
How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease

 , ,
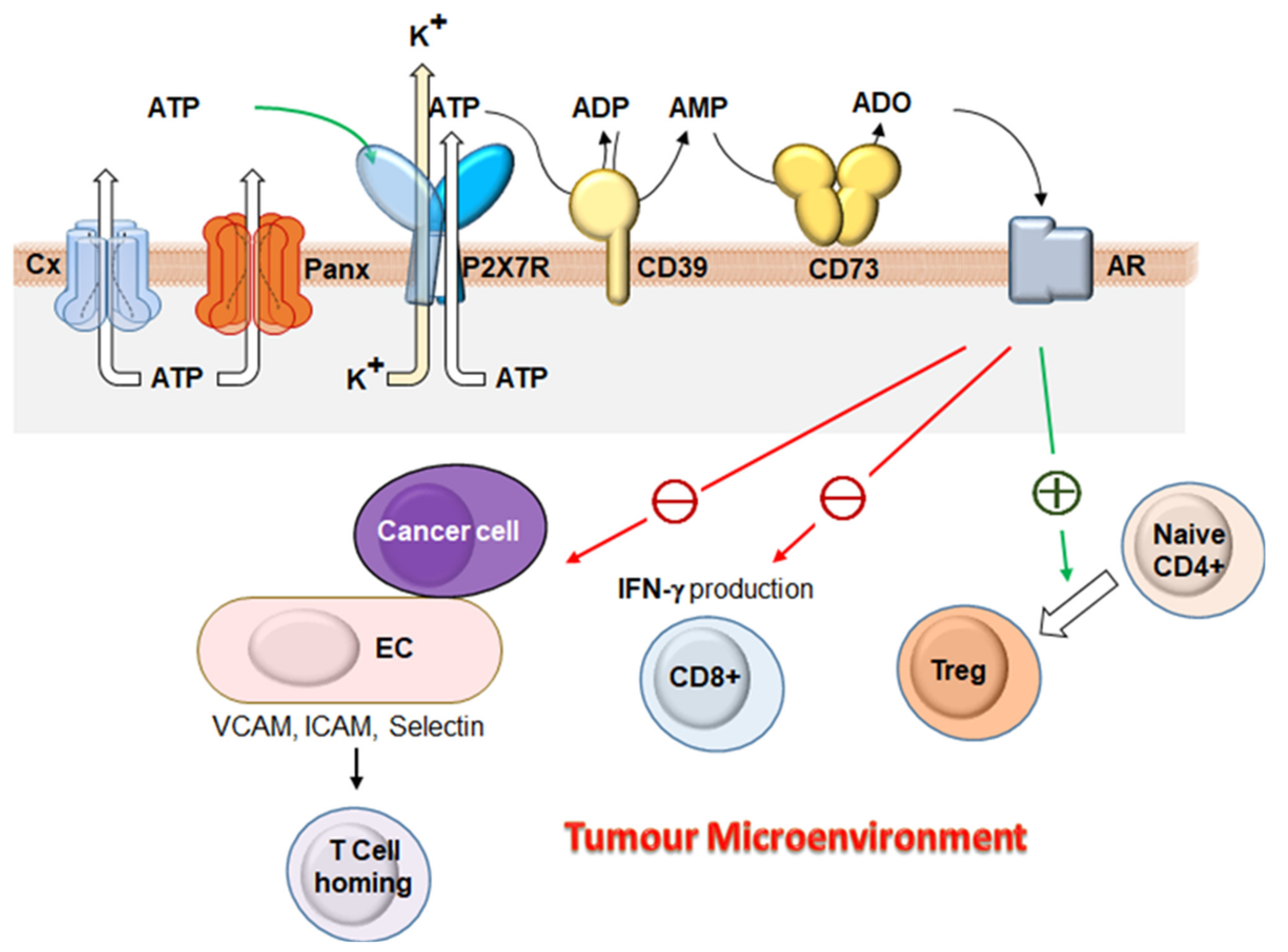
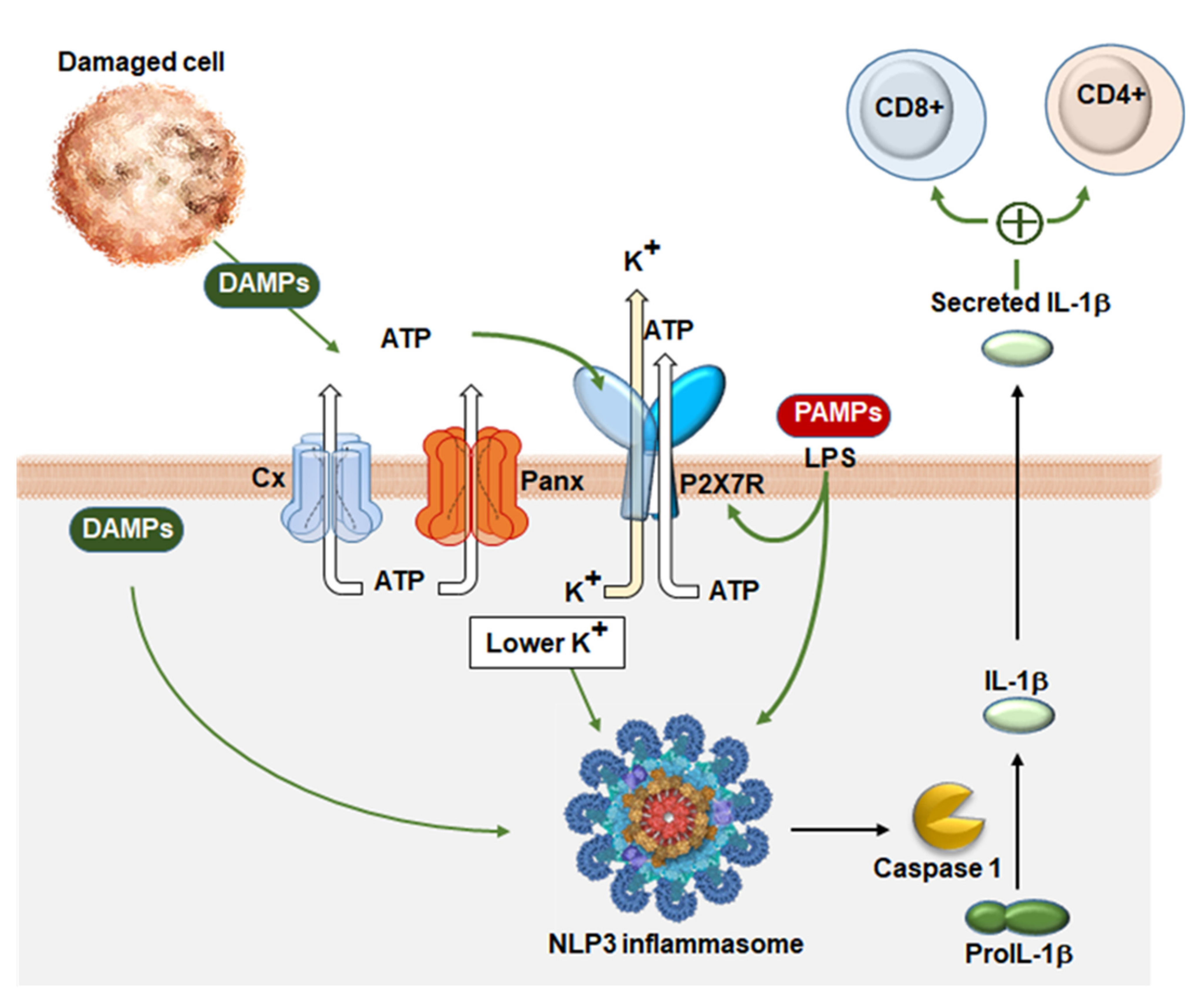
, , 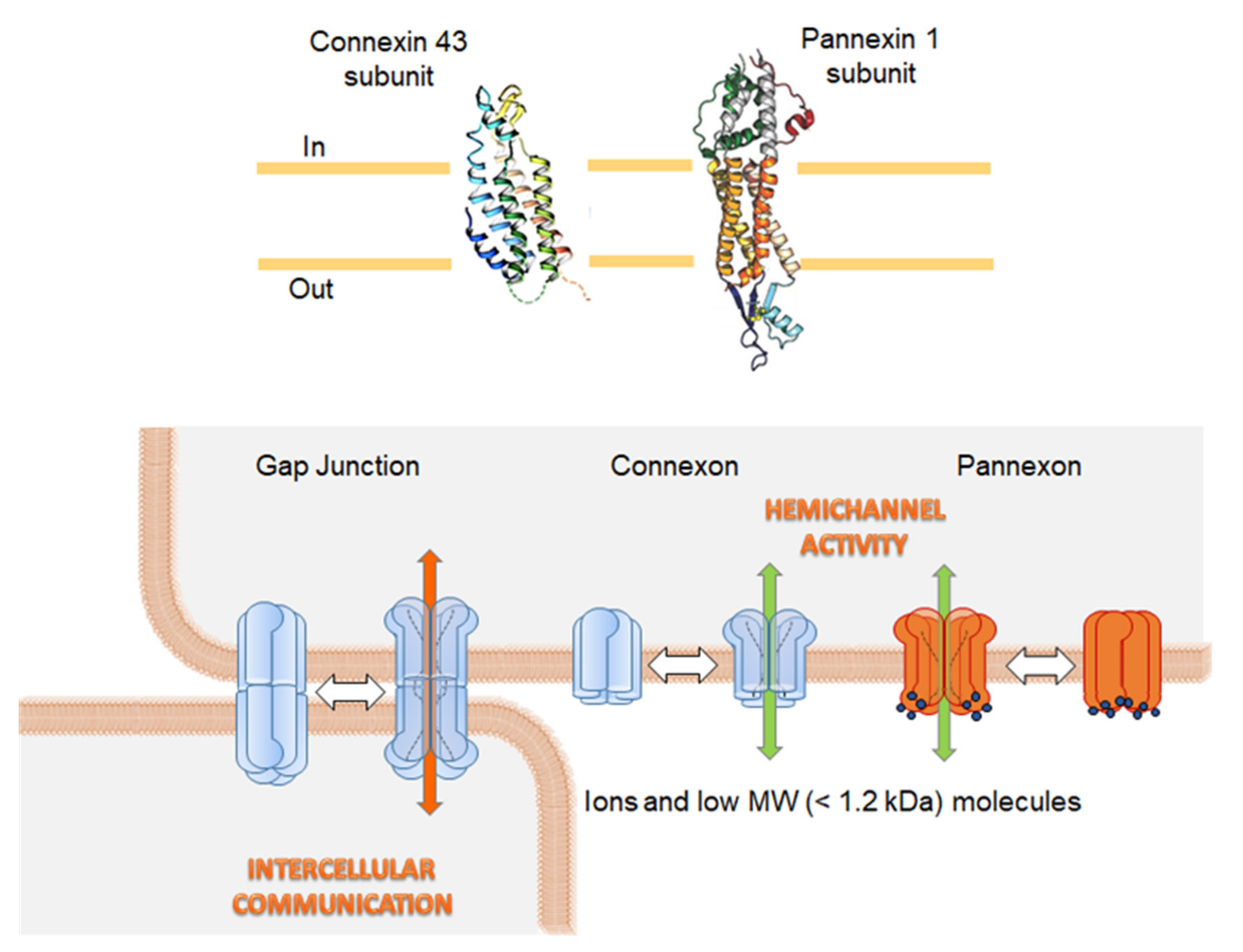{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Search Strategy
3. Activating Invasion and Metastasis
3.1. Role of GJIC and HCs in Cancer Cell Invasiveness
3.2. Role of GJIC and HCs in the TME ATP/Adenosine Modulation
4. Avoiding Immune Destruction
4.1. Immune Evasion and Cell Deaths: Therapeutic Strategies
4.2. The Role of Cytokines and GJIC in the TME
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54–86. [Google Scholar] [CrossRef] [PubMed]
- Garber, J.E.; Offit, K. Hereditary cancer predisposition syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Nishida-Aoki, N.; Gujral, T.S. Emerging approaches to study cell-cell interactions in tumor microenvironment. Oncotarget 2019, 10, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totland, M.Z.; Rasmussen, N.L.; Knudsen, L.M.; Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: Physiological and pathophysiological implications. Cell Mol. Life Sci. 2020, 77, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. Introduction: Connexins, pannexins and their channels as gatekeepers of organ physiology. Cell Mol. Life Sci. 2015, 72, 2775–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, S.R.; Naus, C.C. The pannexins: Past and present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchin, Y.; Kelmanson, I.; Matz, M.; Lukyanov, K.; Usman, N.; Lukyanov, S. A ubiquitous family of putative gap junction molecules. Curr. Biol. 2000, 10, R473–R474. [Google Scholar] [CrossRef] [Green Version]
- Penuela, S.; Bhalla, R.; Gong, X.Q.; Cowan, K.N.; Celetti, S.J.; Cowan, B.J.; Bai, D.; Shao, Q.; Laird, D.W. Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 2007, 120, 3772–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Hondt, C.; Ponsaerts, R.; De Smedt, H.; Bultynck, G.; Himpens, B. Pannexins, distant relatives of the connexin family with specific cellular functions? Bioessays 2009, 31, 953–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbe, M.T.; Monyer, H.; Bruzzone, R. Cell-cell communication beyond connexins: The pannexin channels. Physiology 2006, 21, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; Hormuzdi, S.G.; Barbe, M.T.; Herb, A.; Monyer, H. Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13644–13649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shestopalov, V.I.; Panchin, Y. Pannexins and gap junction protein diversity. Cell Mol. Life Sci. 2008, 65, 376–394. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Leybaert, L.; Naus, C.C.; Saez, J.C. Connexin and pannexin hemichannels in brain glial cells: Properties, pharmacology, and roles. Front. Pharmacol. 2013, 4, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Sinovas, A.; Sanchez, J.A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Connexin and pannexin as modulators of myocardial injury. Biochim. Biophys. Acta 2012, 1818, 1962–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarenkova, H.P.; Shah, S.B.; Shestopalov, V.I. The two faces of pannexins: New roles in inflammation and repair. J. Inflamm. Res. 2018, 11, 273–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Vanden Abeele, F.; Bidaux, G.; Gordienko, D.; Beck, B.; Panchin, Y.V.; Baranova, A.V.; Ivanov, D.V.; Skryma, R.; Prevarskaya, N. Functional implications of calcium permeability of the channel formed by pannexin 1. J. Cell Biol. 2006, 174, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Langlois, S.; Xiang, X.; Young, K.; Cowan, B.J.; Penuela, S.; Cowan, K.N. Pannexin 1 and pannexin 3 channels regulate skeletal muscle myoblast proliferation and differentiation. J. Biol. Chem. 2014, 289, 30717–30731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarenkova, H.P.; Shestopalov, V.I. The role of pannexin hemichannels in inflammation and regeneration. Front. Physiol. 2014, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zefferino, R.; Piccoli, C.; Gioia, S.D.; Capitanio, N.; Conese, M. Gap Junction Intercellular Communication in the Carcinogenesis Hallmarks: Is This a Phenomenon or Epiphenomenon? Cells 2019, 8, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.V.; Jiang, J.X.; Mesnil, M. Connexins and Pannexins: Important Players in Tumorigenesis, Metastasis and Potential Therapeutics. Int. J. Mol. Sci. 2018, 19, 1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, V.I.; Goldberg, P.L.; Minnear, F.L.; Heimark, R.L.; Vincent, P.A. Rearrangement of adherens junctions by transforming growth factor-beta1: Role of contraction. Am. J. Physiol. 1999, 276, L582–L595. [Google Scholar] [PubMed]
