
Deciphering the Role of Wnt and Rho Signaling Pathway in iPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling

 , , , , ,
, , , , ,  ,
, 
Abstract
:1. Introduction
2. Results
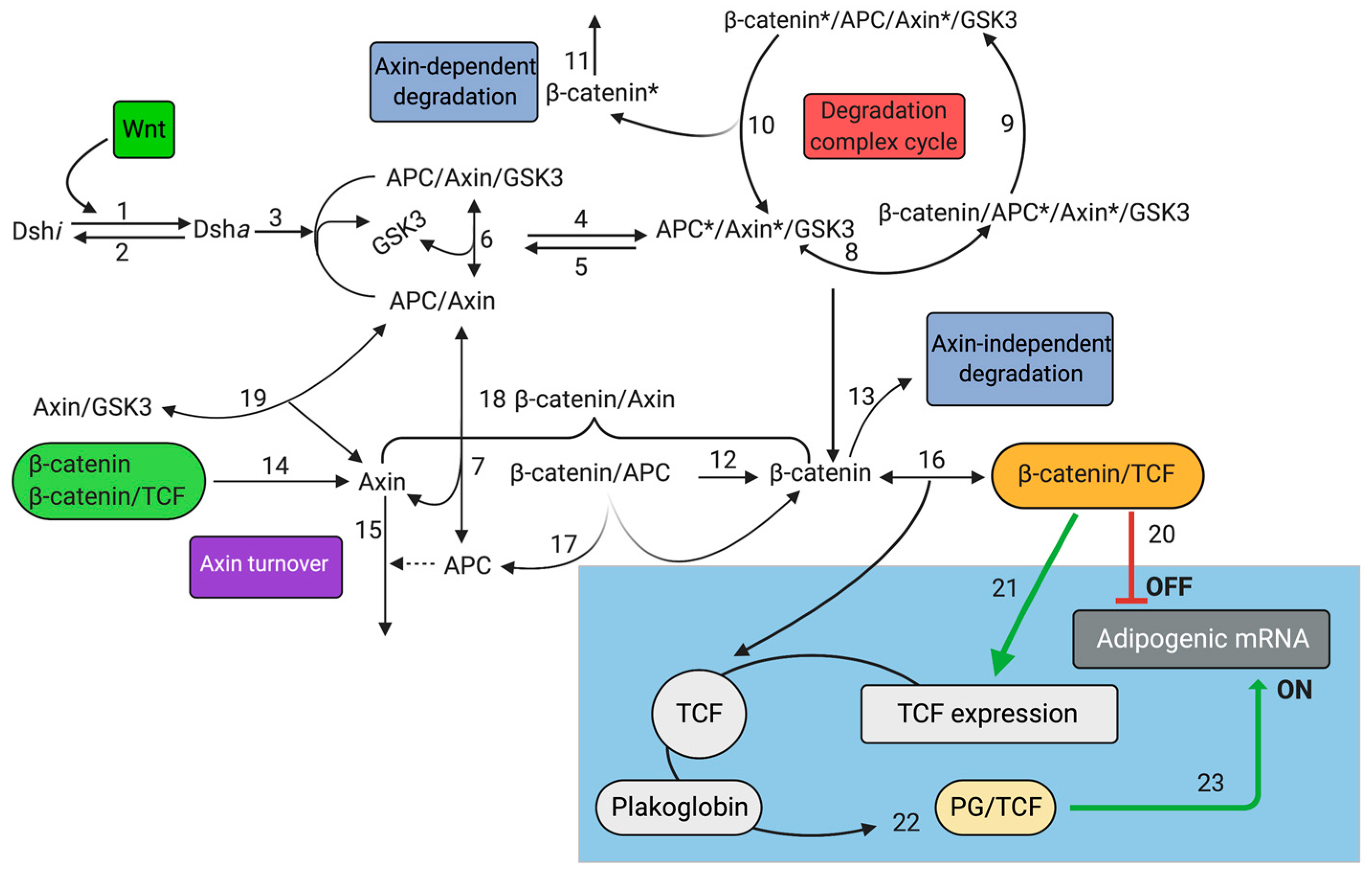
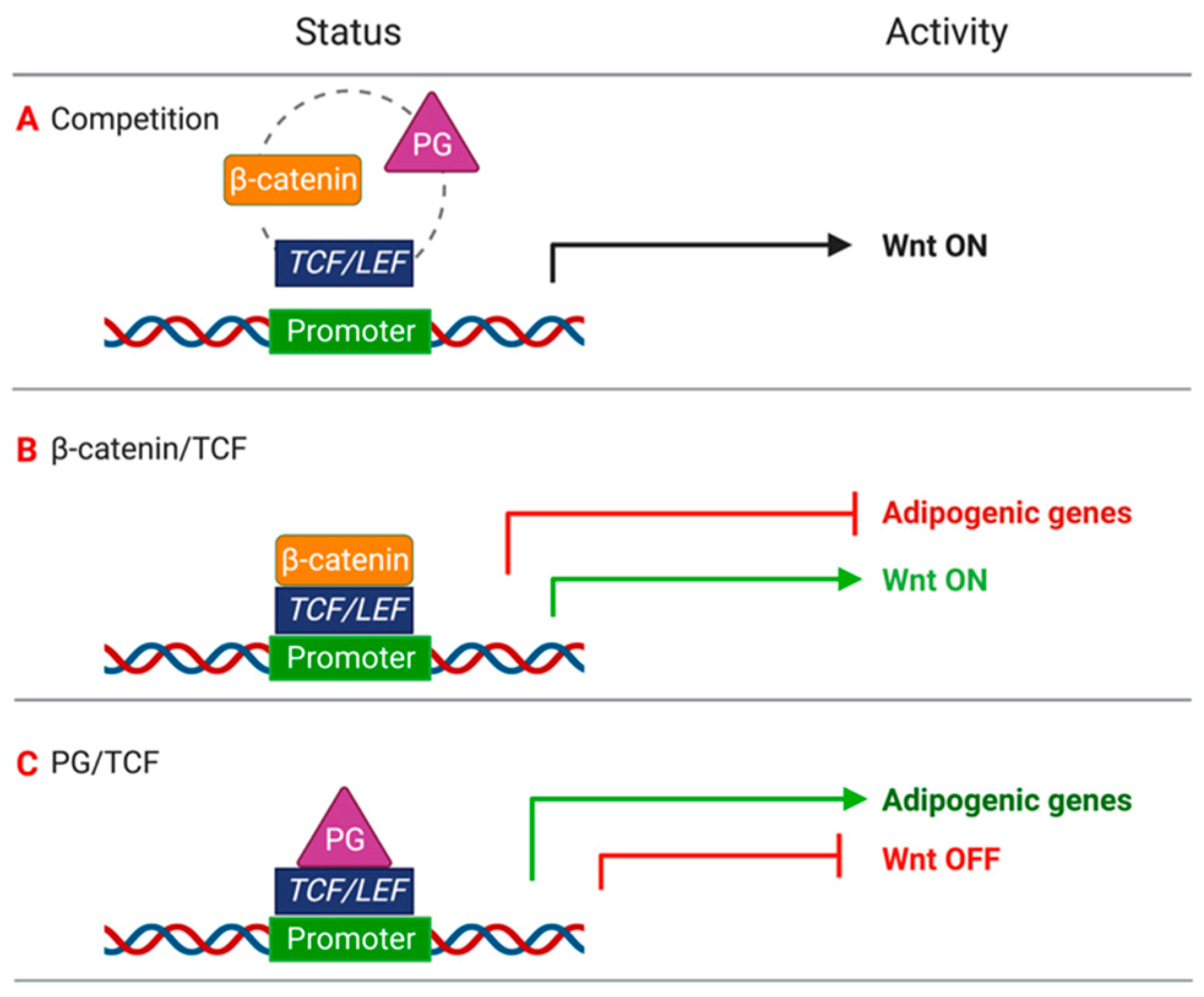
2.1. Mathematical Model of Wnt/β-Catenin Pathway
