19, 1825–1835. [Google Scholar] [CrossRef]
- Ceholski, D.K.; Turnbull, I.C.; Pothula, V.; Lecce, L.; Jarrah, A.A.; Kho, C.; Lee, A.; Hadri, L.; Costa, K.D.; Hajjar, R.J.; et al. CXCR4 and CXCR7 play distinct roles in cardiac lineage specification and pharmacologic β-adrenergic response. Stem Cell Res. 2017, 23, 77–86. [Google Scholar] [CrossRef]
- Wang, E.R.; A Jarrah, A.; Benard, L.; Chen, J.; Schwarzkopf, M.; Hadri, L.; Tarzami, S.T. Deletion of CXCR4 in cardiomyocytes exacerbates cardiac dysfunction following isoproterenol administration. Gene Ther. 2014, 21, 496–506. [Google Scholar] [CrossRef]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194.e4. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Pletscher-Frankild, S.; Pallejà, A.; Tsafou, K.; Binder, J.X.; Jensen, L.J. DISEASES: Text mining and data integration of disease–gene associations. Methods 2015, 74, 83–89. [Google Scholar] [CrossRef]
- Sharma, A.; Li, G.; Rajarajan, K.; Hamaguchi, R.; Burridge, P.W.; Wu, S.M. Derivation of Highly Purified Cardiomyocytes from Human Induced Pluripotent Stem Cells Using Small Molecule-modulated Differentiation and Subsequent Glucose Starvation. J. Vis. Exp. 2015, 18, 52628. [Google Scholar] [CrossRef]
- Schneider, M.D. Upstairs, Downstairs: Atrial and Ventricular Cardiac Myocytes from Human Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 151–152. [Google Scholar] [CrossRef]
- Kolanowski, T.J.; Antos, C.L.; Guan, K. Making human cardiomyocytes up to date: Derivation, maturation state and perspectives. Int. J. Cardiol. 2017, 241, 379–386. [Google Scholar] [CrossRef]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Xu, X.Q.; Soo, S.Y.; Sun, W.; Zweigerdt, R. Global Expression Profile of Highly Enriched Cardiomyocytes Derived from Human Embryonic Stem Cells. Stem Cells 2009, 27, 2163–2174. [Google Scholar] [CrossRef]
- Gupta, M.K.; Illich, D.J.; Gaarz, A.; Matzkies, M.; Nguemo, F.; Pfannkuche, K.; Liang, H.; Classen, S.; Reppel, M.; Schultze, J.L.; et al. Global transcriptional profiles of beating clusters derived from human induced pluripotent stem cells and embryonic stem cells are highly similar. BMC Dev. Biol. 2010, 10, 98. [Google Scholar] [CrossRef]
- Synnergren, J.; Åkesson, K.; Dahlenborg, K.; Vidarsson, H.; Améen, C.; Steel, D.; Lindahl, A.; Olsson, B.; Sartipy, P. Molecular Signature of Cardiomyocyte Clusters Derived from Human Embryonic Stem Cells. Stem Cells 2008, 26, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Cooke, M.P.; Walker, J.R.; HogenEsch, J.B.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Kubalak, S.; Miller-Hance, W.; O’Brien, T.; Dyson, E.; Chien, K. Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J. Biol. Chem. 1994, 269, 16961–16970. [Google Scholar] [CrossRef]
- O’Brien, T.X.; Lee, K.J.; Chien, K.R. Positional specification of ventricular myosin light chain 2 expression in the primitive murine heart tube. Proc. Natl. Acad. Sci. USA 1993, 90, 5157–5161. [Google Scholar] [CrossRef]
- Tanwar, V.; Bylund, J.B.; Hu, J.; Yan, J.; Walthall, J.M.; Mukherjee, A.; Heaton, W.H.; Wang, W.-D.; Potet, F.; Rai, M.; et al. Gremlin 2 promotes differentiation of embryonic stem cells to atrial fate by activation of the JNK signaling pathway. Stem Cells 2014, 32, 1774–1788. [Google Scholar] [CrossRef] [PubMed]
