
Non-Coding RNAs Set a New Phenotypic Frontier in Prostate Cancer Metastasis and Resistance


1
Department of Urology, The Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
2
Department of Oncological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(4), 2100; https://doi.org/10.3390/ijms22042100
Submission received: 20 January 2021
/
Revised: 8 February 2021
/
Accepted: 10 February 2021
/
Published: 20 February 2021
(This article belongs to the Special Issue Cancer Cell Invasion and Metastases 2.0)
Abstract
:Prostate cancer (PCa) mortality remains a significant public health problem, as advanced disease has poor survivability due to the development of resistance in response to both standard and novel therapeutic interventions. Therapeutic resistance is a multifaceted problem involving the interplay of a number of biological mechanisms including genetic, signaling, and phenotypic alterations, compounded by the contributions of a tumor microenvironment that supports tumor growth, invasiveness, and metastasis. The androgen receptor (AR) is a primary regulator of prostate cell growth, response and maintenance, and the target of most standard PCa therapies designed to inhibit AR from interacting with androgens, its native ligands. As such, AR remains the main driver of therapeutic response in patients with metastatic castration-resistant prostate cancer (mCRPC). While androgen deprivation therapy (ADT), in combination with microtubule-targeting taxane chemotherapy, offers survival benefits in patients with mCRPC, therapeutic resistance invariably develops, leading to lethal disease. Understanding the mechanisms underlying resistance is critical to improving therapeutic outcomes and also to the development of biomarker signatures of predictive value. The interconversions between epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-epithelial transition (MET) navigate the prostate tumor therapeutic response, and provide a novel targeting platform in overcoming therapeutic resistance. Both microRNA (miRNA)- and long non-coding RNA (lncRNA)-mediated mechanisms have been associated with epigenetic changes in prostate cancer. This review discusses the current evidence-based knowledge of the role of the phenotypic transitions and novel molecular determinants (non-coding RNAs) as contributors to the emergence of therapeutic resistance and metastasis and their integrated predictive value in prostate cancer progression to advanced disease.
1. Introduction
1.1. Prostate Cancer: The Clinical Challenge
Prostate cancer (PCa) remains among the most significant public health concerns in the United States, predicted to account for approximately 13.09% of all new cancer cases diagnosed and lead to 34,130 deaths in 2021 [1]. It is the third most commonly diagnosed malignancy in the US behind breast and lung cancers [2] and is the most commonly diagnosed cancer in men [3]. Since the advent of prostate specific antigen (PSA) screening, there has been a significant shift in the stage at which patients are diagnosed, as many more men are now discovering and treating their disease while it is still locally confined [4]. While five-year survival rates for patients with localized PCa approach 98–100%, survival rates dramatically decrease once the cancer has metastasized to distant sites [5]. Localized primary PCa is effectively treated with either surgery (radical prostatectomy) or radiation therapy, or a combination of the two [6]. For recurrent or metastatic disease, androgen deprivation therapy (ADT) is the primary standard of care and works by either lowering androgen levels via blocking the production of the hormone centrally, or by inhibiting the binding of androgens to their receptors peripherally [7]. ADT targets the androgen receptor (AR) signaling network that is essential to the proper functioning, growth and maintenance of prostate cells [8]. Androgen deprivation can be achieved surgically (via orchiectomy) or pharmacologically with the chronic administration of luteinizing hormone releasing-hormone (LHRH) agonists [9] or AR blockers such as bicalutamide, flutamide and nilutamide [6]. Although many patients will initially respond to ADT, most patients will eventually develop castration-resistant prostate cancer (CRPC) and ultimately metastatic lethal disease, despite having castrate levels of androgens [10].
1.2. Mechanisms of Therapeutic Resistance
The selective pressure that ADT imposes upon treatment-naïve cancer leads to the emergence of the androgen-independent state. Androgen-independent cancer cells rely on a host of alternative, adaptive mechanisms that are designed to compensate for the lack of androgen stimulation to sustain growth [11]. AR-dependent mechanisms span multiple levels of the androgen axis and affect the synthesis, utilization and signaling of androgens. These mechanisms include the altered intratumoral production of androgens, AR amplification and overexpression, AR point mutations, and the expression of constituently-active AR splice-variants [12]. Although the primary effect of ADT is the reduction in androgen levels to castrate levels, it has been shown that patients with CRPC have persistently elevated levels of androgens that may be derived from the production of dihydrotestosterone (DHT) intratumorally [13]. Prostate cancer cells are also able to convert weaker androgens produced by the adrenal glands, such as androstenedione and dehydroepiandrosterone (DHEA), into testosterone and DHT, which have a higher affinity for AR [14]. Importantly, several studies have revealed that even low levels of androgens are enough to significantly amplify AR in vivo [15,16,17] and that amplified AR can, in turn, efficiently utilize and augment this signal [18]. AR sensitivity to low levels of androgens may also be heightened by the presence of coactivators that enhance AR function [19,20]. One study that examined samples of PCa bone metastases from treated patients found that AR was amplified in 53% of samples [21]. In contrast, AR amplification was rarely detected in patients with primary prostate tumors [22]. Furthermore, through promiscuous AR activation, ligands aside from androgens, testosterone and its derivatives can activate AR. This process occurs as a result of mutations in the AR gene which alter the ligand binding domain (LBD) [23]. AR mutations are rare in early disease but are frequently seen in CRPC [24], and these mutations allow for receptor activation by other steroid hormones such as estrogen, progesterone, and glucocorticoids [25,26]. Certain mutations can also provide resistance against the effects of AR inhibitors; for example, the T877A mutation protects AR against a major active metabolite of flutamide, hydroxyflutamide, [11,27] while the W741C/L mutation protects against bicalutamide [11,28]. Coactivator proteins can also increase AR responsiveness to alternative ligands, such as coactivator ARA70 that increases AR sensitivity to estradiol [19,29]. Several growth factors, including insulin-like growth factor 1 (IGF-1) [30], human epidermal growth factor receptor 2 (HER2/neu) [31], keratinocyte growth factor (KGF), and epidermal growth factor (EGF), can also activate AR [19,32]. In addition, interleukins IL-6 and IL-8 are capable of stimulating AR (IL-6) [33,34,35] and promoting AR transcriptional activity (IL-8) [34]. AR splice variants (AR-Vs) represent another major functional adaption of AR and mechanism of resistance to ADT [11,36]. Some of these variants are constitutively active and can transcribe AR target genes in the absence of natural AR ligands [11,19,37].
Evasion of apoptosis control including the activation of PTEN/PI3K/AKT survival signaling is an important contributor and advanced metastatic disease (mCRPC) [11]. PTEN (phosphatase and tensin homolog) is a tumor suppressor that dephosphorylates PIP3 and negatively regulates the phosphoinositide 3-kinase (PI3K) pathway [8] that, when lost, leads to constitutive activation of PI3K, which acts downstream to activate protein kinase B (AKT) and mTOR (mammalian target of rapamycin) signaling [8,38] pathways that are involved in the regulation of cell growth, survival, and proliferation [39] and protein synthesis [38], respectively. Loss of PTEN is a major event in prostate cancer pathogenesis and appears to play a role in the development of CRPC [8,40]. Deletions or inactivating mutations of PTEN have been observed in approximately 20% of primary prostate cancer samples and in over 50% of cases of CRPC [41,42]. While elegant studies by different groups have demonstrated that negative reciprocal feedback between PI3K and AR signaling pathways can act as an AR-independent mechanism to develop CRPC in PTEN-deficient models of PCa [40,43], there is evidence to suggest a functional interplay between the PI3K/AKT pathways and AR, wherein AR and AKT work synergistically to promote PCa initiation and progression [44]. The NF-κB transcription factor signaling pathway also prominently contributes to CRPC, as NF-κB sustains AR activity [11] and significantly increases AR at both mRNA and protein levels [45].
Enzalutamide and abiraterone are the leading second-generation antiandrogens FDA-approved for the treatment of CRPC [46]. Enzalutamide acts by binding to the LBD of AR, and blocks androgen-AR complex nuclear translocation, thereby preventing the binding of this complex to DNA [6]. Abiraterone targets the biosynthesis of testosterone by inhibiting the cytochrome p450 enzyme 17R-hydroxylase-17,20-lyase (CYP17), an enzyme found in the testes and adrenal glands [6]. Several clinical trials (as described in Table 1) have shown the efficacy of these drugs in that enzalutamide improved overall survival (OS) and radiographic progression free survival (PFS) in patients with CRPC who had received chemotherapy [47], as well as in those who were chemotherapy-naïve [48], while abiraterone treatment combined with prednisone improved overall survival in chemotherapy-naïve men with mCRPC [49]. Resistance to both drugs invariably develops, however [50], and is attributed to various mechanisms; thus, a missense mutation in the LBD of AR, F876L, induces a switch in the properties of enzalutamide from antagonist action to agonist action at the AR exchange [51], whereas T878A and L702H point mutations emerge in response to abiraterone treatment [52]. Since abiraterone is often given with prednisone (Table 1), and the L702H mutation allows for the stimulation of AR via glucocorticoids, this mutation promotes resistance to abiraterone via an enhanced sensitivity to prednisone [53,54]. AR splice variants have also been implicated in therapeutic resistance to abiraterone and enzalutamide, especially AR-V7. AR-V7 is capable of ligand-independent activation and is abundant in CRPC [11]. A landmark study by Antonarakis et al. established that AR-V7 in circulating tumor cells from patients with CRPC was associated with resistance to antiandrogens [55]. Additional mechanisms of resistance include the F877L mutation of the LBD of AR, another mutation which converts the antagonist effects of enzalutamide into agonist effects [24], and upregulation of CYP17 [53,56].
2. The Phenotypic Landscape of Advanced Prostate Cancer
2.1. Therapy-Induced Neuroendocrine Differentiation in Resistant Prostate Cancer
One mechanism of resistance that bypasses treatment to androgen axis targeting by becoming “androgen indifferent [71]” with lethal consequences is the transdifferentiation of CRPC to neuroendocrine prostate cancer (NEPC) [72]. NEPC is poorly differentiated, progresses rapidly, and metastases to visceral organs [73], with a median patient survival of 7 months [72]. It is characterized by the absence of AR [74] and the expression of neuroendocrine markers such as synaptophysin (SYP), chromogranin A (CHGA) and enolase 2 (ENO2) [73,74]. While NEPC rarely arises de novo and accounts for less than 2% of all primary prostate cancer diagnoses [71], therapy-induced NEPC (t-NEPC) can develop from divergent clonal evolution from CRPC in response to the selective pressure of ADT [72,73] and has a much higher incidence, ranging from 17–30% [73]. Furthermore, the prevalence of t-NEPC is expected to rise with the continued use of potent, second-generation antiandrogens [72]. Evidence supporting the transdifferentiation of CRPC includes the similar frequency of transmembrane protease serine 2-v ets erythroblastosis virus E26 oncogene homolog (TMPRSS-ERG) translocations in adenocarcinoma and t-NEPC [75], and the concordance of TMPRSS-ERG translocation status in the foci of both tumor types in mixed adenocarcinoma and t-NEPC [76], as well as the significant overlap in the somatic copy number alterations between CRPC and t-NEPC [77,78]. The molecular drivers of NEPC development include the cooperation between N-myc proto-oncogene protein (N-MYC) and PTEN loss or AKT1 overexpression [79], as well as AURKA (Aurora Kinase A) overexpression [76], TP53 mutation and loss of RB1 (Retinoblastoma 1) [78]. Recent work by Rotinen et al. [80] has identified the ONECUT2 (OC2) transcription factor as a targetable master regulator of lethal PCa that is also associated with the development of neuroendocrine differentiation (NED) in CRPC, as OC2 functions as a survival factor that promotes metastasis, regulates AR, and activates genes promoting neuronal differentiation in mCRPC [80]. Treatment options for t-NEPC are currently limited to platinum-based chemotherapies (cisplatin and carboplatin [72,73]), docetaxel, and etoposide [72] (a topoisomerase II inhibitor [81]), with a median survival of 7–15 months [73,82,83,84].
Taxane-based chemotherapies such as docetaxel (1st line chemotherapy) and cabazitaxel (2nd line chemotherapy) [85] are some of the only drugs available to treat advanced, metastatic prostate cancer, while novel immunotherapies [85], radioactive agents [85], and small-molecule inhibitors targeting upregulated cancer signaling pathways [86] recently provided substantial clinical promise to overcome lethal disease. Taxane chemotherapeutics exert their action on three fronts: they (a) interfere with cell division—by binding to tubulins and microtubules [87] and stabilizing microtubules from depolymerization, taxane drugs prevent cancerous cells from leaving the G2 phase and advancing to the mitotic phase of the cell cycle [88]. They also (b) promote the phosphorylation and deactivation of B-cell lymphoma 2 (BCL-2), an anti-apoptotic protein that suppresses apoptosis, by binding and inhibiting the activation of pro-apoptotic factors such as Bcl-2-like protein 4 (BAX), BCL-2 associated agonist of cell death (BAD) and BH3 interacting-domain death agonist (BID) [88], ultimately leading to apoptosis [87]. Finally, taxanes (c) inhibit AR signaling and activity as AR translocation to the nucleus is facilitated by microtubules, [89] and 1st line taxane chemotherapy downregulates AR transcriptional activity [90]. While docetaxel and cabazitaxel can improve overall survival [90], resistance eventually develops, mainly involving multidrug resistance effectors such as P-gp (P-glycoprotein) and ATP-binding cassette sub-family C member 4 (ABCC4) [91,92]. These proteins form efflux pumps that actively transport chemotherapeutic drugs out of affected cells [91,92]. Cabazitaxel has a lower affinity for P-gp compared to docetaxel, which enables it to be effective in docetaxel-resistant tumors [92,93]. Significantly enough, the antiandrogens bicalutamide and enzalutamide are also able to reduce the activity of P-gp [94] and ABCC4 [95] (Table 2).