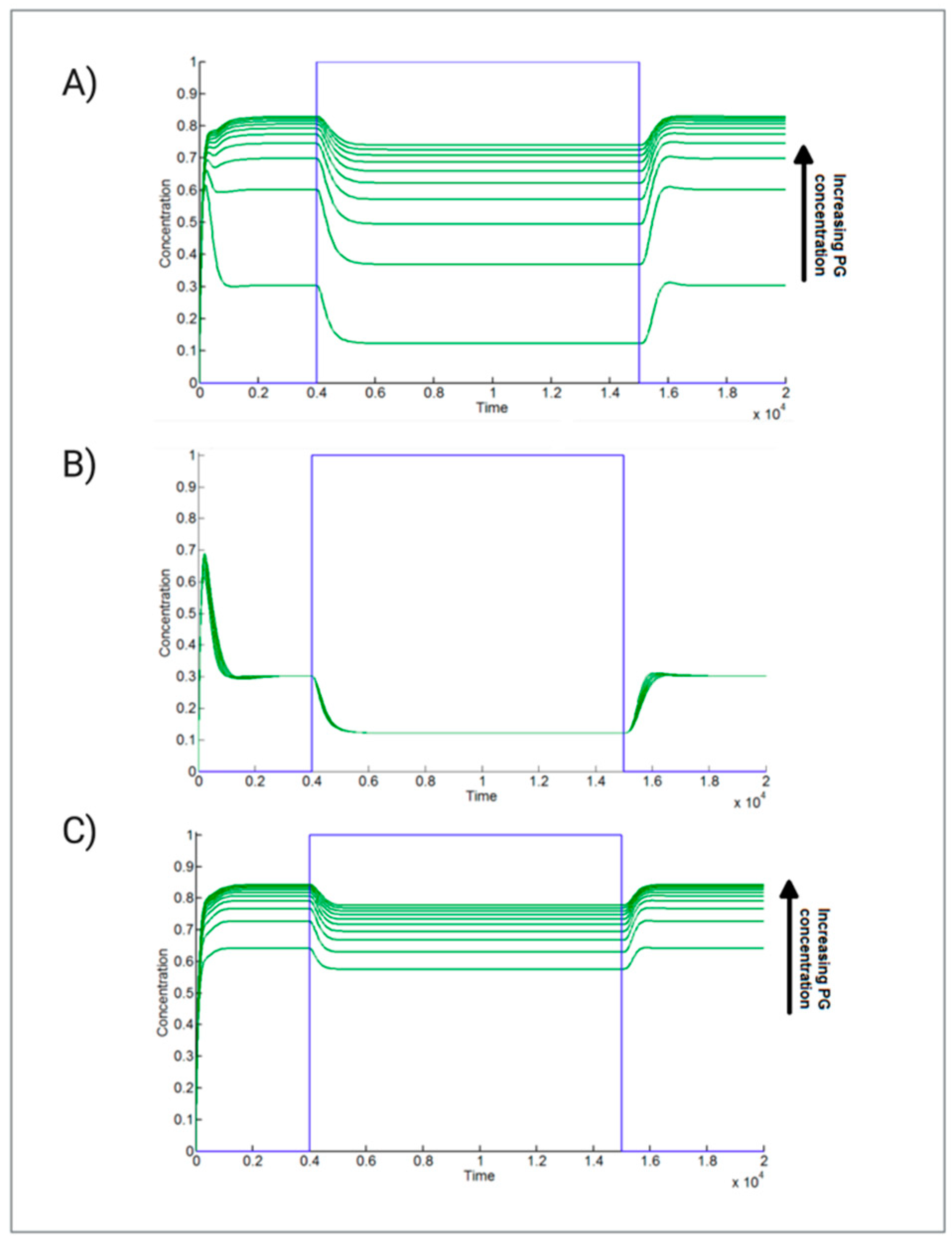
2.2. Adipogenesis Activation Is Robust against Parameter Perturbations in the Presence of High Levels of PG
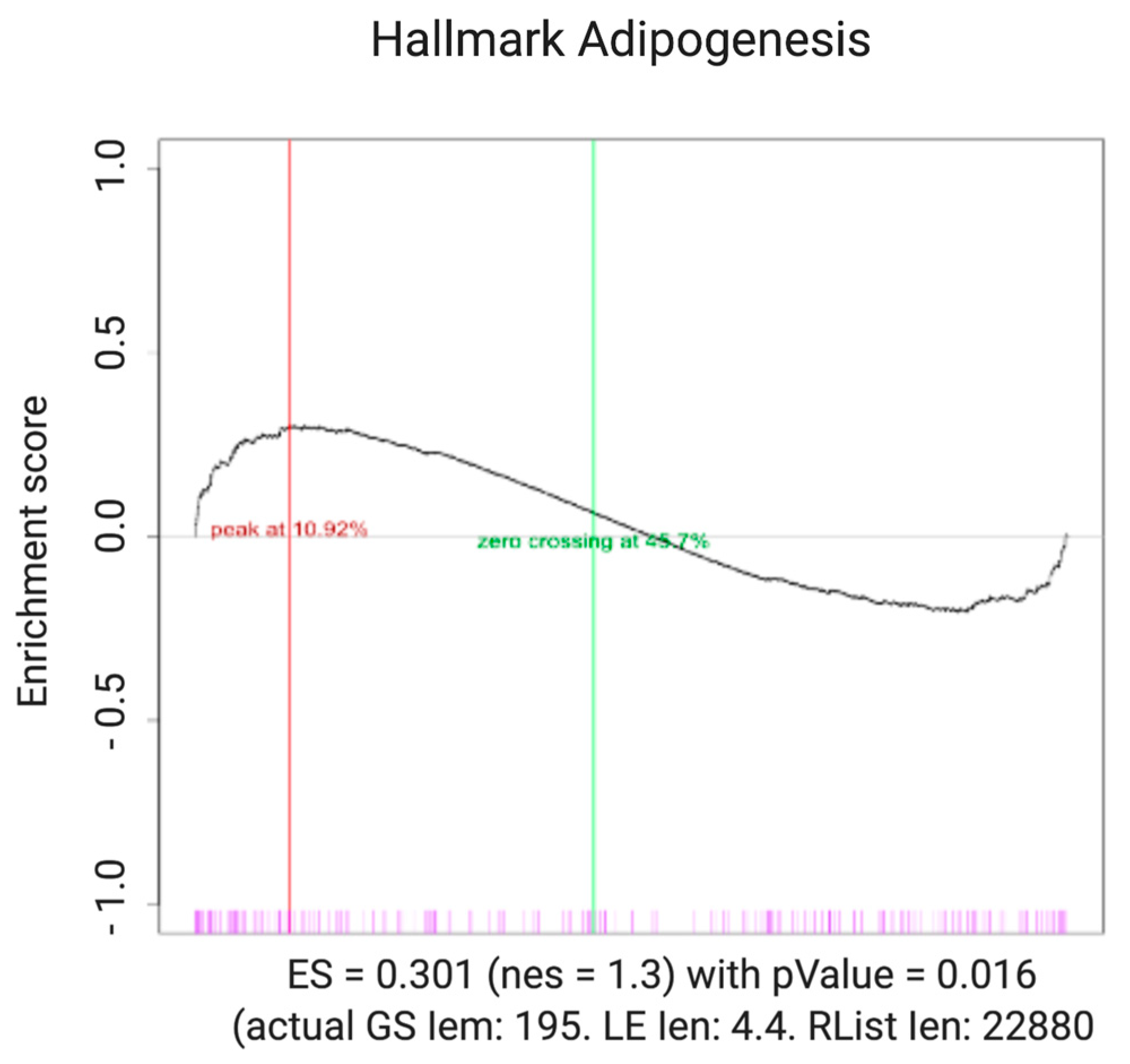
2.3. Pathways Associated with Adipogenesis Are Statistically Upregulated in ARVC
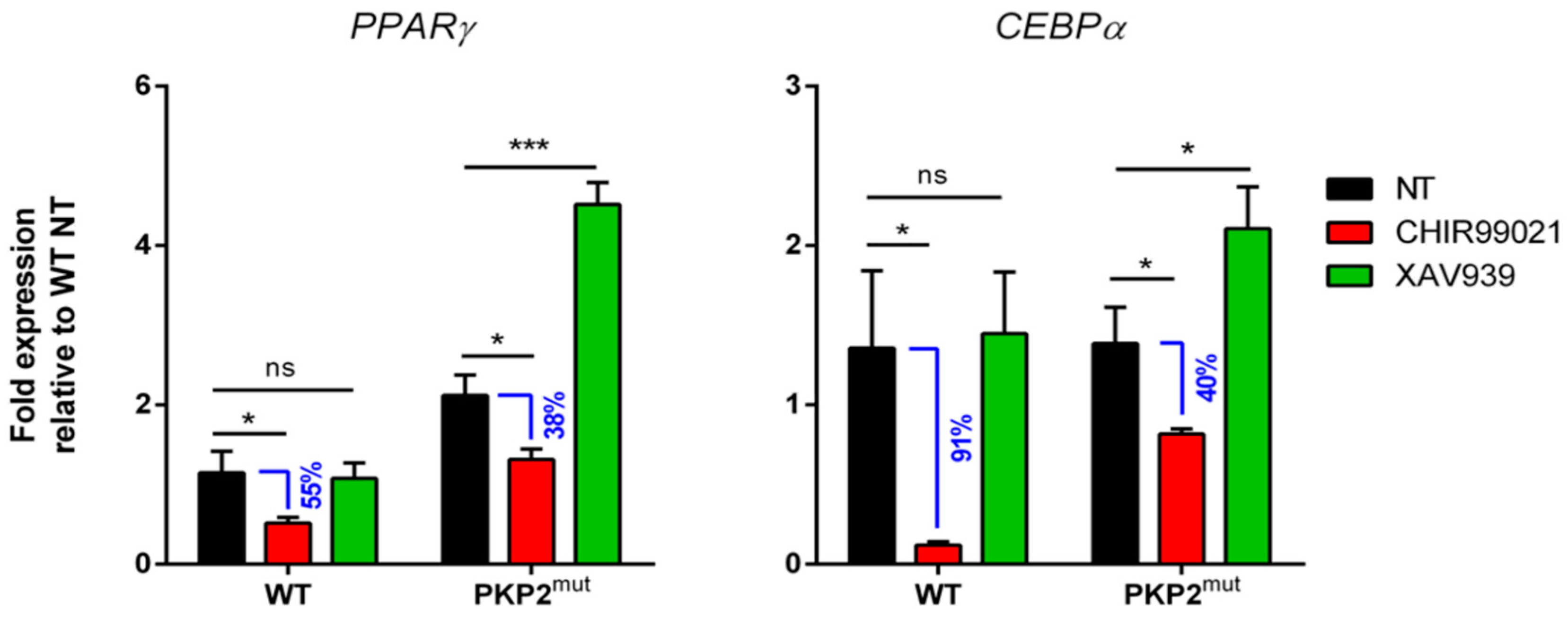