- Koibuchi, N.; Chin, M.T. CHF1/Hey2 Plays a Pivotal Role in Left Ventricular Maturation through Suppression of Ectopic Atrial Gene Expression. Circ. Res. 2007, 100, 850–855. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Van Rooij, E.; Qi, X.; Richardson, J.A.; Srivastava, D.; Nakagawa, O.; Olson, E.N. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc. Natl. Acad. Sci. USA 2007, 104, 7975–7980. [Google Scholar] [CrossRef]
- Fischer, A.; Gessler, M. Hey genes in cardiovascular development. Trends Cardiovasc. Med. 2003, 13, 221–226. [Google Scholar] [CrossRef]
- Kokubo, H.; Tomita-Miyagawa, S.; Hamada, Y.; Saga, Y. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Development 2007, 134, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Wong, C.K.; Tsang, S.Y. Differential gene expressions in atrial and ventricular myocytes: Insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. Physiol. 2010, 299, C1234–C1249. [Google Scholar] [CrossRef]
- Tu, C.; Chao, B.S.; Wu, J.C. Strategies for Improving the Maturity of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2018, 123, 512–514. [Google Scholar] [CrossRef]
- Kannan, S.; Kwon, C. Regulation of cardiomyocyte maturation during critical perinatal window. J. Physiol. 2019, 598, 2941–2956. [Google Scholar] [CrossRef]
- Huang, C.Y.; Maia-Joca, R.P.M.; Ong, C.S.; Wilson, I.; DiSilvestre, D.; Tomaselli, G.F.; Reich, D.H. Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J. Mol. Cell. Cardiol. 2020, 138, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson-Bouchard, K.; Ma, S.P.; Yeager, K.; Chen, T.; Song, L.; Sirabella, D.; Morikawa, K.; Teles, D.; Yazawa, M.; Vunjak-Novakovic, G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nat. Cell Biol. 2018, 556, 239–243. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, G.J.; Butcher, J. Naturally Engineered Maturation of Cardiomyocytes. Front. Cell Dev. Biol. 2017, 5, 50. [Google Scholar] [CrossRef]
- Goversen, B.; van der Heyden, M.A.; van Veen, T.A.; de Boer, T.P.; van der Heyden, M.A.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on I K1. Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Boutjdir, M. Gene Expression of SERCA2a and L- and T-type Ca Channels during Human Heart Development. Pediatr. Res. 2001, 50, 569–574. [Google Scholar] [CrossRef]
- Ii, G.W.D.; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Sim, C.B.; Ziemann, M.; Kaspi, A.; Harikrishnan, K.N.; Ooi, J.; Khurana, I.; Chang, L.; Hudson, J.E.; El-Osta, A.; Porrello, E.R. Dynamic changes in the cardiac methylome during postnatal development. FASEB J. 2014, 29, 1329–1343. [Google Scholar] [CrossRef]
- Uosaki, H.; Cahan, P.; Lee, D.I.; Wang, S.; Miyamoto, M.; Fernandez, L.; Kass, D.A.; Kwon, C. Transcriptional Landscape of Cardiomyocyte Maturation. Cell Rep. 2015, 13, 1705–1716. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Ang, Y.-S.; Radzinsky, E.; Zhou, P.; Huang, Y.; Elfenbein, A.; Foley, A.; Magnitsky, S.; Srivastava, D. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018, 173, 104–116.e12. [Google Scholar] [CrossRef]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta Biomembr. 2018, 1860, 40–47. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; Van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpón, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 Insufficiency Leads to Atrial Electrical and Structural Remodeling Linked to Arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef]