- Shinto, O.; Yashiro, M.; Kawajiri, H.; Shimizu, K.; Shimizu, T.; Miwa, A.; Hirakawa, K. Inhibitory effect of a TGFbeta receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous gastric cancer cells. Br. J. Cancer 2010, 102, 844–851. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, L.; Shi, C.; Sun, H.; Wang, J.; Li, R.; Zou, Z.; Ran, X.; Su, Y. TGF-beta-induced miR-21 negatively regulates the antiproliferative activity but has no effect on EMT of TGF-beta in HaCaT cells. Int. J. Biochem. Cell Biol. 2012, 44, 366–376. [Google Scholar] [CrossRef]
- Zeng, Z.; Sarbassov dos, D.; Samudio, I.J.; Yee, K.W.; Munsell, M.F.; Ellen Jackson, C.; Giles, F.J.; Sabatini, D.M.; Andreeff, M.; Konopleva, M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007, 109, 3509–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiem, S.; Pierce, T.P.; Palmieri, M.; Putoczki, T.L.; Buchert, M.; Preaudet, A.; Farid, R.O.; Love, C.; Catimel, B.; Lei, Z.; et al. mTORC1 inhibition restricts inflammation-associated gastrointestinal tumorigenesis in mice. J. Clin. Investig. 2013, 123, 767–781. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wang, C.; Huang, D.Y.; Zhang, Y.; Xu, J.; Kolesnikov, S.S.; Sung, K.L.; Zhao, H. TGF-beta1 induces the different expressions of lysyl oxidases and matrix metalloproteinases in anterior cruciate ligament and medial collateral ligament fibroblasts after mechanical injury. J. Biomech. 2013, 46, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Pez, F.; Dayan, F.; Durivault, J.; Kaniewski, B.; Aimond, G.; Le Provost, G.S.; Deux, B.; Clezardin, P.; Sommer, P.; Pouyssegur, J.; et al. The HIF-1-inducible lysyl oxidase activates HIF-1 via the Akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 2011, 71, 1647–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, K.; Hall, A. GSK-3beta sets Snail’s pace. Nat. Cell Biol. 2004, 6, 913–915. [Google Scholar] [CrossRef]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Noren, N.K.; Liu, B.P.; Burridge, K.; Kreft, B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 2000, 150, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Muerdter, C.P.; Knickerbocker, A.D.; Walsh, R.M.; Zepeda-Rivera, M.A.; Depner, K.H.; Sangesland, M.; Cisneros, T.B.; Kim, J.Y.; Sanchez-Vazquez, P.; et al. Cdc42 GTPase and Rac1 GTPase act downstream of p120 catenin and require GTP exchange during gastrulation of zebrafish mesoderm. Dev. Dyn. 2012, 241, 1545–1561. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Anastasiadis, P.Z. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J. Cell Biol. 2006, 174, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semina, E.V.; Rubina, K.A.; Rutkevich, P.N.; Voyno-Yasenetskaya, T.A.; Parfyonova, Y.V.; Tkachuk, V.A. T-cadherin activates Rac1 and Cdc42 and changes endothelial permeability. Biochemistry 2009, 74, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, K.; Kulkarni, S.; Du, X.; Goll, D.E.; Saido, T.C.; Fox, J.E. Evidence that beta3 integrin-induced Rac activation involves the calpain-dependent formation of integrin clusters that are distinct from the focal complexes and focal adhesions that form as Rac and RhoA become active. J. Cell Biol. 2000, 151, 685–696. [Google Scholar] [CrossRef]
- Migeotte, I.; Omelchenko, T.; Hall, A.; Anderson, K.V. Rac1-dependent collective cell migration is required for specification of the anterior-posterior body axis of the mouse. PLoS Biol. 2010, 8, e1000442. [Google Scholar] [CrossRef] [Green Version]
- Hills, C.E.; Siamantouras, E.; Smith, S.W.; Cockwell, P.; Liu, K.K.; Squires, P.E. TGFbeta modulates cell-to-cell communication in early epithelial-to-mesenchymal transition. Diabetologia 2012, 55, 812–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, C.C.; Zeitz, M.J.; Calhoun, P.J.; Lamouille, S.; Smyth, J.W. Altered translation initiation of Gja1 limits gap junction formation during epithelial-mesenchymal transition. Mol. Biol. Cell 2018, 29, 797–808. [Google Scholar] [CrossRef]