2.2. Epithelial-to-Mesenchymal Transition (EMT) Navigates the Tumor Microenvironment and Contributes to Metastasis and CRPC
Impairing the emergence of therapeutically-resistant mCRPC is critical in overcoming lethal disease and improving patient survival [102,103]. Metastasis is the cause of more than 90% of cancer-related deaths, and the majority of prostate cancer-related deaths [104]. Metastasis involves the cooperation of multiple cellular processes, including the dissociation cancer epithelial cells from the primary tumor site, detachment from the extracellular matrix (ECM), resistance to anoikis, invasion, migration into blood or lymph vasculature (intravasation), circulation, extravasation, and finally, the colonization of secondary sites [102,104,105,106]. Epithelial-to-mesenchymal transition (EMT) is an essential phenotypic process functionally linked to cancer metastasis [107] as cancerous epithelial cells can reactivate and exploit the EMT program to acquire a more mesenchymal phenotype, which dramatically increases their invasiveness and metastatic potential [108]. Several transcriptional regulators of EMT in PCa, including zinc finger protein SNAI1 (SNAIL), zinc finger protein SNAI2 (SLUG), ZEB1/2, and twist-related protein 1 (TWIST) [108,109], are under the control of diverse growth factor signaling mechanisms, such as transforming growth factor-β (TGF-β) [109]. EMT is a transient reversible process, as cells with a mesenchymal phenotype can re-acquire epithelial properties (mesenchymal-to-epithelial transition (MET)), which further increases the intrigue of EMT-related effectors as potential actionable targets to impair the metastatic journey.
The prostate gland is composed of two main cell types: the glandular epithelium and the stroma [110]. Prostate carcinogenesis involves the malignant transformation of epithelial cells, supported by a reactive stroma that aids in the growth and development of the tumor [111]. The epithelial compartment contains the glandular cells of the prostate [112] and consists of a layer of basal epithelial cells that rest on a basement membrane, intermediate cells, neuroendocrine cells, and luminal epithelial cells that are secretory in nature lining the prostatic lumen [8,113,114]. Stromal tissue is connective tissue [112] and mesenchymal in origin, composed mostly of smooth muscle cells and fibroblasts [115], but also contains myofibroblasts, an extracellular matrix (ECM) composed of laminin and collagen, immune cells, nerves, and vasculature [116]. In response to androgens, and through interactions mitigated by AR [111], prostatic stroma engages in a signaling exchange with the epithelial compartment [117], which also induces the differentiation of smooth muscle cells in the stroma [118]. A multilayered forum of crosstalk between stromal and epithelial components within the prostate microenvironment contributes to tumor growth and progression to metastasis [119]. In the efferent pathway, cancer cells release soluble factors such as TGF-β and platelet-derived growth factor (PDGF) that cause changes within the stroma and activate it, known as a reactive response, whereas in the afferent pathway, cancer cells respond to and are influenced by the reactive stroma [119] towards the development of prostatic intraepithelial neoplasia (PIN) [120,121,122]. Myofibroblasts develop from their fibroblast precursors, and their phenotype is marked by the expression of vimentin, an intermediate filament that is upregulated in poorly differentiated prostate cancer and in bone metastases [123]. Myofibroblasts contribute to ECM remodeling [111,124] by secreting ECM components such as collagen I, collagen III, fibronectin isoforms, tenascin, and versican [120], as well as enzymes that help degrade the ECM—proteases such as urokinase-type plasminogen activator and matrix metalloproteases (MMPs) that cause the breakdown of basement membrane [120,124,125]. Myofibroblasts promote invasion via loss of E-cadherin [111], a transmembrane cell–cell adhesion molecule [126], and upregulation of vimentin [127] to enhance prostate tumor epithelial cell invasion and migration in metastatic prostate cancer [123].
TGF-β is a multifunctional soluble factor cytokine that has been extensively studied in prostate carcinogenesis [119] via its functional contribution to the regulation of cell proliferation, differentiation, ECM production, cell motility, migration, and apoptosis [119,128]. While part of a large superfamily of cytokines, the TGF-β subgroup consists of three isoforms [119] (TGF-β1, TGF-β2 and TGF-β3 [129]), which signal through transmembrane type I (TβRI) and type II (TβRII) receptors [119]. Signaling is initiated by the binding of activated TGF-β ligands, which bring together receptor serine/theonine kinases, the TβRI and TβRII receptors, to form a complex [130,131]. TβRII receptors activate the TβRI receptors via phosphorylation, which promotes the binding of receptor-regulated Smads (R-Smads) [130,131]. R-Smads are then phosphorylated and released from the receptor complex, where they translocate to the nucleus to bind with Smad proteins and a variety of cofactors to initiate target gene transcription [131]. Depending on ligand abundance and activity, the composition of receptor complexes, and a host of other factors, TGF-β signaling can generate hundreds of different cell-specific responses [131,132,133]. TGF-β receptor complexes may also, in certain cell types, signal through Smad-independent means, further enhancing the nuance and complexity of TGF-β signaling [131]. TGF-β can either suppress or promote tumorigenesis [134]; in early stage disease, TGF-β inhibits cellular proliferation and promotes apoptosis [128], whereas in advanced disease it functionally switches to promote metastasis [128]. This functional switch is explained by its mediation through either Smad-dependent or -independent pathways [128]. TGF-β pro-apoptotic and anti-proliferative activity is Smad-dependent and governed by Smad control of c-Myc and cyclin-dependent kinase inhibitors [128,130,135]. TGF-β signaling can also transactivate AR; Kang et al. [136] demonstrated that Smad-3, a downstream mediator in the TGF-β signaling, functions as a coregulator of AR [136]. TGF-β promotes prostate cancer progression by inducing angiogenesis and EMT [119,128], both integral processes to metastasis.
EMT and its reversible counterpart MET are critical phenotypic processes involved in embryonic gastrulation, regulation of stem cell pluripotency [137,138], remodeling of the cytoskeleton and the disruption of cell–cell adhesion and cell polarity [139]. Additionally, mesenchymal cells have an increased resistance to apoptosis and produce a higher quantity of ECM components [138]. In contrast, epithelial cells are polarized, uniform, fixed and rigid, and adhere tightly to neighboring cells and to the matrix [138,140]. The pathophysiologic byproduct of EMT related to tumorigenesis (known as Type 3 EMT; EMT related to embryo/organ development and wound healing and are denoted Type 1 and 2 EMT, respectively [140]) is that the basal surface of the epithelial cell loses adherence to its closely associated basement membrane, rendering it free to invade local structures and migrate to distant sites [140]. In doing so, these cells in transition acquire mesenchymal phenotypic markers such as vimentin and desmin [140,141]. In the context of tumor progression, EMT is not a terminal, unidirectional process that commences with epithelial cells acquiring phenotypic features that eventually make it fully mesenchymal, starting with local invasion and ending with metastasis to distant sites. Rather, cancer cells which metastasized undergo phenotypic reversal to their epithelial traits, such re-expressing E-cadherin [138], in order to colonize secondary sites [140] through MET. The bidirectional nature of EMT-MET is an attractive therapeutic target [142] to impair metastasis.
EMT is characterized by the loss of the transmembrane cell–cell adhesion molecule E-cadherin, as well as occludins and claudins that are essential to maintaining the integrity of a stable epithelium [126,138,143]. This loss of E-cadherin is under multitier regulatory control in both physiologic and pathophysiologic EMT [140,144,145]. At the transcriptional level, repressors such as SNAIL [146], SLUG [147], ZEB family [148], and TWIST [149] downregulate the expression of E-cadherin through binding to the E-box region of the E-cadherin promoter [150]. While these transcription factors coordinate to upregulate the expression of one another in a complex pattern [151], their overall activation is mediated by TGF-β [140,151]. Decreased E-cadherin is often succeeded by N-cadherin, a protein that increases the migratory and invasive capacity of tumor cells [138,152]. E-cadherin loss directly promotes metastasis by facilitating the dissociation of cancer cells from the tumor mass [145,153]. Post-translationally, E-cadherin is anchored to the actin cytoskeleton as part of a complex with β-catenin [138,154]. β-catenin is a protein that serves multiple functions, one of which is as a critical component in the canonical Wnt signaling pathway [154,155], which plays a role in embryologic development, stem cell maintenance, and when mutated or dysregulated, contributes to tumorigenesis [156]. Loss of E-cadherin renders β-catenin free to translocate to the cytoplasm and participate in the Wnt signaling cascade [157]. β-catenin can also bind to cytosolic AR, which upon translocation into the nucleus increases AR transcriptional activity [138]. Of clinical relevance are the associations between co-expressed AR/β-catenin and Gleason grade 4–5 tumors and higher PSA levels [158], and the enrichment of AR and Wnt signaling in patients with early stage cancer [159].
Metastasis and colonization of distant sites by cancer cells requires overcoming anoikis, or detachment-induced cell death [102]. Cancerous cells must lose their adhesive properties and be able to detach from the ECM, evade apoptosis, and have the capacity for anchorage-independent growth at secondary sites [102]. As cells detach, anoikis is activated via the ECM-integrin cell survival pathway and by the mitochondrial mediated pathway [102]. Resistance to anoikis promotes prostate tumor migration, invasion, and metastasis [108]. Drivers of this event include the overexpression of galectin proteins (especially Galectin-3) [108,160], the activation of TRrkB (a neurotrophic tyrosine kinase) with its ligand brain-derived neurotrophic factor (BDNF), the upregulation of caveolin-1, and an increase in IGF-1 signaling [108].
2.3. Integrated Targeting of Non-Coding RNAs with EMT to Overcome Therapeutic Resistance in Advanced Prostate Cancer
Non-coding RNAs (ncRNAs) are molecules that are transcribed but not translated into protein products, serving the function of altering gene expression at the transcriptional, translational, and post-translational levels [161,162]. Non-coding RNAs are generated from intergenic sequences, from the introns of protein-coding genes or from antisense strands [161], and are broadly characterized by size as either small (<200 nucleotides) or long (>200 nucleotides) ncRNAs [161]. Non-coding RNAs function as regulatory molecules that mediate a wide array of cellular processes such as chromatin remodeling, transcription and post-transcriptional modifications [163], and as such, certain ncRNAs are known to be capable of functioning as oncogenes or tumor suppressors [164]. As it is estimated that over 90% of the human genome encodes for non-protein coding RNAs, and that close to 75% of those genes encode for ncRNAs [165], it is reasonable to assume that ncRNAs play a far more important—and far more complex—role in regulating gene expression in cancer than we currently realize. Excitingly, several ncRNAs have been seen to hold tremendous potential, or already serve as diagnostic or prognostic biomarkers for PCa, while other ncRNAs appear highly attractive as targets for therapeutic intervention [161]. Small non-coding RNAs include microRNAs (miRNAs), PIWI-interacting RNAs (piRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and transfer RNA-derived small RNAs (tsRNAs), among others [164,166,167], while long non-coding RNAs (lncRNAs) include antisense RNAs, sense intronic RNAs, pseudogenes and circular RNAs (circRNAs) [164,166,168]. In PCa, evidence is continuing to mount which reveals the role that several kinds of both small and long ncRNAs have in regulating EMT and metastasis.
MicroRNAs are short (19–25 nucleotide) ncRNAs that regulate post-transcriptional gene expression by either targeting mRNAs for cleavage or by repressing their translation, interacting with the 3′- untranslated regions (UTRs) of target mRNAs [169,170,171]. They are among the most extensively studied and well-known of the ncRNAs in cancer and have repeatedly been implicated for their roles in regulating EMT in PCa [172,173]. MicroRNAs can be oncogenic or tumor-suppressive, and regulate EMT in PCa by either directly inhibiting EMT-related transcription factors or cytoskeletal components or by regulating the signaling pathways involved in EMT [173]. The miR-200 family of miRNAs (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) are important negative regulators of metastasis via EMT inhibition that are all downregulated in PCa [173]. Thus, miR-200 inhibits ZEB1, ZEB2, and SLUG expression in PC3 cells [173,174]. In another study, Liu et al. had observed that both miR-1 and miR-200b target SLUG, and that SLUG also acts as a repressor of miR-1 and miR-200b transcription, suggesting that SLUG and miR-1/miR-200b regulate one another in a feedback loop that amplifies EMT [173,175]. Importantly, the overexpression of either miRNA leads to decreased SLUG, reduced growth and invasion and the reversal of EMT to MET in human PCa cell lines [173,175]. Reversing EMT via the reintroduction of EMT-suppressive miRNAs has been observed by other investigative groups, including work performed by Basu et al., wherein Hsa-miR-200c was overexpressed in hypoPC3 cells and was found to reverse the EMT markers vimentin, ZEB1, and SLUG [176]. The overexpression of miR-21 in PC3 cells leads to increased expression of vimentin and N-cadherin, and the downregulation of E-cadherin [173,177]. More recent studies [178] identified two onco-miRNAs, 181a-5p and 181b-5p, as contributors to migration and invasion inhibitory protein (MIIP), a protein with tumor suppressor functions [178]. MIIP was shown to inhibit these two miRNAs, which normally act to suppress KLF17 [178], a protein that has been shown to inhibit EMT in other human cancers [179,180]. MIIP inhibition of miRNA 181a-5p and 181b-5p also leads to reduced expression of SNAIL and TWIST, and knockdown of MIIP promoted tumor growth or osteolytic bone lesions [178], as well as AR activity [181]. Specifically, miRNA-299, located at chromosome 14q32.31, is one of a large cluster of miRNAs that have been implicated in the progression of prostate cancer [182,183]. The overexpression of miR-299 was shown to inhibit the expression of SLUG and TGF-β3 and increase E-cadherin expression, correlating with low AR levels and consequential reduced cell migration and proliferation [182]. Another emerging protagonist is miRNA-539, which was found to act by inhibiting the expression of the oncogene DLX1, leading to downstream reduction in levels of SNAIL, SLUG and vimentin, as well as inhibiting the TGF-β/Smad axis [184]. Other miRNAs linked to EMT markers inhibit the invasion of prostate cancer cells through the TGF-β/Smad axis, including miR15a/16 [185], as the overexpression of miR15a/16 leads to decreased invasiveness of LNCaP cells and the downregulation of SNAIL and TWIST [185]. Excitedly, some miRNAs have been seen to have a positive effect on overcoming therapeutic resistance, as miR27-b and miR34-a suppression of ZEB1 was shown to correlate with overcoming docetaxel resistance in vitro [186].