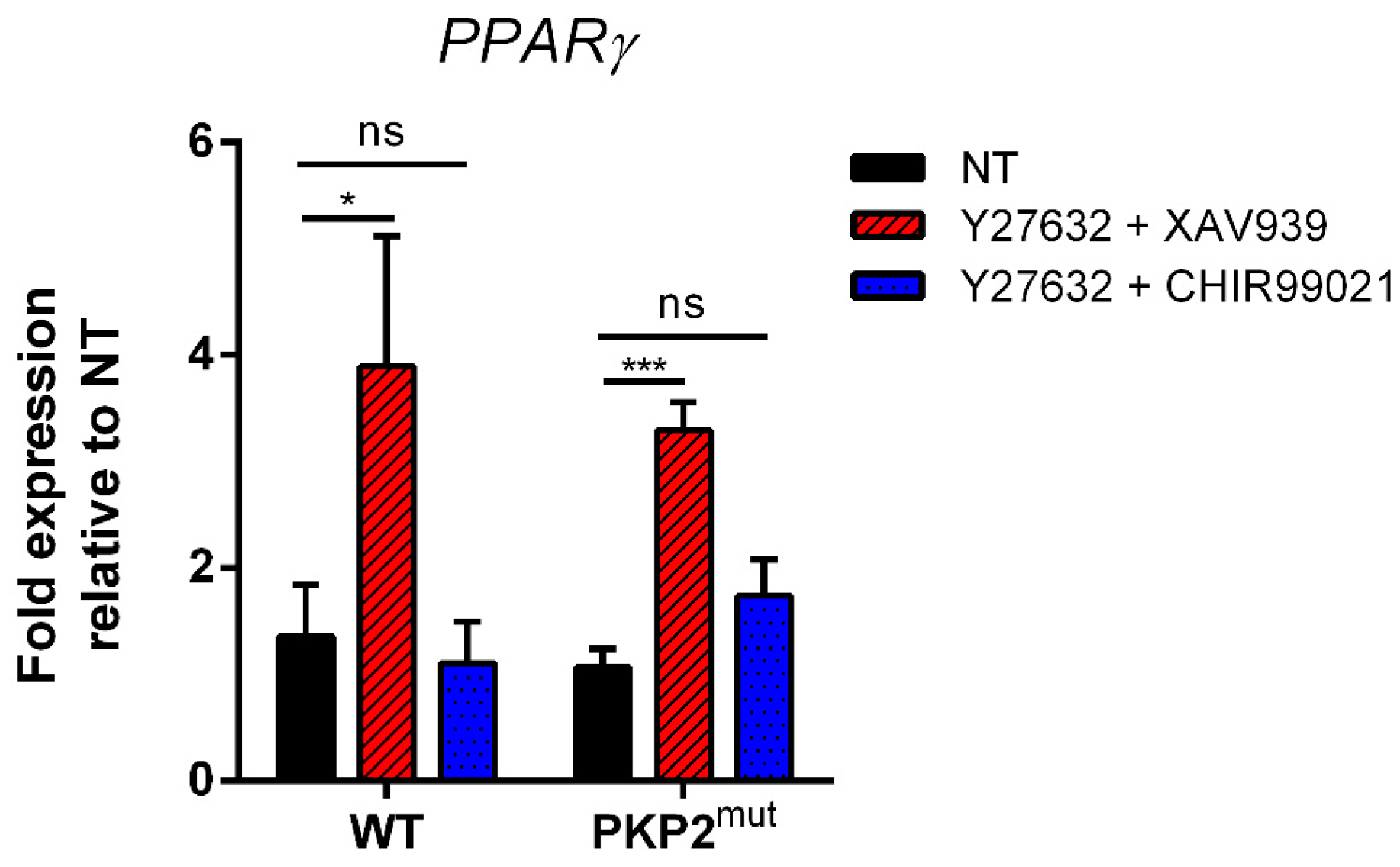
2.4. Biological Validation of the Trend of “Adipogenic mRNA” in PKP2mut CMs during Wnt/β-Catenin Pathway Modulation
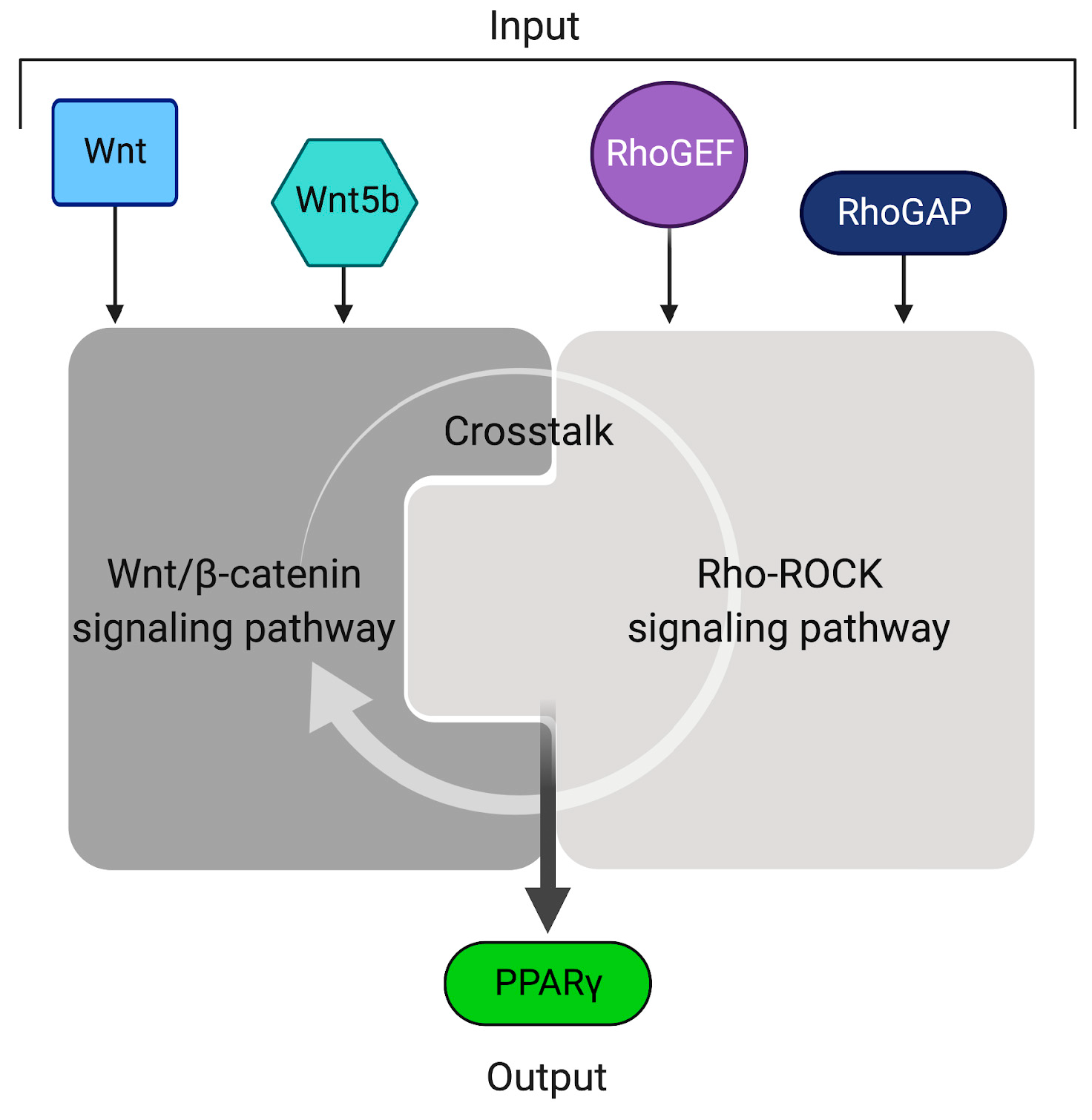
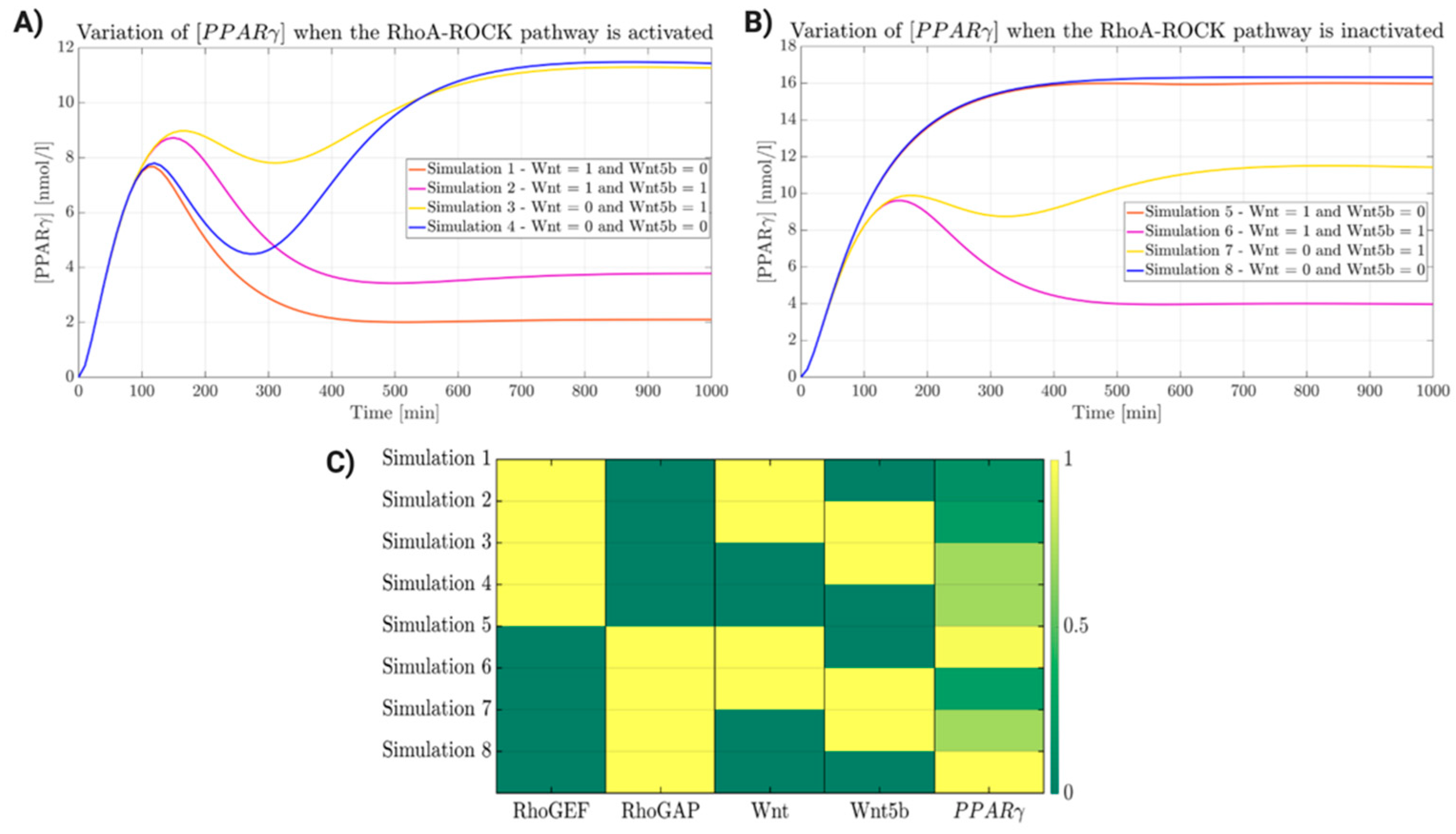
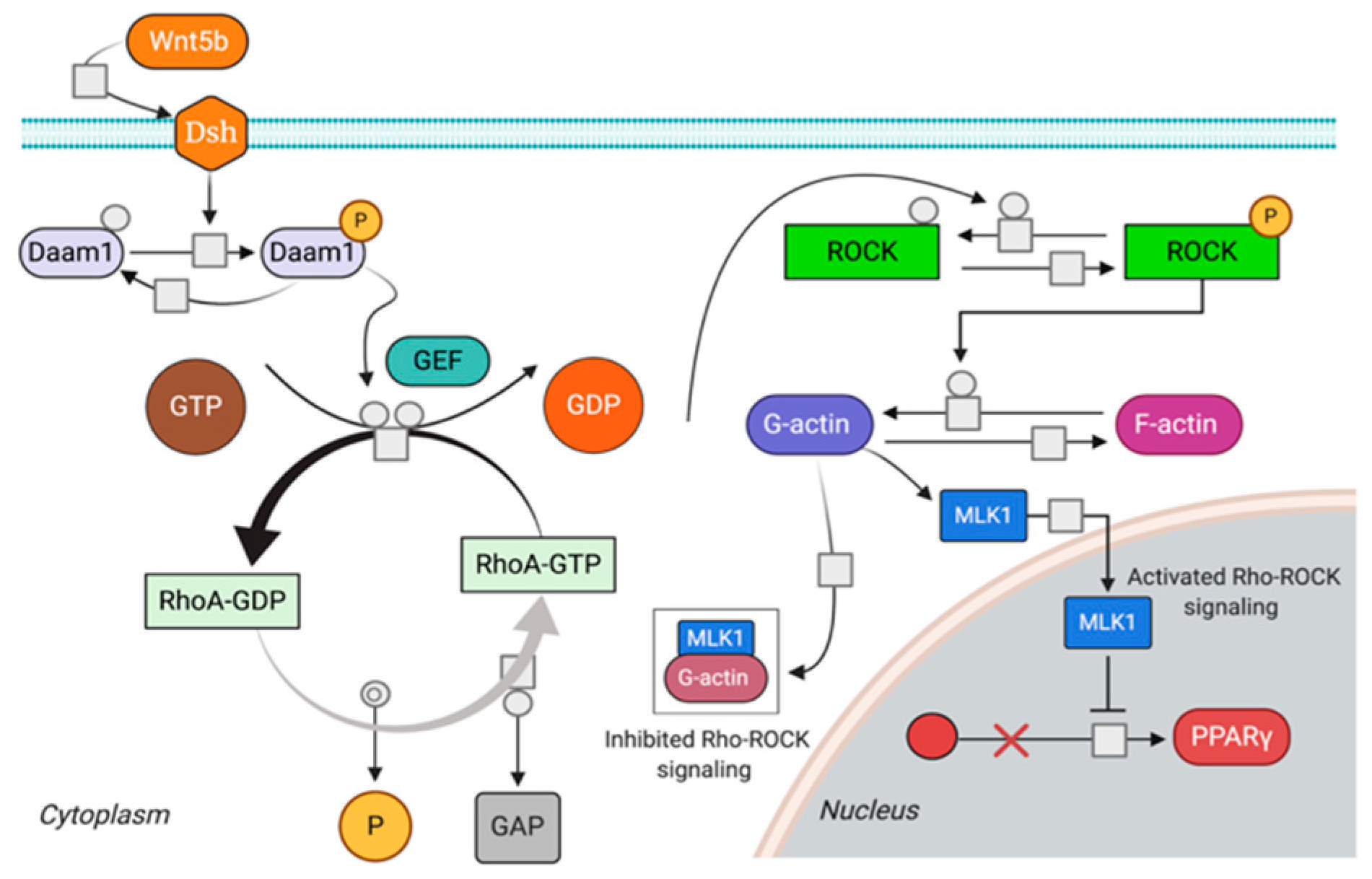
2.5. Wnt/β-Catenin and Rho-ROCK Integrative Model