- Fukuda, S.; Akiyama, M.; Harada, H.; Nakahama, K.I. Effect of gap junction-mediated intercellular communication on TGF-beta induced epithelial-to-mesenchymal transition. Biochem. Biophys. Res. Commun. 2019, 508, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; Pijnappels, D.A.; van Oorschot, A.A.; Winter, E.M.; de Vries, A.A.; van Tuyn, J.; Braun, J.; Maas, S.; Schalij, M.J.; Atsma, D.E.; et al. Epithelial-to-mesenchymal transformation alters electrical conductivity of human epicardial cells. J. Cell Mol. Med. 2011, 15, 2675–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLachlan, E.; Shao, Q.; Wang, H.L.; Langlois, S.; Laird, D.W. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006, 66, 9886–9894. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Luo, Y.; Mao, N.; Huang, G.; Teng, C.; Wang, H.; Wu, J.; Liao, X.; Yang, J. Cancer-Associated Fibroblasts Accelerate Malignant Progression of Non-Small Cell Lung Cancer via Connexin 43-Formed Unidirectional Gap Junctional Intercellular Communication. Cell Physiol. Biochem. 2018, 51, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Katoh, F.; Kataoka, T.R.; Okada, M.; Tsubota, N.; Asada, H.; Yoshikawa, K.; Maeda, S.; Kitamura, Y.; Yamasaki, H.; et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J. Clin. Invest. 2000, 105, 1189–1197. [Google Scholar] [CrossRef] [Green Version]
- Asencio-Barria, C.; Defamie, N.; Saez, J.C.; Mesnil, M.; Godoy, A.S. Direct Intercellular Communications and Cancer: A Snapshot of the Biological Roles of Connexins in Prostate Cancer. Cancers 2019, 11, 1370. [Google Scholar] [CrossRef] [Green Version]
- Tu, M.T.; Luo, S.F.; Wang, C.C.; Chien, C.S.; Chiu, C.T.; Lin, C.C.; Yang, C.M. P2Y(2) receptor-mediated proliferation of C(6) glioma cells via activation of Ras/Raf/MEK/MAPK pathway. Br. J. Pharmacol. 2000, 129, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimovskaya, E.V.; Woodward, H.N.; Tucker, D.A.; Stenmark, K.R. Extracellular ATP is a pro-angiogenic factor for pulmonary artery vasa vasorum endothelial cells. Angiogenesis 2008, 11, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Falzoni, S.; Donvito, G.; Di Virgilio, F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013, 3, 20120101. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F. Purines, purinergic receptors, and cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Campos-Contreras, A.D.R.; Diaz-Munoz, M.; Vazquez-Cuevas, F.G. Purinergic Signaling in the Hallmarks of Cancer. Cells 2020, 9, 1612. [Google Scholar] [CrossRef]
- De Andrade Mello, P.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Adenosine receptors as drug targets. Exp. Cell Res. 2010, 316, 1284–1288. [Google Scholar] [CrossRef] [Green Version]
- Bours, M.J.; Dagnelie, P.C.; Giuliani, A.L.; Wesselius, A.; Di Virgilio, F. P2 receptors and extracellular ATP: A novel homeostatic pathway in inflammation. Front. Biosci. Schol. Ed. 2011, 3, 1443–1456. [Google Scholar] [PubMed]
- Gordon, J.L. Extracellular ATP: Effects, sources and fate. Biochem. J. 1986, 233, 309–319. [Google Scholar] [CrossRef]
- Riteau, N.; Gasse, P.; Fauconnier, L.; Gombault, A.; Couegnat, M.; Fick, L.; Kanellopoulos, J.; Quesniaux, V.F.; Marchand-Adam, S.; Crestani, B.; et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Plesner, L. Ecto-ATPases: Identities and functions. Int. Rev. Cytol. 1995, 158, 141–214. [Google Scholar]
- Burnstock, G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol. Rev. 2006, 58, 58–86. [Google Scholar] [CrossRef] [PubMed]
- Rayah, A.; Kanellopoulos, J.M.; Di Virgilio, F. P2 receptors and immunity. Microbes Infect. 2012, 14, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colgan, S.P.; Eltzschig, H.K. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu. Rev. Physiol. 2012, 74, 153–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Corriden, R.; Insel, P.A. Basal release of ATP: An autocrine-paracrine mechanism for cell regulation. Sci. Signal. 2010, 3, re1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabirov, R.Z.; Dutta, A.K.; Okada, Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J. Gen. Physiol. 2001, 118, 251–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebert, E.M.; Zsembery, A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim. Biophys. Acta 2003, 1615, 7–32. [Google Scholar] [CrossRef] [Green Version]