Extracellular Vesicles Deliver Critical Cargo
Exosomal miRNAs (exomiRNAs) have also been linked to EMT in PCa. Exosomes are a group of extracellular vesicles that were discovered in 1969 by H.C. Anderson, and have long been studied for their role in the intercellular transport of small molecules and trafficking within the tumor microenvironment [187,188]. Exosomes are produced by all cell types, range in size from 40 nm–150 nm and contain DNA, RNA and proteins [189]. Exosomal miRNAs are among the cargo of these vesicles that have been studied for their role in cancer progression [190], including PCa [191]. Exosomal miR-26a was found to downregulate N-cadherin and vimentin, while increasing E-cadherin levels in LNCaP cells [192]. Another exosomal miRNA, miR-1246, was found to be downregulated in prostate cancer cells, but was found to inhibit tumor growth in vivo while inhibiting N-cadherin and vimentin levels in vitro, suggesting a protective role against disease progression for this miRNA [193].
As there are at least 30 different miRNAs known to interact with EMT pathways and components in PCa [173], not all can be described in detail here; however, it is clear that miRNAs are emerging as key regulators of EMT in PCa. A schematic illustration of significant miRNAs that negatively regulate phenotypic EMT in PCa is shown in Figure 1.
P-element-induced wimpy testis (PIWI)-interacting RNAs (piRNAs) are a relatively new class of ncRNA, having been discovered in 2006; however, there is some evidence to suggest their role in contributing to EMT in prostate cancer [194]. These small ncRNAs are single-stranded, 24–32 nucleotides in length and interact with PIWI-proteins to form piRNA-PIWI complexes, which are involved in germline development, stem cell maintenance, epigenetic regulation and translation control [194,195,196]. In cancer, overexpression of PIWI proteins PIWIL1 and PIWIL2 has been observed in several types of carcinoma, including breast, esophageal, gastric, ovarian and colorectal cancers [194]. In prostate cancer, Yang et al. observed increased expression of PIWIL2 in malignant prostate specimens compared to non-malignant adjacent tissues [194,197], while the silencing of PIWIL2 led to decreased cell invasion and migration in PC3 prostate cancer cells [194,197]. Furthermore, loss of PIWIL2 impacted EMT effectors, reduced the expression of N-cadherin, TWIST, and vimentin, and increased E-cadherin levels [194,197]. Of translational significance is evidence that a three-piRNA signature (hsa_pir_000627, hsa_pir_005553 and hsa_pir_019346) is associated with clinical biochemical recurrence [198]. More recently, using small RNA sequencing Zhang et al. found two piRNAs, piR-001773 and piR-017184, to be upregulated in PCa, their expression correlating with Gleason score and pathological stage [199], and increased expression of piR-001773 and piR-017184 promoted the invasion and migration of androgen-independent prostate cancer cells [199]. Thus, compelling evidence supports the regulatory role of PIWI-piRNA complexes and piRNAs in EMT, with enhanced clinical relevance in PCa (Figure 1).
Small nucleolar RNAs (snoRNAs) are small (60–300 nucleotide) ncRNAs that are actually processed intron fragments, essentially recycled by cells to be used in pre-RNA processing [200,201]. SnoRNAs typically form complexes with catalytic ribonucleoproteins and function as guide RNAs that base-pair with the complementary RNA sequences that are to be modified [200,201]. SnoRNAs are usually found within the nucleoli, where they are responsible for post-transcriptionally modifying and maturing many other types of RNA, and are divided into two classes depending on the types of modifications they perform (C/D box for 2′-O-ribose methylation and H/ACA box for pseudouridylation) [201]. SnoRNAs have in recent years become increasingly implicated in tumorigenesis and cancer, with evidence building in support for their roles in contributing to EMT in prostate cancer and prostate cancer progression [202]. Sequencing of the small RNA transcriptome of normal and malignant tissues revealed that snoRNA-derived RNAs (sdRNAs) are highly upregulated in PCa and account for over one third of the differentially expressed small RNAs in tumor tissue compared to normal adjacent tissues [203]. SdRNAs derived from C/D box snoRNAs were also found to be associated with the development of metastatic disease [203]. In a separate study, Crea et al. had found that SNORA55 was upregulated in metastatic vs. non-metastatic paired PCa xenografts, and that it could also predict shorter relapse-free survival [203]. Silencing of SNORA55 led to reduced proliferation and migration in PCa cell lines [204]. In 2018, Yi et al. found that H/ACA snoRNA SNORA42 was upregulated in PCa cell lines and tissue samples, and that the overexpression of SNORA42 inhibited apoptosis and increased cell proliferation, migration and invasion [202]. Additionally, PC3 and DU145 cells transiently-transfected with SNORA42 were found to have increased expression of vimentin, N-cadherin and ZEB1 with decreased expression of E-cadherin, while small interfering RNA (siRNA) knockdown of SNORA42 led to a reversal of this phenotype, with decreased vimentin, N-cadherin and ZEB1, paralleled by an increased expression of E-cadherin [202].
Long non-coding RNAs (lncRNAs, those ncRNAs that are >200 nucleotides in length) are another major class of ncRNAs known to be involved in regulating EMT and prostate cancer progression. They are structurally similar to protein coding genes in several respects, yet they possess no open reading frames, have fewer exons and are generally expressed at lower levels than their protein coding counterparts [161,164]. Compared to smaller ncRNAs, lncRNAs are able to fold into secondary and tertiary structures [162] and exhibit far greater functional diversity [164]. LncRNAs can regulate gene expression at the epigenetic, transcriptional, and post-transcriptional levels, and can either operate near their own sites of transcription (i.e., cis-acting) or act in distant genomic or cellular locations relative to where they were transcribed (i.e., trans-acting) [164]. Their regulatory mechanistic repertoire includes the ability to guide chromatin modifiers to specific genomic locations (to activate or suppress transcription), alter pre-mRNA splicing, inhibit mRNA translation, and act as decoys to displace transcriptional repressors or as scaffolds for multiple protein complexes to interact with one another [205,206]. One of the first lncRNAs to be described in PCa was prostate cancer gene expression marker 1 (PCGEM1), a lncRNA that inhibits apoptosis and promotes cell proliferation in vitro via enhanced androgen-dependent gene transcription [161]. Amongst the lncRNAs most characterized as clinically relevant is prostate cancer antigen 3 (PCA3), a unique, atypically alternatively spliced lncRNA mapped to the long arm of human chromosome 9q21–22 [207] and overexpressed in >95% of primary prostate tumors [161,208]. PCA3 is the most specific prostate cancer molecule currently known to date, and is used as a diagnostic biomarker for PCa in the US, Europe and Canada [207]. Functional loss of PCA3 increases the expression of SLUG, SNAIL, and E-cadherin in LNCaP cells [209]. Some lncRNAs act by competitively binding to miRNAs, while others act independently of miRNAs. Specifically, ZNFX1 antisense RNA 1 (ZFAS1) [210] and small nucleolar RNA host gene 3 (SNHG3) [211] have been shown to bind miRNAs that inhibit EMT and promote the apoptosis of prostate cancer cells. LncRNA SNHG7 was also suggested to promote EMT in prostate cancer via binding to miRNA-324-3p, as well as through the Wnt pathway [212], while the lncRNA E3 ubiquitin-protein ligase (CHFR) was found to act through multiple pathways via miR-10b to promote EMT in PC3 cells, mainly through the GSK/AKT and NF-κB pathways [213]. In oral squamous cell carcinoma, the downstream targets of lncRNAs include the PI3K/AKT pathway, under the regulation of lncRNA metastasis associated lung adenocarcinoma transcript 1 (MALAT1) [214]. In the same study, it was also shown that MALAT1 modulation of the PI3K/AKT pathway was associated with EMT induction [214]. In prostate cancer, the loss of MALAT1 impedes the growth of PCa xenografts [215] and reduces cell proliferation and migration, while it promotes apoptosis in AR-negative prostate cancer cells [216]. VIM antisense RNA 1 (VIM-AS1) increases N-cadherin and vimentin while downregulating E-cadherin in promoting prostate cancer EMT [217].
Circular RNAs (circRNAs) have also been linked to EMT and PCa progression, although the evidence supporting these roles for circRNAs in PCa is continuing to emerge. Circular RNAs are closed loop sequences of RNA that lack 5′ or 3′ ends, and have the ability to affect gene expression by binding to miRNA (acting as miRNA sponges), RNA binding proteins, and protein kinases, among other components [218]. Dai et al. found that the circRNA myosin light chain kinase (MYLK) was significantly upregulated in both bladder and prostate cancers, and that it promoted cancer progression via the downregulation of miRNA-29a expression [219]. In PCa, circular RNA17 has been found to be inversely correlated to prostate cancer aggressiveness and enzalutamide resistance [220]. One circRNA, circSMAD2, plays a role in attenuating EMT in prostate cancer cells (Figure 1). Han et al. demonstrated that circSMAD2 levels were low in prostate cancer cells and that circSMAD2 upregulation led to the inhibition of invasion and EMT through miR-9 [221].
2.4. Epigenetic Regulation by ncRNAs Contributes to EMT and Disease Progression
Epigenetic modifications are diverse, and include covalent modifications to DNA (i.e., acetylation, methylation, phosphorylation) as well as post-translational modifications to histones [206,222]. An altered epigenetic landscape both results from and contributes to cancer, a landscape that can be actively shaped from the participation of ncRNAs [206]. Dysregulated ncRNA expression is associated with the development of tumors and can influence epigenetic modifications; however, interestingly enough, ncRNA dysregulation appears to primarily result from epigenetic changes [206].
MicroRNA regulation of the epigenome occurs via their post-transcriptional silencing of epigenetic modifiers such as histone deacetylases (HDACs), histone methyltransferases (HMTs) and DNA methyltransferases (DNMTs) [206]. An important example of miRNA epigenetic regulation in prostate cancer is miR-101 regulation of enhancer of zeste homolog 2 (EZH2) [223]. EZH2 is a catalytic subunit that is part of the chromatin-modifying, epigenetic modulator polycomb repressor complex 2 (PRC2), and is overexpressed in PCa and associated with metastatic and neuroendocrine disease [223,224,225]. In fact, EZH2 is thought to be a master regulator of NEPC reprogramming and is overly expressed in the vast majority (87%) of NEPC patients [225]. miR-101 negatively regulates EZH2, and the downregulation of miR-101, which is frequently seen in PCa, may be directly responsible for the upregulation of EZH2 [223,226]. Functional restoration of miR-101 expression inhibits EZH2 and decreases cell proliferation and tumor invasiveness [223,226]. Mechanistically, the expression of members of the miR-200 family (including miR-205) that regulate EMT in PCa are themselves epigenetically regulated [223]. miR-205 expression, for example, is downregulated in PCa via hypermethylation of its promoter, and is associated with resistance to chemotherapy. Significantly, miR-200c and miR-141 are also downregulated in androgen-independent prostate cancer cells and contain a hypermethylated CpG promoter, but not in androgen-sensitive cells, in which the promotor region of these genes remains unmethylated [223,227,228].
LncRNA epigenetic regulation also occurs through their direct interactions with epigenetic modifiers [206]. The lncRNA second chromosome locus associated with prostate-1 (SChLAP1) is found to be overexpressed in PCa, with significantly increased levels in metastatic tumors [206,229]. Mechanistically, SChLAP1 interacts with and antagonizes SWItch/Sucrose Non-Fermentable (SWI/SNF), a chromatin remodeling complex that exhibits tumor-suppressive activity, thus SChLAP1 overexpression promotes cell invasion and metastasis [206,229]. The lncRNA HOXD cluster anti-sense RNA 1 (HOXD-AS1) is also overexpressed in PCa and highly expressed in CRPC cells, and correlates with Gleason score and metastasis [230]. Recent mechanistic insights revealed that HOXD-AS1 recruits WD repeat-containing protein 5 (WDR5), a key subunit of the lysine-specific methyltransferase 2A (MLL1) chromatin remodeling complex, and regulates target gene transcription via mediating histone H3 lysine 4 tri-methylation (H3K4me3) to promote chemo-resistance of human prostate cancer cells [230].
The concerted involvement of so many various noncoding RNAs and other molecular species in epigenetic gene regulation can be utilized to generate clinically-useful epigenetic noncoding RNA signatures with prognostic or diagnostic value [231]. While few to no epigenetic biomarkers exist that can identify aggressive phenotypes, epigenetic biomarkers are emerging that can, for example, predict clinically significant cancer in patients on active surveillance (AS) [231]. The development and progression of PCa are frequently associated with epigenetic changes such as global DNA hypomethylation, and the hypermethylation of genes such as GSTP1 (glutathione S-transferase Pi 1) and HOXD8, and the dysregulation of ncRNAs such as miR-129a (decreased expression) and miR-18a (increased expression) [231]. Table 3 summarizes current evidence supporting the functional contributions of ncRNAs to EMT and defining their potential clinical value as biomarkers in prostate cancer progression.