- Pérez-Hernández, M.; Matamoros, M.; Barana, A.; Amorós, I.; Gómez, R.; Núñez, M.; Sacristán, S.; Pinto, Á.; Fernández-Avilés, F.; Tamargo, J.; et al. Pitx2c increases in atrial myocytes from chronic atrial fibrillation patients enhancing IKs and decreasing ICa,L. Cardiovasc. Res. 2015, 109, 431–441. [Google Scholar] [CrossRef]
- Syeda, F.; Holmes, A.P.; Yu, T.Y.; Tull, S.; Kuhlmann, S.M.; Pavlovic, D.; Betney, D.; Riley, G.; Kucera, J.P.; Jousset, F.; et al. PITX2 Modulates Atrial Membrane Potential and the Antiarrhythmic Effects of Sodium-Channel Blockers. J. Am. Coll. Cardiol. 2016, 68, 1881–1894. [Google Scholar] [CrossRef]
- Syeda, F.; Kirchhof, P.; Fabritz, L. PITX2 -dependent gene regulation in atrial fibrillation and rhythm control. J. Physiol. 2017, 595, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; Boogaard, M.V.D.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5 -dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8, 354ra115. [Google Scholar] [CrossRef]
- Hilton, T.; Gross, M.K.; Kioussi, C. Pitx2-dependent Occupancy by Histone Deacetylases Is Associated with T-box Gene Regulation in Mammalian Abdominal Tissue*. J. Biol. Chem. 2010, 285, 11129–11142. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.; Wang, J.; Li, X.; Cao, H.; Gao, S.; Moreno, M.; Amendt, B.A. A Pituitary Homeobox 2 (Pitx2):microRNA-200a-3p:β-catenin Pathway Converts Mesenchymal Cells to Amelogenin-expressing Dental Epithelial Cells. J. Biol. Chem. 2014, 289, 27327–27341. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Cabrera, C.P.; Finlay, M.; Lall, K.; Nobles, M.; Schilling, R.J.; Wood, K.; Mein, C.A.; Barnes, M.R.; Munroe, P.B.; et al. Differentially expressed genes for atrial fibrillation identified by RNA sequencing from paired human left and right atrial appendages. Physiol. Genom. 2019, 51, 323–332. [Google Scholar] [CrossRef]
- Christophersen, I.E.; Olesen, M.S.; Liang, B.; Andersen, M.N.; Larsen, A.P.; Nielsen, J.B.; Haunsø, S.; Olesen, S.-P.; Tveit, A.; Svendsen, J.H.; et al. Genetic variation in KCNA5: Impact on the atrial-specific potassium current IKur in patients with lone atrial fibrillation. Eur. Hearth J. 2012, 34, 1517–1525. [Google Scholar] [CrossRef]
- Colman, M.A.; Ni, H.; Liang, B.; Schmitt, N.; Zhang, H. In silico assessment of genetic variation in KCNA5 reveals multiple mechanisms of human atrial arrhythmogenesis. PLoS Comput. Biol. 2017, 13, e1005587. [Google Scholar] [CrossRef]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef]
- Yang, T.; Yang, P.; Roden, D.M.; Darbar, D. Novel KCNA5 mutation implicates tyrosine kinase signaling in human atrial fibrillation. Heart Rhythm 2010, 7, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, J.; Lin, X.; Yang, Y.; Hong, K.; Wang, L.; Liu, J.; Li, L.; Yan, D.; Liang, D.; et al. Novel KCNA5 loss-of-function mutations responsible for atrial fibrillation. J. Hum. Genet. 2009, 54, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bai, Y.; Li, N.; Ye, W.; Zhang, M.; Greene, S.B.; Tao, Y.; Chen, Y.; Wehrens, X.H.T.; Martin, J.F. Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 9181–9186. [Google Scholar] [CrossRef]
- Wang, J.; Klysik, E.; Sood, S.; Johnson, R.L.; Wehrens, X.H.T.; Martin, J.F. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. USA 2010, 107, 9753–9758. [Google Scholar] [CrossRef]