- Reisin, I.L.; Prat, A.G.; Abraham, E.H.; Amara, J.F.; Gregory, R.J.; Ausiello, D.A.; Cantiello, H.F. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J. Biol. Chem. 1994, 269, 20584–20591. [Google Scholar] [CrossRef]
- Romanello, M.; Pani, B.; Bicego, M.; D’Andrea, P. Mechanically induced ATP release from human osteoblastic cells. Biochem. Biophys. Res. Commun. 2001, 289, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Bodin, P.; Burnstock, G. Purinergic signalling: ATP release. Neurochem. Res. 2001, 26, 959–969. [Google Scholar] [CrossRef]
- D’Hondt, C.; Ponsaerts, R.; De Smedt, H.; Vinken, M.; De Vuyst, E.; De Bock, M.; Wang, N.; Rogiers, V.; Leybaert, L.; Himpens, B.; et al. Pannexin channels in ATP release and beyond: An unexpected rendezvous at the endoplasmic reticulum. Cell Signal. 2011, 23, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Falzoni, S.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol. Biol. Cell 2005, 16, 3659–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Seror, C.; Metivier, D.; Perfettini, J.L.; Zitvogel, L.; et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009, 8, 3723–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar]
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.H.; Mousawi, F.; Yang, X.; Roger, S. ATP-induced Ca(2+)-signalling mechanisms in the regulation of mesenchymal stem cell migration. Cell Mol. Life Sci. 2017, 74, 3697–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, R.C.; Taylor, A.F.; Rogers, J.R.; Donahue, H.J. ATP release mediates fluid flow-induced proliferation of human bone marrow stromal cells. J. Bone Miner. Res. 2007, 22, 589–600. [Google Scholar] [CrossRef]
- Sun, D.; Junger, W.G.; Yuan, C.; Zhang, W.; Bao, Y.; Qin, D.; Wang, C.; Tan, L.; Qi, B.; Zhu, D.; et al. Shockwaves induce osteogenic differentiation of human mesenchymal stem cells through ATP release and activation of P2X7 receptors. Stem Cells 2013, 31, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.Y. The Role of MSCs in the Tumor Microenvironment and Tumor Progression. Anticancer Res. 2020, 40, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Otsu, K.; Kuruma, A.; Shoji, S.; Yanagida, E.; Muto, Y.; Yoshikawa, F.; Hirayama, Y.; Mikoshiba, K.; Furuichi, T. ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium 2006, 39, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Klepeis, V.E.; Weinger, I.; Kaczmarek, E.; Trinkaus-Randall, V. P2Y receptors play a critical role in epithelial cell communication and migration. J. Cell Biochem. 2004, 93, 1115–1133. [Google Scholar] [CrossRef] [PubMed]
- Ehring, G.R.; Szabo, I.L.; Jones, M.K.; Sarfeh, I.J.; Tarnawski, A.S. ATP-induced CA2+-signaling enhances rat gastric microvascular endothelial cell migration. J. Physiol. Pharmacol. 2000, 51, 799–811. [Google Scholar] [PubMed]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelassi, B.; Chantome, A.; Alcaraz-Perez, F.; Baroja-Mazo, A.; Cayuela, M.L.; Pelegrin, P.; Surprenant, A.; Roger, S. P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 2011, 30, 2108–2122. [Google Scholar] [CrossRef] [Green Version]
- Jelassi, B.; Anchelin, M.; Chamouton, J.; Cayuela, M.L.; Clarysse, L.; Li, J.; Gore, J.; Jiang, L.H.; Roger, S. Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2X7 receptors. Carcinogenesis 2013, 34, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Li, W.H.; Zhang, H.Q.; Liu, Y.; Tian, X.X.; Fang, W.G. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS ONE 2014, 9, e114371. [Google Scholar] [CrossRef] [Green Version]
- Giannuzzo, A.; Pedersen, S.F.; Novak, I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol. Cancer 2015, 14, 203. [Google Scholar] [CrossRef] [Green Version]