2.5. Pharmacologic Targeting of EMT to Overcome Prostate Cancer Resistance
There has been extensive investigation into various therapeutic actionable targets in signaling pathways EMT to MET interconversions. Cajigas-du Ross et al., [246] for example, performed RNA-seq on docetaxel-resistant and -sensitive prostate cancer cells and found that E-cadherin levels were significantly reduced, while there was an increase in vimentin, SNAIL and TWIST levels in docetaxel-resistant prostate cancer cell lines [246]. Moreover, the same group reported robust upregulation of tetraspanin-8 (TSPAN8) as one of several key proteins involved in this process, which encodes proteins involved with cell–cell communication via interaction with integrins [246], suggesting its involvement in the acquisition of chemoresistance in PCa. Xue et al. [247] treated human androgen-independent prostate cancer cells with a combination of zinc and paclitaxel in an attempt to overcome taxane resistance [247]. Zinc was found to increase prostate cell sensitivity to paclitaxel, while the combination of zinc and paclitaxel decreased the expression of TWIST1 and induced apoptosis [247]. Loss of TWIST1 increased the sensitivity of these cells to taxane [247], while increased ZEB1 was detected in response to docetaxel; taken together, this evidence implicates a role for EMT regulators in resistance to taxane chemotherapy [248].
Receptor tyrosine kinases (RTKs) have also been studied for their connection to EMT and taxane resistance in prostate cancer. One such RTK, tyrosine-protein kinase receptor UFO (AXL), has been implicated for its role in resistance to imatinib and erlotinib in leukemia and non-small cell lung cancer, respectively [249,250]. In prostate cancer, AXL was found to be overexpressed in docetaxel-resistant cell lines, and AXL overexpression alone was found sufficient to induce resistance to docetaxel [251]. The inhibition of AXL abated EMT phenotypic features and suppressed tumor proliferation and migration, positing AXL as a possible therapeutic target to overcome docetaxel resistance [251].
The PI3K/AKT survival signaling pathway has also been implicated in shaping the EMT phenotypic landscape within the prostate tumor microenvironment. Chen and colleagues probed the PI3K/AKT pathway using the tumor suppressor inositol polyphosphate 4-phosphatase B (INPP4B) on prostate cancer cells, finding that overexpression of INPP4B led to increased sensitivity to docetaxel [252]. Mechanistically, INPP4B was found to inhibit the PI3K/AKT pathway, as well as upregulate E-cadherin and reduce levels of vimentin, fibronectin, and N-cadherin [252], thus the PI3K/AKT pathway could be a link between docetaxel resistance and EMT. Additionally, pre-clinical models have demonstrated that splice variants of AR, most notably AR-V7, are linked to EMT and mesenchymal phenotypes [253,254]. The EMT transcriptional suppressor SNAIL enables a potential link between full-length AR, AR splice variants and EMT, as increasing levels of SNAIL promote antiandrogen resistance and increased AR activity, whereas the repression of SNAIL re-sensitized resistant prostate cancer cells to enzalutamide [255].
The anoikis-driven antitumor effect of α1-adrenoreceptor antagonists promises a safe-strategy in treating advanced disease—both therapeutically-resistant and castration-sensitive prostate cancer [143,256,257]. Quinazoline-based compounds developed after the pharmacological optimization of α1-adrenoceptor antagonists cause phenotypic reversion of EMT to MET and induce anoikis towards overcoming resistance to AR antiandrogens in pre-clinical models of advanced prostate cancer [143,257,258,259].
3. Conclusions
Since the original work by Charles Huggins in 1941 on the effects of ADT on progression to lethal disease, the emergence of castration resistance in patients with prostate cancer has reinforced the need for understanding actionable drivers of prostate cancer progression beyond AR, its ligands, and downstream targets. Prostate cancer is remarkably heterogenous and driven by a host of molecular factors; evidence-based knowledge of the genomic and molecular underpinnings of PCa has paved the way for personalized treatments and reliable biomarkers with diagnostic or prognostic value. The PARP (poly (adenosine diphosphate (ADP)-ribose) polymerase) inhibitor olaparib and the lncRNA biomarker PCA3 mentioned previously are two such examples. Olaparib, originally used to treat BRCA-driven ovarian cancers [260], was recently FDA approved last year for the treatment of mCRPC in men with alterations in genes involved in homologous recombination repair who failed antiandrogen therapy [70]. PARP is an enzyme involved in multiple DNA repair pathways and in repairing single strand breaks, which eventually lead to cell death if not addressed [261]. Interestingly, and fittingly so, recent mechanistic evidence revealed that the silencing of PARP1 in prostate cancer cells suppresses their growth and induces MET [262].
Non-coding RNAs are as rich and diverse in function as they are in number, and intense efforts pursue their potential to become clinically actionable. One could easily argue that defining the role of each ncRNA as a driver of phenotypic EMT in the context of prostate cancer metastasis and/or therapeutic resistance would lead to the development of novel phenotypic or molecular signatures that yield diagnostic and therapeutic value (summarized on Table 3). Could ncRNA signatures better predict NEPC, therapeutic resistance or metastasis-free survival? Considering the emerging value of ncRNA biomarkers, these genomic regulators are of high potential clinical relevance as prostate-specific signatures with therapeutic targeting value in lethal disease.
Author Contributions
Conceptualization J.A., J.A.S. and N.K.; original draft preparation, J.A.; writing and editing, J.A., J.A.S. and N.K.; supervision, N.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by an NIH/NCI R01 CA232574 grant (N.K.) and the Department of Urology, The Tisch Cancer Institute at Icahn School of Medicine at Mount Sinai, New York.
Acknowledgments
The authors thank BioRender.com for use of their services.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABCC4 | ATP-binding cassette sub-family C member 4 |
| ADT | Androgen Deprivation Therapy |
| AKT | Protein kinase B |
| AKT1 | RAC-alpha serine/threonine-protein kinase |
| AR | Androgen Receptor |
| AR-V | AR splice variant |
| AR-V7 | AR splice variant 7 |
| AS | Active surveillance |
| AXL | Tyrosine-protein kinase receptor UFO |
| AURKA | Aurora Kinase A |
| BAD | BCL-2 associated agonist of cell death |
| BAX | Bcl-2-like protein 4 |
| BCL-2 | B-cell lymphoma 2 |
| BD | Brain-derived neurotrophic factor |
| BID | BH3 interacting-domain death agonist |
| BPH | Benign prostatic hyperplasia |
| CHFR | E3 ubiquitin-protein ligase |
| CHGA | Chromogranin A |
| circRNA | Circular RNA |
| CRPC | Castration-resistant prostate cancer |
| CSPS | Castration-sensitive prostate cancer |
| CYP17 | Cytochrome p450 enzyme 17R-hydroxylase-17,20-lyase |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DLX1 | Homeobox protein DLX-1 |
| DNA | Deoxyribonucleic acid |
| DNMT | DNA methyltransferase |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EMT | Epithelial-to-mesenchymal transition |
| ENO2 | Enolase 2 |
| ExomiRNA | Exosomal miRNA |
| EZH2 | Enhancer of zeste homolog 2 |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSTP1 | Glutathione S-transferase Pi 1 |
| H3K4me3 | Histone H3 lysine 4 tri-methylation |
| HDAC | Histone deacetylase |
| HER2/neu | Human epidermal growth factor receptor 2 |
| HMT | Histone methyltransferase |
| HOXD-AS1 | HOXD cluster anti-sense RNA 1 |
| IGF-1 | Insulin-like growth factor 1 |
| IGFBP-3 | Insulin-like growth factor-binding protein 3 |
| INPP4B | Inositol polyphosphate 4-phosphatase B |
| KGF | Keratinocyte growth factor |
| KLF17 | Kruppel Like Factor 17 |
| LBD | Ligand binding domain |
| LH | Luteinizing hormone |
| LHRH | Luteinizing hormone releasing-hormone |
| lncRNA | Long non-coding RNA |
| MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| mCRPC | Metastatic castration-resistant prostate cancer |
| mCSPC | Metastatic castration-sensitive prostate cancer |
| MET | Mesenchymal-to-epithelial transition |
| MFS | Metastasis-free survival |
| MIIP | Migration and invasion inhibitory protein |
| miRNA | microRNA |
| MLL1 | Lysine-specific methyltransferase 2A |
| MMP | Matrix metalloprotease |
| mTOR | Mammalian target of rapamycin |
| MYLK | Myosin light chain kinase |
| N-MYC | N-myc proto-oncogene protein |
| ncRNA | Non-coding RNA |
| NED | Neuroendocrine differentiation |
| NEPC | Neuroendocrine prostate cancer |
| nmCRPC | Non-metastatic castration-resistant prostate cancer |
| nmCSPC | Non-metastatic castration-sensitive prostate cancer |
| OC2 | ONECUT2 transcription factor |
| OS | Overall survival |
| P-gp | P-glycoprotein |
| PAP | Prostatic acid phosphatase |
| PARP | Poly (adenosine diphosphate (ADP)-ribose) polymerase |
| PCa | Prostate cancer |
| PCA3 | Prostate cancer antigen 3 |
| PCGEM1 | Prostate cancer gene expression marker 1 |
| PD1 | Programmed cell death protein 1 |
| PDGF | Platelet-derived growth factor |
| PFS | Progression-free survival |
| PI3K | Phosphoinositide 3-kinase |
| PIN | Prostatic intraepithelial neoplasia |
| piRNA | PIWI-interacting RNAs |
| PIWI | P-element-induced wimpy testis |
| PIWIL1 | Piwi-like protein 1 |
| PIWIL2 | Piwi-like protein 2 |
| PRC2 | Polycomb repressor complex 2 |
| PSA | Prostate specific antigen |
| PTEN | Phosphatase and tensin homolog |
| R-Smad | Receptor-regulated Smad protein |
| RB1 | Retinoblastoma 1 |
| RNA | Ribonucleic acid |
| RTK | Receptor tyrosine kinase |
| SChLAP1 | Second chromosome locus associated with prostate-1 |
| sdRNA | SnoRNA-derived RNA |
| siRNA | Small interfering RNA |
| SLUG | Zinc finger protein SNAI2 |
| SNAIL | Zinc finger protein SNAI1 |
| SNHG3 | Small nucleolar RNA host gene 3 |
| SNHG7 | Small nucleolar RNA host gene 7 |
| snoRNA | Small nucleolar RNA |
| snRNA | Small nuclear RNA |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| SYP | Synaptophysin |
| t-NEPC | Therapy-induced NEPC |
| TβRI | Type I transforming growth factor-β receptor |
| TβRII | Type II transforming growth factor-β receptor |
| TGF-β | Transforming growth factor-β |
| TGF-β2 | Transforming growth factor-β2 |
| TGF-β3 | Transforming growth factor-β3 |
| TMPRSS-ERG | Transmembrane protease serine 2-v‑ets erythroblastosis virus E26 oncogene homolog |
| TP53 | Tumor protein 53 |
| TrkB | Tropomyosin receptor kinase B |
| TSP-1 | Thrombospondin 1 |
| TSPAN8 | Tetraspanin-8 |
| tsRNA | Transfer RNA-derived small RNAs |
| TWIST | Twist-related protein 1 |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
| VIM-AS1 | VIM antisense RNA 1 |
| WDR5 | WD repeat-containing protein 5 |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
| ZEB2 | Zinc finger E-box-binding homeobox 2 |
| ZFAS1 | ZNFX1 antisense RNA 1 |
| ZO-1 | Zona occludens-1 |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Common Cancer Types. Available online: https://www.cancer.gov/types/common-cancers (accessed on 11 February 2020).