- Bakker, M.L.; Boink, G.J.J.; Boukens, B.J.; Verkerk, A.O.; van den Boogaard, M.; den Haan, A.D.; Hoogaars, W.M.H.; Buermans, H.P.; De Bakker, J.M.T.; Seppen, J.M.; et al. T-box transcription factor TBX3 reprogrammes mature cardiac myocytes into pacemaker-like cells. Cardiovasc. Res. 2012, 94, 439–449. [Google Scholar] [CrossRef]
- DeHaan, R.L.; Eichna, L.W. Differentiation of the Atrioventricular Conducting System of the Heart. Circulation 1961, 24, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.M.H.; Engel, A.; Brons, J.F.; Verkerk, A.O.; De Lange, F.J.; Wong, L.Y.E.; Bakker, M.L.; Clout, D.E.; Wakker, V.; Barnett, P.; et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007, 21, 1098–1112. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, J.K.; Ohgi, M.; Koshiba-Takeuchi, K.; Shiratori, H.; Sakaki, I.; Ogura, K.; Saijoh, Y.; Ogura, T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development 2003, 130, 5953–5964. [Google Scholar] [CrossRef]
- Franco, D.; Sedmera, D.; Lozano-Velasco, E. Multiple Roles of Pitx2 in Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2017, 4, 16. [Google Scholar] [CrossRef]
- Cai, H.; Li, Z.; Goette, A.; Mera, F.; Honeycutt, C.; Feterik, K.; Wilcox, J.N.; Dudley, S.C.; Harrison, D.G.; Langberg, J.J. Downregulation of Endocardial Nitric Oxide Synthase Expression and Nitric Oxide Production in Atrial Fibrillation. Circulation 2002, 106, 2854–2858. [Google Scholar] [CrossRef]
- Ferro, D.; Loffredo, L.; Polimeni, L.; Fimognari, F.; Villari, P.; Pignatelli, P.; Fuster, V.; Violi, F. Soluble CD40 Ligand Predicts Ischemic Stroke and Myocardial Infarction in Patients with Nonvalvular Atrial Fibrillation. Arter. Thromb. Vasc. Biol. 2007, 27, 2763–2768. [Google Scholar] [CrossRef]
- Iwasaki, Y.-K.; Nishida, K.; Kato, T.; Nattel, S. Atrial Fibrillation Pathophysiology. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- Li-Saw-Hee, F.; Blann, A.; Gurney, D.; Lip, G. Plasma von Willebrand factor, fibrinogen and soluble P-selectin levels in paroxysmal, persistent and permanent atrial fibrillation. Effects of cardioversion and return of left atrial function. Eur. Heart J. 2001, 22, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Chiba, K.; Iwasaki, Y.-K.; Katsouras, G.; Shi, Y.-F.; Blostein, M.D.; Khairy, P.; Guerra, P.G.; Dubuc, M.; Tardif, J.-C.; et al. Atrial Fibrillation-Associated Remodeling Does Not Promote Atrial Thrombus Formation in Canine Models. Circ. Arrhythmia Electrophysiol. 2012, 5, 1168–1175. [Google Scholar] [CrossRef]
- Violi, F.; Loffredo, L. Thromboembolism or Atherothromboembolism in Atrial Fibrillation? Circ. Arrhythmia Electrophysiol. 2012, 5, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Developed with the special contribution of the European Heart Rhythm Association (EHRA); Camm, A.J.; Kirchhof, P.; Lip, G.Y.; Schotten, U.; Savelieva, I.; Ernst, S.; Van Gelder, I.C.; Al-Attar, N.; Hindricks, G.; et al. Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur. Heart J. 2010, 31, 2369–2429. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Biological Process | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| Heart development (GO:0007507) | 66/149 | 4.09 × 10−26 | 2.09 × 10−22 |
| Extracellular matrix organization (GO:0030198) | 75/229 | 6.62 × 10−20 | 1.69 × 10−16 |