- Salvestrini, V.; Zini, R.; Rossi, L.; Gulinelli, S.; Manfredini, R.; Bianchi, E.; Piacibello, W.; Caione, L.; Migliardi, G.; Ricciardi, M.R.; et al. Purinergic signaling inhibits human acute myeloblastic leukemia cell proliferation, migration, and engraftment in immunodeficient mice. Blood 2012, 119, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Li, W.H.; Qiu, Y.; Zhang, H.Q.; Liu, Y.; You, J.F.; Tian, X.X.; Fang, W.G. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 2013, 109, 1666–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Eun, S.Y.; Lee, J.S.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2 receptor activation by nucleotides released from highly metastatic breast cancer cells increases tumor growth and invasion via crosstalk with endothelial cells. Breast Cancer Res. 2014, 16, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Seo, J.; Eun, S.Y.; Joo, Y.N.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. P2Y2 R activation by nucleotides promotes skin wound-healing process. Exp. Dermatol. 2014, 23, 480–485. [Google Scholar] [CrossRef]
- Xie, R.; Xu, J.; Wen, G.; Jin, H.; Liu, X.; Yang, Y.; Ji, B.; Jiang, Y.; Song, P.; Dong, H.; et al. The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J. Biol. Chem. 2014, 289, 19137–19149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadet, S.; Jelassi, B.; Wannous, R.; Angoulvant, D.; Chevalier, S.; Besson, P.; Roger, S. The activation of P2Y2 receptors increases MCF-7 breast cancer cells migration through the MEK-ERK1/2 signalling pathway. Carcinogenesis 2014, 35, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Simek, J.; Thompson, R.J. Regulation of pannexin channels by post-translational modifications. FEBS Lett. 2014, 588, 1411–1415. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef] [PubMed]
- Wilkaniec, A.; Gassowska, M.; Czapski, G.A.; Cieslik, M.; Sulkowski, G.; Adamczyk, A. P2X7 receptor-pannexin 1 interaction mediates extracellular alpha-synuclein-induced ATP release in neuroblastoma SH-SY5Y cells. Purinergic Signal. 2017, 13, 347–361. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, P.; Surprenant, A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J. Biol. Chem. 2007, 282, 2386–2394. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 Receptor Primes IL-1beta and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front. Cell Neurosci. 2017, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xing, Y.; Mao, L.; Luo, Y.; Kang, L.; Meng, G. Pannexin-1 influences peritoneal cavity cell population but is not involved in NLRP3 inflammasome activation. Protein Cell 2013, 4, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.; Zetterqvist, A.V.; Wright, W.R.; Kirkby, N.S.; Mitchell, J.A.; Paul-Clark, M.J. Differential role of pannexin-1/ATP/P2X7 axis in IL-1beta release by human monocytes. FASEB J. 2017, 31, 2439–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.W.; Demarco, B.; Broz, P. Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur. J. Immunol. 2020, 50, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Furlow, P.W.; Zhang, S.; Soong, T.D.; Halberg, N.; Goodarzi, H.; Mangrum, C.; Wu, Y.G.; Elemento, O.; Tavazoie, S.F. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 2015, 17, 943–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Callegari, M.G.; Ferrari, D.; Bolognesi, C.; Minelli, M.; Wieckowski, M.R.; Pinton, P.; Rizzuto, R.; Di Virgilio, F. Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol. Biol. Cell 2005, 16, 3260–3272. [Google Scholar] [CrossRef] [Green Version]
- Baricordi, O.R.; Melchiorri, L.; Adinolfi, E.; Falzoni, S.; Chiozzi, P.; Buell, G.; Di Virgilio, F. Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J. Biol. Chem. 1999, 274, 33206–33208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Ferrari, D.; Los, M.; Bauer, M.K.; Vandenabeele, P.; Wesselborg, S.; Schulze-Osthoff, K. P2Z purinoreceptor ligation induces activation of caspases with distinct roles in apoptotic and necrotic alterations of cell death. FEBS Lett. 1999, 447, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Trosko, J.E. What Can Chemical Carcinogenesis Shed Light on the LNT Hypothesis in Radiation Carcinogenesis? Dose Response 2019, 17, 1559325819876799. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. Immunological surveillance in neoplasia. Transplant. Rev. 1971, 7, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Targeting adenosine in cancer immunotherapy: A review of recent progress. Expert Rev. Anticancer Ther. 2017, 17, 527–535. [Google Scholar] [CrossRef]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef]