- Houts, P.S.; Lenhard, R.; Varricchio, C. ACS Cancer Facts and Figures. Cancer Pract. 2000, 8, 105–108. [Google Scholar] [CrossRef]
- Catalona, W.J.; Smith Ds Fau-Ratliff, T.L.; Ratliff Tl Fau-Basler, J.W.; Basler, J.W. Detection of organ-confined prostate cancer is increased through prostate-specific antigen-based screening. JAMA 1993, 270, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol 2019, 9, 801. [Google Scholar] [CrossRef]
- Huggins, C.; Stevens, R.E., Jr.; Hodges, C.V. Studies on Prostatic Cancer: II. The Effects of Castration on Advanced Carcinoma of the Prostate Gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Schrecengost, R.; Knudsen, K.E. Molecular pathogenesis and progression of prostate cancer. Semin. Oncol. 2013, 40, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Hotte, S.J.; Saad, F. Current management of castrate-resistant prostate cancer. Curr. Oncol. 2010, 17 (Suppl 2), S72–S79. [Google Scholar] [CrossRef]
- Nakazawa, M.; Paller, C.; Kyprianou, N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [Green Version]
- Gregory, C.W.; He, B.; Johnson, R.T.; Ford, O.H.; Mohler, J.L.; French, F.S.; Wilson, E.M. A Mechanism for Androgen Receptor-mediated Prostate Cancer Recurrence after Androgen Deprivation Therapy. Cancer Res. 2001, 61, 4315. [Google Scholar]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.A.-O.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Fau-Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef]
- Nakazawa, M.; Antonarakis, E.S.; Luo, J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm Cancer 2014, 5, 265–273. [Google Scholar] [CrossRef]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef]
- Djusberg, E.; Jernberg, E.; Thysell, E.; Golovleva, I.; Lundberg, P.; Crnalic, S.; Widmark, A.; Bergh, A.; Brattsand, M.; Wikstrom, P. High levels of the AR-V7 Splice Variant and Co-Amplification of the Golgi Protein Coding YIPF6 in AR Amplified Prostate Cancer Bone Metastases. Prostate 2017, 77, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, G.; Greenberg, N.M.; Scher, H.I.; Harris, J.M.; Marshall, V.R.; Tilley, W.D. Collocation of androgen receptor gene mutations in prostate cancer. Clin. Cancer Res. 2001, 7, 1273–1281. [Google Scholar] [PubMed]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper GG, J.M.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Bohl, C.E.; Miller, D.D.; Chen, J.; Bell, C.E.; Dalton, J.T. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol Chem. 2005, 280, 37747–37754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, C.E.; Gao, W.; Miller, D.D.; Bell, C.E.; Dalton, J.T. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 6201–6206. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.; Miyamoto, H.; Shima, H.; Chang, C. From estrogen to androgen receptor: A new pathway for sex hormones in prostate. Proc. Natl. Acad. Sci. USA 1998, 95, 5527–5532. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.D.; Haugk, K.; Woodke, L.; Nelson, P.; Coleman, I.; Plymate, S.R. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J. Cell. Biochem. 2006, 99, 392–401. [Google Scholar] [CrossRef]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar] [PubMed]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobisch, A.; Eder, I.E.; Putz, T.; Horninger, W.; Bartsch, G.; Klocker, H.; Culig, Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998, 58, 4640–4645. [Google Scholar] [PubMed]
- Lamont, K.R.; Tindall, D.J. Minireview: Alternative Activation Pathways for the Androgen Receptor in Prostate Cancer. Mol. Endocrinol. 2011, 25, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, K.; Neuwirt, H.; Cavarretta, I.T.; Bektic, J.; Steiner, H.; Dietrich, H.; Moser, P.L.; Fuchs, D.; Hobisch, A.; Culig, Z. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr. Relat. Cancer 2009, 16, 155–169. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J.; Brinkmann, A.O. Androgen Physiology: Receptor and Metabolic Disorders. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Xin, L.; Teitell, M.A.; Lawson, D.A.; Kwon, A.; Mellinghoff, I.K.; Witte, O.N. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 7789–7794. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Altuwaijri, S.; Deng, F.; Chen, L.; Lal, P.; Bhanot, U.K.; Korets, R.; Wenske, S.; Lilja, H.G.; Chang, C.; et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 2009, 175, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caram, M.E.V.; Kaufman, S.R.; Modi, P.K.; Herrel, L.; Oerline, M.; Ross, R.; Skolarus, T.A.; Hollenbeck, B.K.; Shahinian, V. Adoption of Abiraterone and Enzalutamide by Urologists. Urology 2019, 131, 176–183. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Buttigliero, C.; Tucci, M.; Bertaglia, V.; Vignani, F.; Bironzo, P.; Di Maio, M.; Scagliotti, G.V. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 2015, 41, 884–892. [Google Scholar] [CrossRef]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L Mutation in Androgen Receptor Confers Genetic and Phenotypic Resistance to MDV3100 (Enzalutamide). Cancer Discov. 2013, 3, 1030. [Google Scholar] [CrossRef] [Green Version]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015, 7, 312re310. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S. Current understanding of resistance to abiraterone and enzalutamide in advanced prostate cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 316–319. [Google Scholar]
- Maughan, B.L.; Antonarakis, E.S. Androgen pathway resistance in prostate cancer and therapeutic implications. Expert Opin. Pharmacother. 2015, 16, 1521–1537. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Boudadi, K.; Antonarakis, E.S. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez Soto, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in Metastatic Prostate Cancer without Previous Chemotherapy. N. Engl. J. Med. 2012, 368, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Tannock, I.F.; Osoba, D.; Stockler, M.R.; Ernst, D.S.; Neville, A.J.; Moore, M.J.; Armitage, G.R.; Wilson, J.J.; Venner, P.M.; Coppin, C.M.; et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: A Canadian randomized trial with palliative end points. J. Clin. Oncol. 1996, 14, 1756–1764. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, G.; Yau, T.C.; Chiu, J.W.; Tse, E.; Kwong, Y.L. Pembrolizumab (Keytruda). Hum. Vaccin. Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef] [Green Version]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Dong, X.; Gleave, M. Molecular model for neuroendocrine prostate cancer progression. BJU Int. 2018, 122, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.K.; Chugh, N.; Tripathi, M. Neuroendocrine Differentiation of Prostate Cancer-An Intriguing Example of Tumor Evolution at Play. Cancers 2019, 11, 1405. [Google Scholar] [CrossRef] [Green Version]
- Vellky, J.E.; Ricke, W.A. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia 2020, 22, 566–575. [Google Scholar] [CrossRef]
- Lotan, T.L.; Gupta, N.S.; Wang, W.; Toubaji, A.; Haffner, M.C.; Chaux, A.; Hicks, J.L.; Meeker, A.K.; Bieberich, C.J.; De Marzo, A.M.; et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod. Pathol. 2011, 24, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yanez, A.; Hazelett, D.J.; et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med. 2018, 24, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Cattrini, C.; Capaia, M.; Boccardo, F.; Barboro, P. Etoposide and topoisomerase II inhibition for aggressive prostate cancer: Data from a translational study. Cancer Treat. Res. Commun. 2020, 25, 100221. [Google Scholar] [CrossRef]
- Sargos, P.; Ferretti, L.; Gross-Goupil, M.; Orre, M.; Cornelis, F.; Henriques de Figueiredo, B.; Houédé, N.; Merino, C.; Roubaud, G.; Dallaudiére, B.; et al. Characterization of prostate neuroendocrine cancers and therapeutic management: A literature review. Prostate Cancer Prostatic Dis. 2014, 17, 220–226. [Google Scholar] [CrossRef]
- Wang, H.T.; Yao, Y.H.; Li, B.G.; Tang, Y.; Chang, J.W.; Zhang, J. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: Factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 2014, 32, 3383–3390. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [Green Version]
- Ingrosso, G.; Detti, B.; Scartoni, D.; Lancia, A.; Giacomelli, I.; Baki, M.; Carta, G.; Livi, L.; Santoni, R. Current therapeutic options in metastatic castration-resistant prostate cancer. Semin. Oncol. 2018, 45, 303–315. [Google Scholar] [CrossRef]
- Morgan, T.M.; Koreckij, T.D.; Corey, E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/mTOR pathway. Curr. Cancer Drug Targets 2009, 9, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Ojima, I.; Lichtenthal, B.; Lee, S.; Wang, C.; Wang, X. Taxane anticancer agents: A patent perspective. Expert Opin. Ther. Pat. 2016, 26, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienta, K.J. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin. Oncol. 2001, 28, 3–7. [Google Scholar] [CrossRef]
- Bai, S.; Zhang, B.Y.; Dong, Y. Impact of taxanes on androgen receptor signaling. Asian J. Androl. 2019, 21, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Lohiya, V.; Aragon-Ching, J.B.; Sonpavde, G. Role of Chemotherapy and Mechanisms of Resistance to Chemotherapy in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, B.; Li, W. Taxane resistance in castration-resistant prostate cancer: Mechanisms and therapeutic strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef]
- Abidi, A. Cabazitaxel: A novel taxane for metastatic castration-resistant prostate cancer-current implications and future prospects. J. Pharmacol. Pharmacother. 2013, 4, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Kartner, N.; Riordan, J.R.; Ling, V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983, 221, 1285–1288. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Armstrong, C.; Lou, W.; Sandher, A.; Gao, A.C. Antiandrogens Inhibit ABCB1 Efflux and ATPase Activity and Reverse Docetaxel Resistance in Advanced Prostate Cancer. Clin. Cancer Res. 2015, 21, 4133–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-F.; Ji, H.-H.; Zhang, Z.-L.; Zhang, T.-T.; Gan, W.; Zhang, S.-F. Targeting MRP4 expression by anti-androgen treatment reverses MRP4-mediated docetaxel resistance in castration-resistant prostate cancer. Oncol. Lett. 2017, 14, 1748–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [Green Version]
- Rhea, L.P.; Mendez-Marti, S.; Aragon-Ching, J.B. Darolutamide for treatment of castration-resistant prostate cancer. Drugs Today 2020, 56, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef]
- Thara, E.; Dorff, T.B.; Pinski, J.K.; Quinn, D.I. Vaccine therapy with sipuleucel-T (Provenge) for prostate cancer. Maturitas 2011, 69, 296–303. [Google Scholar] [CrossRef]
- Handy, C.E.; Antonarakis, E.S. Sipuleucel-T for the treatment of prostate cancer: Novel insights and future directions. Future Oncol 2018, 14, 907–917. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Mol. Aspects Med. 2010, 31, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: Lethal “Liaisons” during Cancer Progression. Crit Rev. Oncog. 2016, 21, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Li, H.; Zhou, J.; Chen, X.; Tang, D.G. Cellular determinants and microenvironmental regulation of prostate cancer metastasis. Semin. Cancer Biol. 2017, 44, 83–97. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Rennebeck, G.; Martelli, M.; Kyprianou, N. Anoikis and Survival Connections in the Tumor Microenvironment: Is There a Role in Prostate Cancer Metastasis? Cancer Res. 2005, 65, 11230. [Google Scholar] [CrossRef] [Green Version]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. Adv. Exp. Med. Biol 2018, 1095, 101–110. [Google Scholar] [CrossRef]
- Verze, P.; Cai, T.; Lorenzetti, S. The role of the prostate in male fertility, health and disease. Nat. Rev. Urol. 2016, 13, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Krušlin, B.; Ulamec, M.; Tomas, D. Prostate cancer stroma: An important factor in cancer growth and progression. Bosn J. Basic Med. Sci 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Tyekucheva, S.; Bowden, M.; Bango, C.; Giunchi, F.; Huang, Y.; Zhou, C.; Bondi, A.; Lis, R.; Van Hemelrijck, M.; Andren, O.; et al. Stromal and epithelial transcriptional map of initiation progression and metastatic potential of human prostate cancer. Nat. Commun. 2017, 8, 420. [Google Scholar] [CrossRef] [Green Version]
- De Marzo, A.M.; Meeker, A.K.; Epstein, J.I.; Coffey, D.S. Prostate stem cell compartments: Expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am. J. Pathol 1998, 153, 911–919. [Google Scholar] [CrossRef]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate cancer stem cells: Deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef]
- Kassen, A.; Sutkowski, D.M.; Ahn, H.; Sensibar, J.A.; Kozlowski, J.M.; Lee, C. Stromal cells of the human prostate: Initial isolation and characterization. Prostate 1996, 28, 89–97. [Google Scholar] [CrossRef]
- Nieto, C.M.; Rider, L.C.; Cramer, S.D. Influence of stromal-epithelial interactions on androgen action. Endocr. Relat. Cancer 2014, 21, T147–T160. [Google Scholar] [CrossRef] [Green Version]
- Berry, P.A.; Maitland, N.J.; Collins, A.T. Androgen receptor signalling in prostate: Effects of stromal factors on normal and cancer stem cells. Mol. Cell. Endocrinol. 2008, 288, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Cunha, G.R.; Ricke, W.; Thomson, A.; Marker, P.C.; Risbridger, G.; Hayward, S.W.; Wang, Y.Z.; Donjacour, A.A.; Kurita, T. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J. Steroid Biochem. Mol. Biol. 2004, 92, 221–236. [Google Scholar] [CrossRef]