| Cardiac muscle tissue morphogenesis (GO:0055008) | 27/36 | 1.43 × 10−19 | 2.43 × 10−16 |
| Circulatory system development (GO:0072359) | 48/109 | 3.09 × 10−19 | 3.94 × 10−16 |
| Ventricular cardiac muscle tissue morphogenesis (GO:0055010) | 24/34 | 1.56 × 10−16 | 1.60 × 10−13 |
| Muscle contraction (GO:0006936) | 49/137 | 3.45 × 10−15 | 2.94 × 10−12 |
| Myofibril assembly (GO:0030239) | 26/47 | 5.10 × 10−14 | 3.72 × 10−11 |
| Cardiac ventricle morphogenesis (GO:0003208) | 24/43 | 3.69 × 10−13 | 2.35 × 10−10 |
| Regulation of heart contraction (GO:0008016) | 36/95 | 2.15 × 10−12 | 1.22 × 10−9 |
| cardiac atrium morphogenesis (GO:0003209) | 11/17 | 1.22 × 10−7 | 1.27 × 10−5 |
| Human Tissue/Cell Types | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| Human Gene Atlas (up-regulated genes in human tissues from BioGPS) | |||
| Uterus | 94/405 | 5.11 × 10−13 | 2.15 × 10−11 |
| Placenta | 58/201 | 1.17 × 10−13 | 9.85 × 10−12 |
| Heart | 88/415 | 1.23 × 10−10 | 3.43 × 10−9 |
| Smooth Muscle | 78/363 | 7.28 × 10−10 | 1.53 × 10−8 |
| Cardiac Myocytes | 62/273 | 4.36 × 10−9 | 7.33 × 10−8 |
| GTEx Tissue Samples (up-regulated genes) | |||
| GTEX-X4EP-0326-SM-3P5Z6 heart female 60–69 years | 210/755 | 1.37 × 10−41 | 4.00 × 10−38 |
| GTEX-X62O-0826-SM-46MW8 heart male 50–59 years | 208/766 | 1.79 × 10−39 | 2.62 × 10−36 |
| GTEX-SE5C-0626-SM-2XCDV heart female 40–49 years | 229/906 | 4.75 × 10−38 | 4.62 × 10−35 |
| GTEX-S3XE-0426-SM-3K2AC heart male 50–59 years | 163/547 | 2.78 × 10−36 | 2.03 × 10−33 |
| GTEX-WY7C-1126-SM-3NB3A heart male 50–59 years | 176/637 | 1.88 × 10−34 | 1.09 × 10−31 |
| Diseases | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| ClinVar (2019) Gene Sets | |||
| Cardiomyopathy | 33/48 | 1.13 × 10−21 | 2.06 × 10−19 |
| Primary dilated cardiomyopathy | 23/34 | 2.94 × 10−15 | 2.67 × 10−13 |
| Primary familial hypertrophic cardiomyopathy | 18/22 | 1.24 × 10−14 | 7.54 × 10−13 |
| Familial dilated cardiomyopathy | 21/30 | 1.66 × 10−14 | 7.53 × 10−13 |
| Myofibrillar myopathy | 5/6 | 7.22 × 10−5 | 2.19 × 10−3 |
| COVID-19 Related Cardiomyocyte Gene Set | |||
| Down-regulated by SARS-CoV-2 in cardiomyocytes from GSE150392 | 143/499 | 6.53 × 10−30 | 1.34 × 10−27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Curran, J.E.; Kumar, K.; DeLeon, E.; Leandro, A.C.; Peralta, J.; Williams-Blangero, S.; Blangero, J. Disease Modeling and Disease Gene Discovery in Cardiomyopathies: A Molecular Study of Induced Pluripotent Stem Cell Generated Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 3311. https://doi.org/10.3390/ijms22073311
Kumar S, Curran JE, Kumar K, DeLeon E, Leandro AC, Peralta J, Williams-Blangero S, Blangero J. Disease Modeling and Disease Gene Discovery in Cardiomyopathies: A Molecular Study of Induced Pluripotent Stem Cell Generated Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(7):3311. https://doi.org/10.3390/ijms22073311
Chicago/Turabian StyleKumar, Satish, Joanne E. Curran, Kashish Kumar, Erica DeLeon, Ana C. Leandro, Juan Peralta, Sarah Williams-Blangero, and John Blangero. 2021. "Disease Modeling and Disease Gene Discovery in Cardiomyopathies: A Molecular Study of Induced Pluripotent Stem Cell Generated Cardiomyocytes" International Journal of Molecular Sciences 22, no. 7: 3311. https://doi.org/10.3390/ijms22073311