- Poth, J.M.; Brodsky, K.; Ehrentraut, H.; Grenz, A.; Eltzschig, H.K. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J. Mol. Med. 2013, 91, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Turcotte, M.; Stagg, J. CD73-generated adenosine: Orchestrating the tumor-stroma interplay to promote cancer growth. J. Biomed. Biotechnol. 2012, 2012, 485156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappas, C.M.; Rieger, J.M.; Linden, J. A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J. Immunol. 2005, 174, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A Receptor Stimulation Inhibits TCR-Induced Notch1 Activation in CD8+T-Cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Extracellular adenosine-mediated modulation of regulatory T cells. Front. Immunol. 2014, 5, 304. [Google Scholar] [CrossRef]
- Bouma, M.G.; van den Wildenberg, F.A.; Buurman, W.A. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am. J. Physiol. 1996, 270, C522–C529. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, J.K.; Ridley, A.J. CD73 represses pro-inflammatory responses in human endothelial cells. J. Inflamm. 2010, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A Adenosine Receptor Gene Deletion or Synthetic A2A Antagonist Liberate Tumor-Reactive CD8(+) T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [Green Version]
- Montalban Del Barrio, I.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wockel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages—A self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J. Immunother. Cancer 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef] [Green Version]
- Siemens, D.R.; Hu, N.; Sheikhi, A.K.; Chung, E.; Frederiksen, L.J.; Pross, H.; Graham, C.H. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: Role of nitric oxide. Cancer Res. 2008, 68, 4746–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Buart, S.; Van Pelt, J.; Richon, C.; Hasmim, M.; Leleu, N.; Suchorska, W.M.; Jalil, A.; Lecluse, Y.; El Hage, F.; et al. The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 2009, 182, 3510–3521. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Tittarelli, A.; Janji, B.; Van Moer, K.; Noman, M.Z.; Chouaib, S. The Selective Degradation of Synaptic Connexin 43 Protein by Hypoxia-induced Autophagy Impairs Natural Killer Cell-mediated Tumor Cell Killing. J. Biol. Chem. 2015, 290, 23670–23679. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Bronte, V.; Apolloni, E.; Cabrelle, A.; Ronca, R.; Serafini, P.; Zamboni, P.; Restifo, N.P.; Zanovello, P. Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 2000, 96, 3838–3846. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Velders, M.P.; Sotomayor, E.M.; Kast, W.M. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J. Immunol. 2001, 166, 5398–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005, 65, 11743–11751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef] [PubMed]
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Raffaghello, L.; Chiozzi, P.; Falzoni, S.; Di Virgilio, F.; Pistoia, V. The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res. 2006, 66, 907–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, V.; Protti, M.P. Tumor-derived factors affecting immune cells. Cytokine Growth Factor Rev. 2017, 36, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Dransfield, I.; Gregory, C.; Haslett, C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002, 2, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Szondy, Z.; Sarang, Z.; Kiss, B.; Garabuczi, E.; Koroskenyi, K. Anti-inflammatory Mechanisms Triggered by Apoptotic Cells during Their Clearance. Front. Immunol. 2017, 8, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Massenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The pathological features of regulated necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadzadeh, Z.; Safarzadeh, E.; Safaei, S.; Baradaran, A.; Mohammadi, A.; Hajiasgharzadeh, K.; Derakhshani, A.; Argentiero, A.; Silvestris, N.; Baradaran, B. Current Approaches for Combination Therapy of Cancer: The Role of Immunogenic Cell Death. Cancers 2020, 12, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar]