- Niu, Y.-N.; Xia, S.-J. Stroma-epithelium crosstalk in prostate cancer. Asian J. Androl. 2009, 11, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar] [PubMed]
- San Martin, R.; Barron, D.A.; Tuxhorn, J.A.; Ressler, S.J.; Hayward, S.W.; Shen, X.; Laucirica, R.; Wheeler, T.M.; Gutierrez, C.; Ayala, G.E.; et al. Recruitment of CD34(+) fibroblasts in tumor-associated reactive stroma: The reactive microvasculature hypothesis. Am. J. Pathol 2014, 184, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuxhorn, J.A.; Ayala, G.E.; Rowley, D.R. Reactive stroma in prostate cancer progression. J. Urol. 2001, 166, 2472–2483. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, Q.; Long, X.; Chen, X.; Wang, Y. Vimentin affects the mobility and invasiveness of prostate cancer cells. Cell Biochem. Funct. 2008, 26, 571–577. [Google Scholar] [CrossRef]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb Perspect Med. 2018, 8, a030510. [Google Scholar] [CrossRef]
- Stewart, D.A.; Cooper, C.R.; Sikes, R.A. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol. Endocrinol. 2004, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Tomas, D.; Ulamec, M.; Hudolin, T.; Bulimbašić, S.; Belicza, M.; Krušlin, B. Myofibroblastic stromal reaction and expression of tenascin-C and laminin in prostate adenocarcinoma. Prostate Cancer Prostatic Dis. 2006, 9, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Kyprianou, N. Mechanisms navigating the TGF-beta pathway in prostate cancer. Asian J. Urol. 2015, 2, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, M.M.; Kim, S.K.; Barron, L.; Kodali, R.; Baardsnes, J.; Hinck, C.S.; Krzysiak, T.C.; Henen, M.A.; Pakhomova, O.; Mendoza, V.; et al. Binding Properties of the Transforming Growth Factor-β Coreceptor Betaglycan: Proposed Mechanism for Potentiation of Receptor Complex Assembly and Signaling. Biochemistry 2016, 55, 6880–6896. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Vo, B.T.; Morton, D., Jr.; Komaragiri, S.; Millena, A.C.; Leath, C.; Khan, S.A. TGF-β effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 2013, 154, 1768–1779. [Google Scholar] [CrossRef]
- Guo, Y.; Kyprianou, N. Overexpression of transforming growth factor (TGF) beta1 type II receptor restores TGF-beta1 sensitivity and signaling in human prostate cancer cells. Cell Growth Differ. 1998, 9, 185–193. [Google Scholar] [PubMed]
- Kang, H.Y.; Lin, H.K.; Hu, Y.C.; Yeh, S.; Huang, K.E.; Chang, C. From transforming growth factor-beta signaling to androgen action: Identification of Smad3 as an androgen receptor coregulator in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3018–3023. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M.; Kyprianou, N. Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl. Androl. Urol. 2013, 2, 202–211. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun-Ju Huang, R.; Yo-Yan Huang, T. A new dimension in drug discovery: Reversing epithelial-mesenchymal transition (EMT). Cell Death Dis. 2016, 7, e2417. [Google Scholar] [CrossRef] [Green Version]
- Hensley, P.J.; Desiniotis, A.; Wang, C.; Stromberg, A.; Chen, C.S.; Kyprianou, N. Novel pharmacologic targeting of tight junctions and focal adhesions in prostate cancer cells. PLoS ONE 2014, 9, e86238. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Celiktaş, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S.; et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am. J. Pathol 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Tilló, E.; Lázaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Jin, S.; Li, Y.; Khadaroo, P.A.; Dai, Y.; He, L.; Zhou, D.; Lin, H. Genetic And Epigenetic Regulation Of E-Cadherin Signaling In Human Hepatocellular Carcinoma. Cancer Manag. Res. 2019, 11, 8947–8963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.-H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Multitasking in tumor progression: Signaling functions of cell adhesion molecules. Ann. N. Y. Acad. Sci. 2004, 1014, 58–66. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [Green Version]
- Yardy, G.W.; Brewster, S.F. Wnt signalling and prostate cancer. Prostate Cancer Prostatic Dis. 2005, 8, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Auersperg, N.; Pan, J.; Grove, B.D.; Peterson, T.; Fisher, J.; Maines-Bandiera, S.; Somasiri, A.; Roskelley, C.D. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc. Natl. Acad. Sci. USA 1999, 96, 6249–6254. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.J.; Oh, S.; Lee, G.T.; Chung, J.; Min, K.; Yoon, J.; Kim, W.; Ryu, D.S.; Kim, I.Y.; Kang, D.I. Clinical Significance of Wnt/β-Catenin Signalling and Androgen Receptor Expression in Prostate Cancer. World J. Mens Health 2013, 31, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.J.; Stehr, H.; Rausch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Li, T.; Mo, Z.; Hu, Y.; Yi, Q.; He, R.; Zhu, X.; Zhou, X.; She, S.; Chen, Y. Overexpression of the galectin-3 during tumor progression in prostate cancer and its clinical implications. Int. J. Clin. Exp. Pathol. 2018, 11, 839–846. [Google Scholar] [PubMed]
- Bolton, E.M.; Tuzova, A.V.; Walsh, A.L.; Lynch, T.; Perry, A.S. Noncoding RNAs in prostate cancer: The long and the short of it. Clin. Cancer Res. 2014, 20, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Fu, P.; Qu, L.; Liu, J.; Lin, A. Long Noncoding RNAs, New Critical Regulators in Cancer Immunity. Front. Oncol 2020, 10, 550987. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guisier, F.; Camargo Barros-Filho, M.; Rock, L.D.; Constantino, B.F.; Minatel, B.C.; Sage, A.P.; Marshall, E.A.; Martinez, V.D.; Lam, W.L. Small Noncoding RNA Expression in Cancer. In Gene Expression Profiling in Cancer; Vlachakis, D., Ed.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Hua, J.T.; Chen, S.; He, H.H. Landscape of Noncoding RNA in Prostate Cancer. Trends Genet. 2019, 35, 840–851. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immunol. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Mekhail, S.M.; Yousef, P.G.; Jackinsky, S.W.; Pasic, M.; Yousef, G.M. miRNA in Prostate Cancer: New Prospects for Old Challenges. EJIFCC 2014, 25, 79–98. [Google Scholar]
- Sekhon, K.; Bucay, N.; Majid, S.; Dahiya, R.; Saini, S. MicroRNAs and epithelial-mesenchymal transition in prostate cancer. Oncotarget 2016, 7, 67597–67611. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Li, Y.; Wang, Z.; Banerjee, S.; Ahmad, A.; Kim, H.R.; Sarkar, F.H. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009, 27, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.N.; Yin, J.J.; Abou-Kheir, W.; Hynes, P.G.; Casey, O.M.; Fang, L.; Yi, M.; Stephens, R.M.; Seng, V.; Sheppard-Tillman, H.; et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 2013, 32, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Chaudhary, A.; Chowdhury, P.; Karmakar, D.; Basu, K.; Karmakar, D.; Chatterjee, J.; Sengupta, S. Evaluating the role of hsa-miR-200c in reversing the epithelial to mesenchymal transition in prostate cancer. Gene 2020, 730, 144264. [Google Scholar] [CrossRef] [PubMed]
- Jalava, S.E.; Urbanucci, A.; Latonen, L.; Waltering, K.K.; Sahu, B.; Jänne, O.A.; Seppälä, J.; Lähdesmäki, H.; Tammela, T.L.J.; Visakorpi, T. Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer. Oncogene 2012, 31, 4460–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Yan, F.; Ru, Y.; Xia, M.; Yan, G.; Zhang, M.; Wang, H.; Wu, G.; Yao, L.; Shen, L.; et al. MIIP inhibits EMT and cell invasion in prostate cancer through miR-181a/b-5p-KLF17 axis. Am. J. Cancer Res. 2020, 10, 630–647. [Google Scholar]
- Gumireddy, K.; Li, A.; Gimotty, P.A.; Klein-Szanto, A.J.; Showe, L.C.; Katsaros, D.; Coukos, G.; Zhang, L.; Huang, Q. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nat. Cell Biol. 2009, 11, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zhang, P.; Liangfang, Y.; Wenshe, S.; Wang, H.; Lin, X.; Dai, Y.; Feng, X.H.; Moses, R.; Wang, D.; et al. KLF17 empowers TGF-beta/Smad signaling by targeting Smad3-dependent pathway to suppress tumor growth and metastasis during cancer progression. Cell Death Dis. 2015, 6, e1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Östling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Mäkelä, R.; Hagman, Z.; Edsjö, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, K.; Staklinski, S.; Hasan, M.F.; Ottman, R.; Andl, T.; Berglund, A.E.; Park, J.Y.; Chakrabarti, R. Multifaceted Function of MicroRNA-299-3p Fosters an Antitumor Environment Through Modulation of Androgen Receptor and VEGFA Signaling Pathways in Prostate Cancer. Sci. Rep. 2020, 10, 5167. [Google Scholar] [CrossRef] [Green Version]
- Formosa, A.; Markert, E.K.; Lena, A.M.; Italiano, D.; Finazzi-Agro, E.; Levine, A.J.; Bernardini, S.; Garabadgiu, A.V.; Melino, G.; Candi, E. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 2014, 33, 5173–5182. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Fan, Y.; Yang, A.; Liang, L.; Cao, J. MicroRNA-539 functions as a tumour suppressor in prostate cancer via the TGF-beta/Smad4 signalling pathway by down-regulating DLX1. J. Cell Mol. Med. 2019, 23, 5934–5948. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Chen, F.; Wang, K.; Song, Y.; Fei, X.; Wu, B. miR-15a/miR-16 cluster inhibits invasion of prostate cancer cells by suppressing TGF-beta signaling pathway. Biomed. Pharmacother. 2018, 104, 637–644. [Google Scholar] [CrossRef]
- Zhang, G.; Tian, X.; Li, Y.; Wang, Z.; Li, X.; Zhu, C. miR-27b and miR-34a enhance docetaxel sensitivity of prostate cancer cells through inhibiting epithelial-to-mesenchymal transition by targeting ZEB1. Biomed. Pharmacother. 2018, 97, 736–744. [Google Scholar] [CrossRef]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Hannafon, B.N.; Ding, W.Q. Intercellular communication by exosome-derived microRNAs in cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Ding, M.; Xu, K.; Yang, C.; Mao, L.J. Exosomes in diagnosis and therapy of prostate cancer. Oncotarget 2017, 8, 97693–97700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, X.; Zhu, Z.; Li, W.; Yu, G.; Jia, Z.; Wang, X. Prostate carcinoma cell-derived exosomal MicroRNA-26a modulates the metastasis and tumor growth of prostate carcinoma. Biomed. Pharmacother. 2019, 117, 109109. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef] [Green Version]
- Litwin, M.; Szczepańska-Buda, A.; Piotrowska, A.; Dzięgiel, P.; Witkiewicz, W. The meaning of PIWI proteins in cancer development. Oncol. Lett. 2017, 13, 3354–3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, T.; Lin, H. The biogenesis and function of PIWI proteins and piRNAs: Progress and prospect. Annu. Rev. Cell Dev. Biol. 2009, 25, 355–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, C.; Wang, J.; Lin, H. Uniting germline and stem cells: The function of Piwi proteins and the piRNA pathway in diverse organisms. Annu Rev. Genet. 2011, 45, 447–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, X.; Song, D.; Wei, J. Piwil2 modulates the invasion and metastasis of prostate cancer by regulating the expression of matrix metalloproteinase-9 and epithelial-mesenchymal transitions. Oncol. Lett. 2015, 10, 1735–1740. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Liang, Y.; Zhang, J.; Hao, Y.; Li, M.; Wen, Z.; Zhao, Y. Transcriptome Analysis Identifies Piwi-Interacting RNAs as Prognostic Markers for Recurrence of Prostate Cancer. Front. Genet. 2019, 10, 1018. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, X.; Li, D.; Han, X. piR-001773 and piR-017184 promote prostate cancer progression by interacting with PCDH9. Cell Signal. 2020, 76, 109780. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.P.; Zhang, B.X.; Chu, L. Small Nucleolar RNAs: Insight Into Their Function in Cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.; Wan, X.; Zhang, Y.; Fu, F.; Zhao, C.; Qin, R.; Wu, H.; Li, Y.; Huang, Y. SNORA42 enhances prostate cancer cell viability, migration and EMT and is correlated with prostate cancer poor prognosis. Int. J. Biochem Cell Biol. 2018, 102, 138–150. [Google Scholar] [CrossRef]
- Martens-Uzunova, E.S.; Hoogstrate, Y.; Kalsbeek, A.; Pigmans, B.; Vredenbregt-van den Berg, M.; Dits, N.; Nielsen, S.J.; Baker, A.; Visakorpi, T.; Bangma, C.; et al. C/D-box snoRNA-derived RNA production is associated with malignant transformation and metastatic progression in prostate cancer. Oncotarget 2015, 6, 17430–17444. [Google Scholar] [CrossRef] [Green Version]
- Crea, F.; Quagliata, L.; Michael, A.; Liu, H.H.; Frumento, P.; Azad, A.A.; Xue, H.; Pikor, L.; Watahiki, A.; Morant, R.; et al. Integrated analysis of the prostate cancer small-nucleolar transcriptome reveals SNORA55 as a driver of prostate cancer progression. Mol. Oncol. 2016, 10, 693–703. [Google Scholar] [CrossRef]
- Tao, B.B.; Liu, X.Q.; Zhang, W.; Li, S.; Dong, D.; Xiao, M.; Zhong, J. Evidence for the association of chromatin and microRNA regulation in the human genome. Oncotarget 2017, 8, 70958–70966. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.P. Non-Coding RNAs as Mediators of Epigenetic Changes in Malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- Lemos, A.E.G.; Matos, A.D.R.; Ferreira, L.B.; Gimba, E.R.P. The long non-coding RNA PCA3: An update of its functions and clinical applications as a biomarker in prostate cancer. Oncotarget 2019, 10, 6589–6603. [Google Scholar] [CrossRef] [Green Version]