- Showalter, A.; Limaye, A.; Oyer, J.L.; Igarashi, R.; Kittipatarin, C.; Copik, A.J.; Khaled, A.R. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine 2017, 97, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, O.; Love Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Metivier, D.; Galluzzi, L.; Perfettini, J.L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- La Sala, A.; Sebastiani, S.; Ferrari, D.; Di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Dendritic cells exposed to extracellular adenosine triphosphate acquire the migratory properties of mature cells and show a reduced capacity to attract type 1 T lymphocytes. Blood 2002, 99, 1715–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gong, L.H.; Zhang, H.Q.; Du, Q.; You, J.F.; Tian, X.X.; Fang, W.G. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 2010, 293, 189–197. [Google Scholar] [CrossRef]
- Ma, Y.; Aymeric, L.; Locher, C.; Mattarollo, S.R.; Delahaye, N.F.; Pereira, P.; Boucontet, L.; Apetoh, L.; Ghiringhelli, F.; Casares, N.; et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J. Exp. Med. 2011, 208, 491–503. [Google Scholar] [CrossRef]
- Frey, B.; Janko, C.; Ebel, N.; Meister, S.; Schlucker, E.; Meyer-Pittroff, R.; Fietkau, R.; Herrmann, M.; Gaipl, U.S. Cells under pressure—Treatment of eukaryotic cells with high hydrostatic pressure, from physiologic aspects to pressure induced cell death. Curr. Med. Chem. 2008, 15, 2329–2336. [Google Scholar] [CrossRef]
- Rozkova, D.; Tiserova, H.; Fucikova, J.; Last’ovicka, J.; Podrazil, M.; Ulcova, H.; Budinsky, V.; Prausova, J.; Linke, Z.; Minarik, I.; et al. FOCUS on FOCIS: Combined chemo-immunotherapy for the treatment of hormone-refractory metastatic prostate cancer. Clin. Immunol. 2009, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Spisek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.W.; Dougherty, T.J. How does photodynamic therapy work? Photochem. Photobiol. 1992, 55, 145–157. [Google Scholar] [CrossRef]
- Shumaker, B.P.; Hetzel, F.W. Clinical laser photodynamic therapy in the treatment of bladder carcinoma. Photochem. Photobiol. 1987, 46, 899–901. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, S.O.; Liu, X.; Owczarczak, B.; Musser, D.A.; Henderson, B.W. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997, 57, 3904–3909. [Google Scholar] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef]
- Korbelik, M.; Zhang, W.; Merchant, S. Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: Surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol. Immunother. 2011, 60, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Romo, D.; Velmurugan, K.; Upham, B.L.; Dwyer-Nield, L.D.; Bauer, A.K. Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model. Cancers 2019, 11, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Couldwell, W.T.; Simard, M.F.; Song, H.; Lin, J.H.; Nedergaard, M. Direct gap junction communication between malignant glioma cells and astrocytes. Cancer Res. 1999, 59, 1994–2003. [Google Scholar]
- Lin, J.H.; Takano, T.; Cotrina, M.L.; Arcuino, G.; Kang, J.; Liu, S.; Gao, Q.; Jiang, L.; Li, F.; Lichtenberg-Frate, H.; et al. Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J. Neurosci. 2002, 22, 4302–4311. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Ladage, K.; Lafenetre, P.; Hinkerohe, D.; Smikalla, D.; Haase, C.G.; Dermietzel, R.; Faustmann, P.M. Intracellular application of TNF-alpha impairs cell to cell communication via gap junctions in glioma cells. J. Neurooncol. 2008, 86, 143–152. [Google Scholar] [CrossRef]
- Zefferino, R.; Leone, A.; Piccaluga, S.; Cincione, R.; Ambrosi, L. Mercury modulates interplay between IL-1beta, TNF-alpha, and gap junctional intercellular communication in keratinocytes: Mitigation by lycopene. J. Immunotoxicol. 2008, 5, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Zefferino, R.; Piccaluga, S.; Lasalvia, M.; D’Andrea, G.; Margaglione, M.; Ambrosi, L. Role of tumour necrosis factor alpha and interleukin 1 beta in promoter effect induced by mercury in human keratinocytes. Int. J. Immunopathol. Pharmacol. 2006, 19, 15–20. [Google Scholar]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fettelschoss, A.; Kistowska, M.; LeibundGut-Landmann, S.; Beer, H.D.; Johansen, P.; Senti, G.; Contassot, E.; Bachmann, M.F.; French, L.E.; Oxenius, A.; et al. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc. Natl. Acad. Sci. USA 2011, 108, 18055–18060. [Google Scholar] [CrossRef] [Green Version]
- Rider, P.; Carmi, Y.; Voronov, E.; Apte, R.N. Interleukin-1alpha. Semin. Immunol. 2013, 25, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Maywald, R.L.; Doerner, S.K.; Pastorelli, L.; De Salvo, C.; Benton, S.M.; Dawson, E.P.; Lanza, D.G.; Berger, N.A.; Markowitz, S.D.; Lenz, H.J.; et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc. Natl. Acad. Sci. USA 2015, 112, E2487–E2496. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhu, L.; Lu, X.; Bian, H.; Wu, X.; Yang, W.; Qin, Q. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem. Biophys. Res. Commun. 2014, 453, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Limoge, M.; Safina, A.; Beattie, A.; Kapus, L.; Truskinovsky, A.M.; Bakin, A.V. Tumor-fibroblast interactions stimulate tumor vascularization by enhancing cytokine-driven production of MMP9 by tumor cells. Oncotarget 2017, 8, 35592–35608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Chantrain, C.F.; Shimada, H.; Jodele, S.; Groshen, S.; Ye, W.; Shalinsky, D.R.; Werb, Z.; Coussens, L.M.; DeClerck, Y.A. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Res. 2004, 64, 1675–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jodele, S.; Chantrain, C.F.; Blavier, L.; Lutzko, C.; Crooks, G.M.; Shimada, H.; Coussens, L.M.; Declerck, Y.A. The contribution of bone marrow-derived cells to the tumor vasculature in neuroblastoma is matrix metalloproteinase-9 dependent. Cancer Res. 2005, 65, 3200–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisha, Y.; Sagi, Y.; Klein, G.; Straussman, R.; Geiger, B. Cooperativity between stromal cytokines drives the invasive migration of human breast cancer cells. Philos Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180231. [Google Scholar] [CrossRef]
- Enns, L.; Ladiges, W. Mitochondrial redox signaling and cancer invasiveness. J. Bioenerg. Biomembr. 2012, 44, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostofa, A.G.; Punganuru, S.R.; Madala, H.R.; Al-Obaide, M.; Srivenugopal, K.S. The Process and Regulatory Components of Inflammation in Brain Oncogenesis. Biomolecules 2017, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, S.; Gupta, A.; Sharma, S.K. Interleukin-6-mediated autocrine growth promotion in human glioblastoma multiforme cell line U87MG. J. Neurochem. 1998, 71, 1837–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Mattox, A.K.; Harward, S.; Adamson, C. Emerging therapeutic targets and agents for glioblastoma migrating cells. Anticancer Agents Med. Chem. 2010, 10, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Dotor, J.; Casares, N.; Bezunartea, J.; Diaz-Valdes, N.; Ruiz, M.; Aranda, F.; Berraondo, P.; Prieto, J.; Lasarte, J.J.; et al. Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 2009, 125, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- De Waal Malefyt, R.; Haanen, J.; Spits, H.; Roncarolo, M.G.; te Velde, A.; Figdor, C.; Johnson, K.; Kastelein, R.; Yssel, H.; de Vries, J.E. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 1991, 174, 915–924. [Google Scholar] [CrossRef] [PubMed]
- See, A.P.; Han, J.E.; Phallen, J.; Binder, Z.; Gallia, G.; Pan, F.; Jinasena, D.; Jackson, C.; Belcaid, Z.; Jeong, S.J.; et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J. Neurooncol. 2012, 110, 359–368. [Google Scholar] [CrossRef]
- Jia, Q.A.; Wang, Z.M.; Ren, Z.G.; Bu, Y.; Xie, X.Y.; Wang, Y.H.; Zhang, L.; Zhang, Q.B.; Xue, T.C.; Deng, L.F.; et al. Herbal compound “Songyou Yin” attenuates hepatoma cell invasiveness and metastasis through downregulation of cytokines secreted by activated hepatic stellate cells. BMC Complement. Altern. Med. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zefferino, R.; Piccoli, C.; Di Gioia, S.; Capitanio, N.; Conese, M. How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease. Int. J. Mol. Sci. 2021, 22, 2550. https://doi.org/10.3390/ijms22052550
Zefferino R, Piccoli C, Di Gioia S, Capitanio N, Conese M. How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease. International Journal of Molecular Sciences. 2021; 22(5):2550. https://doi.org/10.3390/ijms22052550
Chicago/Turabian StyleZefferino, Roberto, Claudia Piccoli, Sante Di Gioia, Nazzareno Capitanio, and Massimo Conese. 2021. "How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease" International Journal of Molecular Sciences 22, no. 5: 2550. https://doi.org/10.3390/ijms22052550