- Bussemakers, M.J.G.; van Bokhoven, A.; Verhaegh, G.W.; Smit, F.P.; Karthaus, H.F.M.; Schalken, J.A.; Debruyne, F.M.J.; Ru, N.; Isaacs, W.B. DD3: A New Prostate-specific Gene, Highly Overexpressed in Prostate Cancer. Cancer Res. 1999, 59, 5975. [Google Scholar] [PubMed]
- Lemos, A.E.; Ferreira, L.B.; Batoreu, N.M.; de Freitas, P.P.; Bonamino, M.H.; Gimba, E.R. PCA3 long noncoding RNA modulates the expression of key cancer-related genes in LNCaP prostate cancer cells. Tumour. Biol. 2016, 37, 11339–11348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Xu, X.; Wang, G. lncRNA ZFAS1 Is Involved in the Proliferation, Invasion and Metastasis of Prostate Cancer Cells Through Competitively Binding to miR-135a-5p. Cancer Manag. Res. 2020, 12, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Xing, Y.; Yang, F.; Sun, Y.; Zhang, S.; Wang, Q.; Zhang, W. LncRNA SNHG3 sponges miR-577 to up-regulate SMURF1 expression in prostate cancer. Cancer Med. 2020, 9, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Hu, H.; Zhou, J. Knockdown of LncRNA SNHG7 inhibited epithelial-mesenchymal transition in prostate cancer though miR-324-3p/WNT2B axis in vitro. Pathol Res. Pract. 2019, 215, 152537. [Google Scholar] [CrossRef]
- Liu, S.; Wang, L.; Li, Y.; Cui, Y.; Wang, Y.; Liu, C. Long non-coding RNA CHRF promotes proliferation and mesenchymal transition (EMT) in prostate cancer cell line PC3 requiring up-regulating microRNA-10b. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Wang, R.; Lu, X.; Yu, R. lncRNA MALAT1 Promotes EMT Process and Cisplatin Resistance of Oral Squamous Cell Carcinoma via PI3K/AKT/m-TOR Signal Pathway. Onco Targets Ther. 2020, 13, 4049–4061. [Google Scholar] [CrossRef]
- Hao, T.; Wang, Z.; Yang, J.; Zhang, Y.; Shang, Y.; Sun, J. MALAT1 knockdown inhibits prostate cancer progression by regulating miR-140/BIRC6 axis. Biomed. Pharmacother. 2020, 123, 109666. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Xu, W.; Du, X.; Hou, J. MALAT1 silencing suppresses prostate cancer progression by upregulating miR-1 and downregulating KRAS. Onco Targets Ther. 2018, 11, 3461–3473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, J.; Liang, S.; Lang, G.; Liu, G.; Liu, P.; Deng, X. Long non-coding RNA VIM-AS1 promotes prostate cancer growth and invasion by regulating epithelial-mesenchymal transition. J. BUON 2019, 24, 2090–2098. [Google Scholar] [PubMed]
- Wan, B.; Liu, B.; Lv, C. Progress of research into circular RNAs in urinary neoplasms. PeerJ 2020, 8, e8666. [Google Scholar] [CrossRef]
- Dai, Y.; Li, D.; Chen, X.; Tan, X.; Gu, J.; Chen, M.; Zhang, X. Circular RNA Myosin Light Chain Kinase (MYLK) Promotes Prostate Cancer Progression through Modulating Mir-29a Expression. Med. Sci. Monit. 2018, 24, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Sun, Y.; Xiang, Z.; Wang, K.; Liu, B.; Xiao, G.; Niu, Y.; Wu, D.; Chang, C. Preclinical study using circular RNA 17 and micro RNA 181c-5p to suppress the enzalutamide-resistant prostate cancer progression. Cell Death Dis. 2019, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Ding, L.; Wei, X.; Fan, L.; Yu, L. circSMAD2 governs migration and epithelial-mesenchymal transition by inhibiting microRNA-9. J. Cell Biochem. 2019. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.K.; Campbell, M.J. The Interactions of microRNA and Epigenetic Modifications in Prostate Cancer. Cancers 2013, 5, 998–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat. Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef]
- Cao, P.; Deng, Z.; Wan, M.; Huang, W.; Cramer, S.D.; Xu, J.; Lei, M.; Sui, G. MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1alpha/HIF-1beta. Mol. Cancer 2010, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, N.; Li, X.; Padi, S.K.; Zhang, Q.; Tang, M.S.; Guo, B. Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010, 1, e105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrba, L.; Jensen, T.J.; Garbe, J.C.; Heimark, R.L.; Cress, A.E.; Dickinson, S.; Stampfer, M.R.; Futscher, B.W. Role for DNA methylation in the regulation of miR-200c and miR-141 expression in normal and cancer cells. PLoS ONE 2010, 5, e8697. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. SChLAP1 Antagonizes the SWI/SNF Complex in Aggressive Prostate Cancer. Cancer Discov. 2013, 3, 1217. [Google Scholar] [CrossRef] [Green Version]
- Gu, P.; Chen, X.; Xie, R.; Han, J.; Xie, W.; Wang, B.; Dong, W.; Chen, C.; Yang, M.; Jiang, J.; et al. lncRNA HOXD-AS1 Regulates Proliferation and Chemo-Resistance of Castration-Resistant Prostate Cancer via Recruiting WDR5. Mol. Ther. 2017, 25, 1959–1973. [Google Scholar] [CrossRef]
- Ferro, M.; Ungaro, P.; Cimmino, A.; Lucarelli, G.; Busetto, G.M.; Cantiello, F.; Damiano, R.; Terracciano, D. Epigenetic Signature: A New Player as Predictor of Clinically Significant Prostate Cancer (PCa) in Patients on Active Surveillance (AS). Int. J. Mol. Sci. 2017, 18, 1146. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wa, Q.; Pan, J.; Peng, X.; Ren, D.; Huang, Y.; Chen, X.; Tang, Y. Downregulation of miR-141-3p promotes bone metastasis via activating NF-κB signaling in prostate cancer. J. Exp. Clin. Cancer Res. 2017, 36, 173. [Google Scholar] [CrossRef] [Green Version]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.F.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Cozar, J.M.; Robles-Fernandez, I.; Rodriguez-Martinez, A.; Puche-Sanz, I.; Vazquez-Alonso, F.; Lorente, J.A.; Martinez-Gonzalez, L.J.; Alvarez-Cubero, M.J. The role of miRNAs as biomarkers in prostate cancer. Mutat. Res. Rev. Mutat. Res. 2019, 781, 165–174. [Google Scholar] [CrossRef]
- Williams, L.V.; Veliceasa, D.; Vinokour, E.; Volpert, O.V. miR-200b inhibits prostate cancer EMT, growth and metastasis. PLoS ONE 2013, 8, e83991. [Google Scholar] [CrossRef] [Green Version]
- Selth, L.A.; Das, R.; Townley, S.L.; Coutinho, I.; Hanson, A.R.; Centenera, M.M.; Stylianou, N.; Sweeney, K.; Soekmadji, C.; Jovanovic, L.; et al. A ZEB1-miR-375-YAP1 pathway regulates epithelial plasticity in prostate cancer. Oncogene 2017, 36, 24–34. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as Prognostic Markers in Castration-resistant Prostate Cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foj, L.; Ferrer, F.; Serra, M.; Arévalo, A.; Gavagnach, M.; Giménez, N.; Filella, X. Exosomal and Non-Exosomal Urinary miRNAs in Prostate Cancer Detection and Prognosis. Prostate 2017, 77, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Ngezahayo, A.; Murua Escobar, H.; Nolte, I. Role of miRNA let-7 and its major targets in prostate cancer. Biomed. Res. Int. 2014, 2014, 376326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Shi, X.-B.; Zou, J.X.; Chen, H.; Zhang, J.; Chen, X.; Luo, J.; et al. MicroRNA let-7c is downregulated in prostate cancer and suppresses prostate cancer growth. PLoS ONE 2012, 7, e32832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, E.J.; Green, W.P.; Buckley, N.E.; McCarthy, H.O. Exploring the Potential of MicroRNA Let-7c as a Therapeutic for Prostate Cancer. Mol. Ther. Nucleic Acids 2019, 18, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Gao, G.; Wang, Z.; Sun, D.; Wei, X.; Ma, Y.; Ding, Y. Long non-coding RNA HOTTIP promotes prostate cancer cells proliferation and migration by sponging miR-216a-5p. Biosci. Rep. 2018, 38, BSR20180566. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Oh, H.; Kim, E.; Ahn, B.; Lee, J.H.; Lee, Y.; Chae, Y.S.; Kang, S.G.; Kim, C.H. Long noncoding RNA HOTTIP overexpression: A potential prognostic biomarker in prostate cancer. Pathol. Res. Pract. 2019, 215, 152649. [Google Scholar] [CrossRef]
- Song, J.; Wu, X.; Ma, R.; Miao, L.; Xiong, L.; Zhao, W. Long noncoding RNA SNHG12 promotes cell proliferation and activates Wnt/β-catenin signaling in prostate cancer through sponging microRNA-195. J. Cell. Biochem. 2019, 120, 13066–13075. [Google Scholar] [CrossRef]
- Cheng, G.; Song, Z.; Liu, Y.; Xiao, H.; Ruan, H.; Cao, Q.; Wang, K.; Xiao, W.; Xiong, Z.; Liu, D.; et al. Long noncoding RNA SNHG12 indicates the prognosis of prostate cancer and accelerates tumorigenesis via sponging miR-133b. J. Cell. Physiol. 2020, 235, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Cajigas-Du Ross, C.K.; Martinez, S.R.; Woods-Burnham, L.; Duran, A.M.; Roy, S.; Basu, A.; Ramirez, J.A.; Ortiz-Hernandez, G.L.; Rios-Colon, L.; Chirshev, E.; et al. RNA sequencing reveals upregulation of a transcriptomic program associated with stemness in metastatic prostate cancer cells selected for taxane resistance. Oncotarget 2018, 9, 30363–30384. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.N.; Yu, B.B.; Liu, Y.N.; Guo, R.; Li, J.L.; Zhang, L.C.; Su, J.; Sun, L.K.; Li, Y. Zinc promotes prostate cancer cell chemosensitivity to paclitaxel by inhibiting epithelial-mesenchymal transition and inducing apoptosis. Prostate 2019, 79, 647–656. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufies, M.; Jacquel, A.; Belhacene, N.; Robert, G.; Cluzeau, T.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011, 2, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Postel-Vinay, S.; Ashworth, A. AXL and acquired resistance to EGFR inhibitors. Nat. Genet. 2012, 44, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Z.; Wang, Z.J.; De, W.; Zheng, M.; Xu, W.Z.; Wu, H.F.; Armstrong, A.; Zhu, J.G. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, H.; Chen, Q. INPP4B reverses docetaxel resistance and epithelial-to-mesenchymal transition via the PI3K/Akt signaling pathway in prostate cancer. Biochem. Biophys. Res. Commun. 2016, 477, 467–472. [Google Scholar] [CrossRef]
- Cottard, F.; Asmane, I.; Erdmann, E.; Bergerat, J.P.; Kurtz, J.E.; Ceraline, J. Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PLoS ONE 2013, 8, e63466. [Google Scholar] [CrossRef]
- Kong, D.; Sethi, S.; Li, Y.; Chen, W.; Sakr, W.A.; Heath, E.; Sarkar, F.H. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate 2015, 75, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Ware, K.E.; Somarelli, J.A.; Schaeffer, D.; Li, J.; Zhang, T.; Park, S.; Patierno, S.R.; Freedman, J.; Foo, W.C.; Garcia-Blanco, M.A.; et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget 2016, 7, 50507–50521. [Google Scholar] [CrossRef]
- Mihalopoulos, M.; Dovey, Z.; Archer, M.; Korn, T.G.; Okhawere, K.E.; Nkemdirim, W.; Funchess, H.; Rambhia, A.; Mohamed, N.; Kaplan, S.A.; et al. Repurposing of alpha1-Adrenoceptor Antagonists: Impact in Renal Cancer. Cancers 2020, 12, 2442. [Google Scholar] [CrossRef] [PubMed]
- Garrison, J.B.; Shaw, Y.-J.; Chen, C.-S.; Kyprianou, N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007, 67, 11344–11352. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Koochekpour, S.; Strup, S.E.; Kyprianou, N. Reversion of epithelial-mesenchymal transition by a novel agent DZ-50 via IGF binding protein-3 in prostate cancer cells. Oncotarget 2017, 8, 78507–78519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, P.J.; Cao, Z.; Pu, H.; Dicken, H.; He, D.; Zhou, Z.; Wang, C.; Koochekpour, S.; Kyprianou, N. Predictive and targeting value of IGFBP-3 in therapeutically resistant prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 188–202. [Google Scholar] [PubMed]
- Moore, K.N.; Birrer, M.J. Administration of the Tablet Formulation of Olaparib in Patients with Ovarian Cancer: Practical Guidance and Expectations. Oncologist 2018, 23, 697–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef]
- Lai, Y.; Kong, Z.; Zeng, T.; Xu, S.; Duan, X.; Li, S.; Cai, C.; Zhao, Z.; Wu, W. PARP1-siRNA suppresses human prostate cancer cell growth and progression. Oncol Rep. 2018, 39, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
microRNA (miRNA) Regulation of Epithelial-to-Mesenchymal Transition (EMT) in Cancer. Over 30 different miRNAs are now known to interact with EMT-related signaling effectors in prostate cancer. This schema reveals the critical miRNAs that negatively regulate EMT, via inhibition of SNAIL, SLUG, ZEB1/2, TWIST and TGF-β. Regulation of EMT is complex, as miRNAs can have multiple targets (miR-299, miR15a/16) or are themselves regulated by EMT components in a feedback loop (miR-1/miR-200b and SLUG). The process of EMT involves the cellular transformation from an epithelial phenotype to a mesenchymal one in preparation for metastasis. Utilizing miRNAs and other non-coding RNAs (ncRNAs) to regulate EMT interconversion to mesenchymal-to-epithelial transition (MET) in order to impair metastasis could be a novel therapeutic strategy to treat advanced or resistant prostate cancer.
Figure 1.
microRNA (miRNA) Regulation of Epithelial-to-Mesenchymal Transition (EMT) in Cancer. Over 30 different miRNAs are now known to interact with EMT-related signaling effectors in prostate cancer. This schema reveals the critical miRNAs that negatively regulate EMT, via inhibition of SNAIL, SLUG, ZEB1/2, TWIST and TGF-β. Regulation of EMT is complex, as miRNAs can have multiple targets (miR-299, miR15a/16) or are themselves regulated by EMT components in a feedback loop (miR-1/miR-200b and SLUG). The process of EMT involves the cellular transformation from an epithelial phenotype to a mesenchymal one in preparation for metastasis. Utilizing miRNAs and other non-coding RNAs (ncRNAs) to regulate EMT interconversion to mesenchymal-to-epithelial transition (MET) in order to impair metastasis could be a novel therapeutic strategy to treat advanced or resistant prostate cancer.


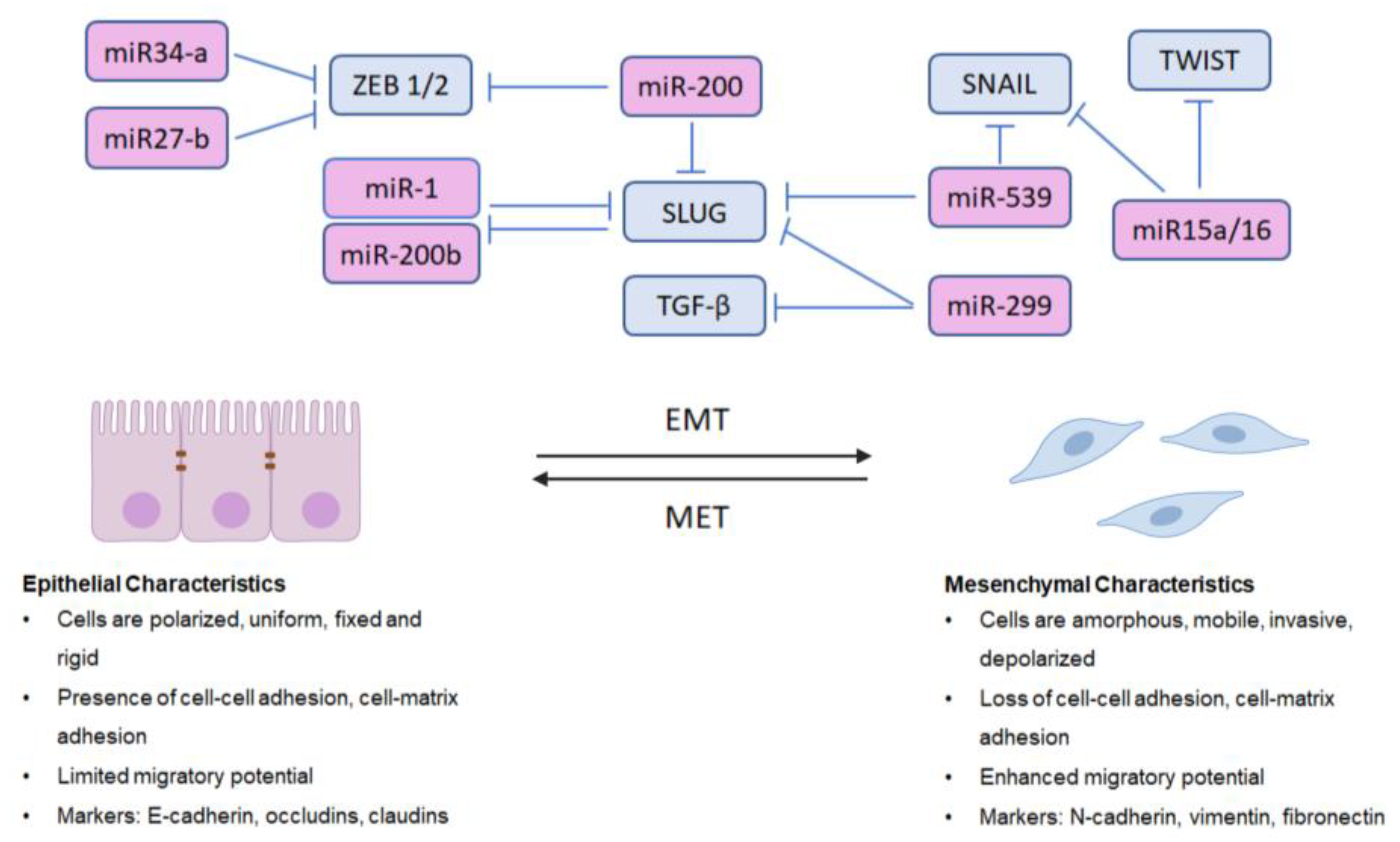{kind=link}
Table 1.
Clinical Significance of Current Treatment Modalities for Advanced Prostate Cancer: Results from Clinical Trials. Clinical efficacy of current treatment modalities for advanced prostate cancer based on results from clinical trials.
Table 1.
Clinical Significance of Current Treatment Modalities for Advanced Prostate Cancer: Results from Clinical Trials. Clinical efficacy of current treatment modalities for advanced prostate cancer based on results from clinical trials.
| Class | Drug | Clinical Significance |
|---|---|---|
| Antiandrogens | Apalutamide (Erleada®) | TITAN: In patients with mCSPC, improved OS and PFS compared with placebo [57] SPARTAN: In patients with nmCRPC, improved MFS compared to placebo [58] |
| Enzalutamide (Xtandi®) | AFFIRM: In patients with mCRPC who failed docetaxel, improved radiographic PFS and OS [47] PREVAIL: In patients with mCRPC who had no prior chemotherapy, improved radiographic PFS and OS [48] PROSPER: In patients with nmCRPC, improved time to metastatic progression or time to death compared with placebo [59] | |
| Darolutamide (Nubequa®) | ARAMIS: In patients with nmCRPC, improved metastasis-free survival compared with placebo [60] | |
| Abiraterone acetate (Zytiga®) | COU-AA-301: In patients with mCRPC after docetaxel, abiraterone + prednisone improved median PFS and OS compared to placebo + prednisone [61] COU-AA-302: In chemotherapy-naïve patients with mCRPC, abiraterone + prednisone improved median radiographic PFS and OS compared to placebo + prednisone [61] LATITUDE: In patients with newly diagnosed high risk CRPC, abiraterone + prednisone + maintenance of traditional ADT improved median radiographic PFS and OS compared with traditional ADT alone [62] | |
| Anthracyclines | Mitoxantrone + prednisone | In patients with symptomatic mCRPC, improved symptoms without difference in OS compared with prednisone alone [63] |
| Taxanes | Docetaxel (Taxotere®) | TAX 327: In chemotherapy-naïve patients with mCRPC, improved OS in docetaxel dosed every 3 weeks compared with weekly docetaxel and mitoxantrone dosed every 3 weeks [64] SWOG 9916: In patients with mCRPC, docetaxel + estramustine improved OS compared with mitoxantrone + prednisone [65] |
| Cabazitaxel (Jevtana®) | TROPIC: In patients with mCRPC who progressed after receiving docetaxel, improved OS and PFS compared with mitoxantrone [66] | |
| Immunotherapy | Sipuleucel-T (Provenge®) | IMPACT: In patients with asymptomatic or minimally symptomatic mCRPC (most were chemotherapy-naïve and did not have visceral metastases), improved OS compared with placebo [67] |
| Pembrolizumab (Keytruda®) | KEYTRUDA: FDA approved in 2017 for patients with microsatellite instability-high or mismatch repair-deficient tumors deemed unresectable or metastatic, including patients who have failed prior treatments [68,69] | |
| PARP inhibitors | Olaparib (Lynparza®) | PROfound: In patients with mCRPC who received enzalutamide or abiraterone, with homologous recombinant repair genetic alterations (BRCA1, BRCA2, ATM), delayed disease progression [70] |
Table 2.
Current Treatment Modalities for Advanced Prostate Cancer and their Mechanisms of Action. Pharmacological targeting of advanced prostate cancer. Pharmacological targeting of advanced prostate cancer: hormone-sensitive prostate cancer (HSPC), castration-resistant prostate cancer (CRPC), and metastatic CRPC.
Table 2.
Current Treatment Modalities for Advanced Prostate Cancer and their Mechanisms of Action. Pharmacological targeting of advanced prostate cancer. Pharmacological targeting of advanced prostate cancer: hormone-sensitive prostate cancer (HSPC), castration-resistant prostate cancer (CRPC), and metastatic CRPC.
| Class | Drug | Disease Stage | Mechanism of Action |
|---|---|---|---|
| Antiandrogens | Apalutamide (Erleada®) | nmCSPC [96] | Blocks AR → inhibits AR translocation to nucleus and prevents binding to DNA [12] |
| Enzalutamide (Xtandi®) | nmCRPC mCRPC with prior chemotherapy mCRPC without prior chemotherapy [96] | Blocks AR → inhibits AR translocation to nucleus and prevents binding to DNA [12] | |
| Darolutamide (Nubequa®) | nmCRPC [97] | Blocks AR → inhibits AR translocation to nucleus and prevents binding to DNA [12] | |
| Abiraterone acetate (Zytiga®) | CSPC mCRPC with prior chemotherapy mCRPC without prior chemotherapy [96] | Inhibits CYP17 → Prevents biosynthesis of androgens [6,12] | |
| Anthracyclines | Mitoxantrone + prednisone | Symptomatic CRPC | Inhibits DNA topoisomerase II → Disrupts DNA synthesis and repair [98] |
| Taxanes | Docetaxel (Taxotere®) | mCRPC [96] | Stabilizes microtubules → inhibits depolymerization → G2M arrest [88] Phosphorylates and deactivates BCL-2 → BCL-2 unable to inhibit pro-apoptotic factors (e.g., BAX, BAD and BID [88]) Impairs nuclear translocation of AR [89] |
| Cabazitaxel (Jevtana®) | mCRPC with prior docetaxel [96] | Stabilizes microtubules → inhibits depolymerization→ G2M arrest [88] Reduced affinity for p-glycoprotein [92] | |
| Immunotherapy | Sipuleucel-T (Provenge®) | Minimally symptomatic mCRPC [96] | Autologous CD54+ dendritic cells processed into fusion protein with PAP and GM-CSF, given as vaccine to generate immune response in prostate microenvironment [99,100] |
| Pembrolizumab (Keytruda®) | Metastatic microsatellite instability-high or mismatch repair deficient solid tumors with progression on prior treatments and no other treatment options [96] | Anti-PD1 monoclonal antibody [68] | |
| PARP inhibitors | Olaparib (Lynparza®) | mCRPC with germline or somatic mutations in BRCA1/BRCA2 or ATM [96] | Inhibits PARP, an enzyme involved in DNA repair [101] |
Table 3.
Diagnostic and Prognostic Biomarker Potential of Several EMT-Associated ncRNAs in Prostate Cancer. Several non-coding RNAs functionally involved in prostate cancer EMT have potential diagnostic and/or prognostic clinical value.
Table 3.
Diagnostic and Prognostic Biomarker Potential of Several EMT-Associated ncRNAs in Prostate Cancer. Several non-coding RNAs functionally involved in prostate cancer EMT have potential diagnostic and/or prognostic clinical value.
| ncRNA | Functional Involvement in EMT | Molecular Target(s) | Clinical Relevance | Potential Diagnostic and Prognostic Value |
|---|---|---|---|---|
| miRNAs | ||||
| miR-141 | Inhibits and/or reverses EMT [232] miR-141 overexpression inhibits metastatic potential, decreases vimentin, fibronectin and increases E-cadherin [232] | NF-κB signaling (TRAF5 and TRAF6) [232] | miR-141 found significantly upregulated in metastatic patients, as well as in patients with PCa compared to those with BPH or healthy donors [233,234] | Diagnostic [233,234] |
| miR-200b | Inhibits and/or reverses EMT [235] miR-200b overexpression decreases tumor growth, invasion and metastasis [235] | ZEB1 and ZEB2 [235] | Highly expressed in metastatic disease [233,234] | Diagnostic [233,234] |
| miR-375 | Inhibits and/or reverses EMT [236] miR-375 overexpression increases Zona occludens-1 (ZO-1), decreases vimentin, fibronectin, invasion and migration [236] | YAP1 [236] | Highly expressed in metastatic disease and associated with poor OS [233,234,237] | Diagnostic [233,234] |
| miR-1246 | Inhibits and/or reverses EMT [193] miR-1246 overexpression decreases EMT and cell proliferation, invasion and migration [193] | N-Cadherin and Vimentin [193] | miR-1246 can distinguish between healthy donors and patients; highly expressed in aggressive tumors and lymph node metastases (selectively released in exosomes) [193] | Diagnostic/prognostic [193] |
| let-7c | Inhibits and/or reverses EMT [238,239] let-7c overexpression decreases cell proliferation, and anchorage-independent growth [240] | HMGA1, HMGA2, MYC, BCL2, Caspase-3 [239,241] | Expression of let-7c associated with advanced clinical stage [234,238] | Prognostic [234,238] |
| lncRNAs | ||||
| HOTTIP | Promotes EMT [242] HOTTIP overexpression increases cell proliferation, invasiveness and migration [242] | miR-216a-5p [242] | High levels associated with poor clinicopathologic features [243] and lymph node metastases [242] | Prognostic [243] |
| SNHG12 | Promotes EMT [244] SNHG12 overexpression increases cell proliferation, invasiveness and migration [244] | miR-195 [244] and miR-133b [245] | Highly expressed in tumor and associated with Gleason score and lymph node metastases [244] | Prognostic [245] |
| snoRNAs | ||||
| SNORA42 | Promotes EMT [202] SNORA42 overexpression increases cell proliferation, invasion and migration [202] | High expression associated with disease progression [202] | Prognostic [202] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Altschuler, J.; Stockert, J.A.; Kyprianou, N. Non-Coding RNAs Set a New Phenotypic Frontier in Prostate Cancer Metastasis and Resistance. Int. J. Mol. Sci. 2021, 22, 2100. https://doi.org/10.3390/ijms22042100
AMA Style
Altschuler J, Stockert JA, Kyprianou N. Non-Coding RNAs Set a New Phenotypic Frontier in Prostate Cancer Metastasis and Resistance. International Journal of Molecular Sciences. 2021; 22(4):2100. https://doi.org/10.3390/ijms22042100
Chicago/Turabian StyleAltschuler, Joshua, Jennifer A. Stockert, and Natasha Kyprianou. 2021. "Non-Coding RNAs Set a New Phenotypic Frontier in Prostate Cancer Metastasis and Resistance" International Journal of Molecular Sciences 22, no. 4: 2100. https://doi.org/10.3390/ijms22042100
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.