2.6. Biological Validation of the Trend of “Adipogenic mRNA” in PKP2mut CMs during Double Inhibition of Wnt/b-Catenin and RhoA-ROCK Pathways Modulation
3. Discussion
4. Materials and Methods
4.1. Canonical Wnt Pathway (CWP)
4.2. Rho Pathway (RKP)
4.3. Crosstalk between Canonical Wnt and Rho-ROCK Pathways
4.4. Cell Culture, Differentiation and Treatments
4.5. Reverse Transcription PCR and Quantitative Real-Time PCR
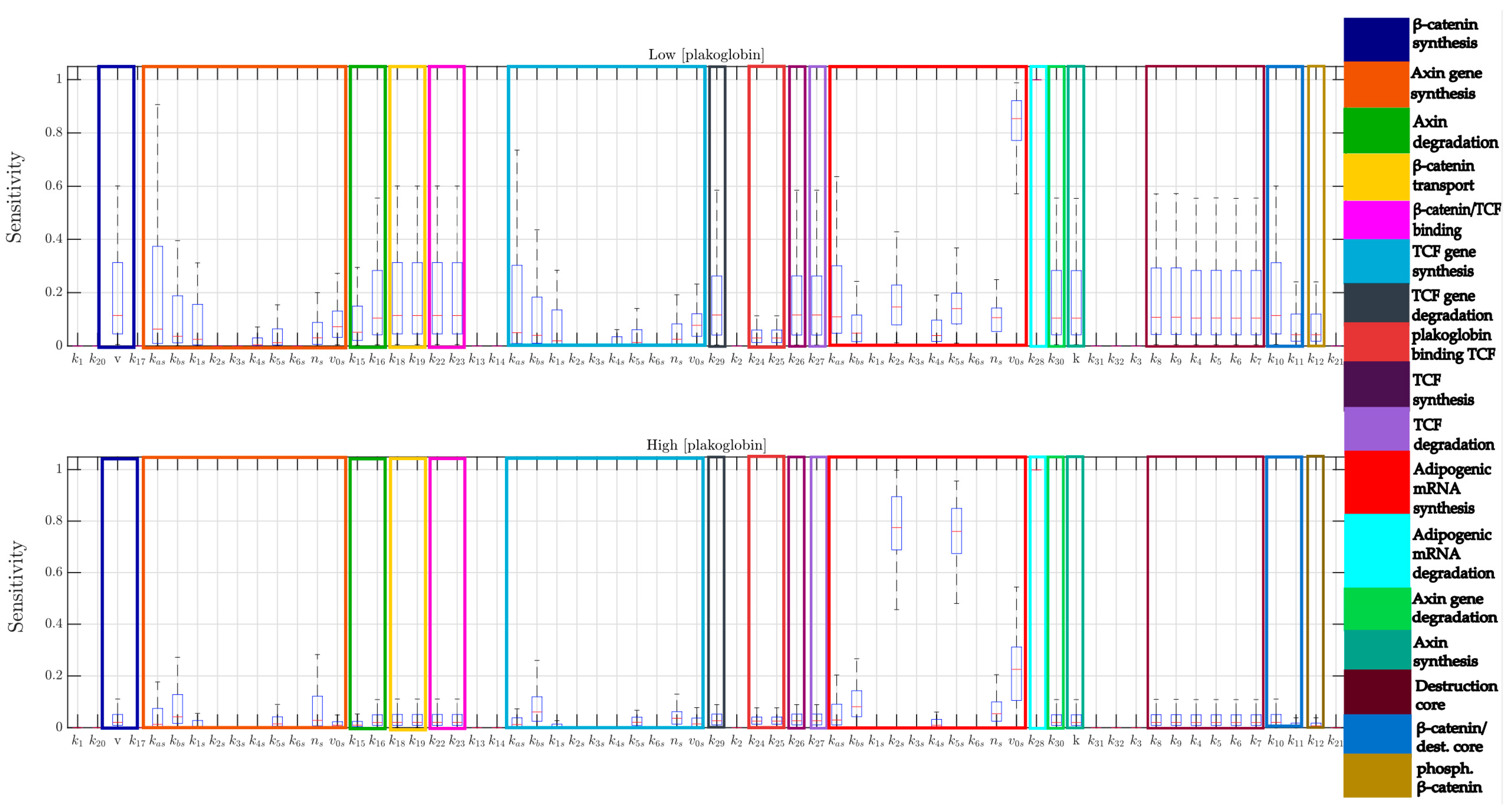
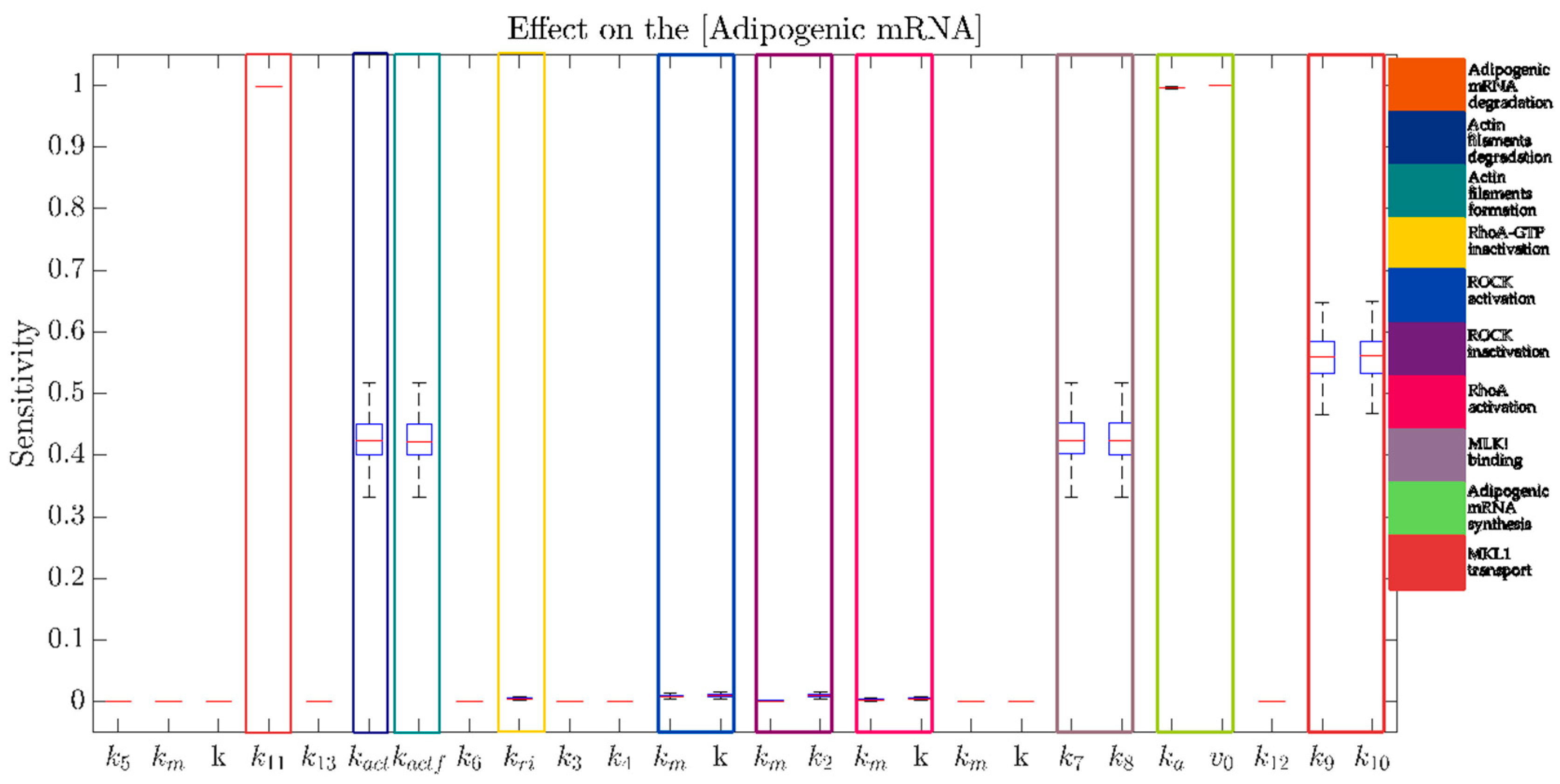
4.6. Multi-Parametric Sensitivity Analysis
- N = 100 random sampled parameter sets are generated from a normal distribution with a mean equal to the nominal value of each parameter, and standard deviation the 10% of this value.
- For each parameter set, the sensitivity of the steady-state value of “Adipogenic mRNA” with respect to each parameter is evaluated.
- Box-plots are used to analyze the distribution of the sensitivity for each parameter.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Procopio, A.; De Rosa, S.; Covello, C.; Merola, A.; Sabatino, J.; De Luca, A.; Liebetrau, C.; Hamm, C.W.; Indolfi, C.; Amato, F.; et al. Estimation of the Acute Myocardial Infarction Onset Time Based on Time-Course Acquisitions. Ann. Biomed. Eng. 2021, 49, 477–486. [Google Scholar] [CrossRef]
- Procopio, A.; Rosa, S.D.; García, M.R.; Covello, C.; Merola, A.; Sabatino, J.; Luca, A.D.; Indolfi, C.; Amato, F.; Cosentino, C. Experimental Modeling and Identification of Cardiac Biomarkers Release in Acute Myocardial Infarction. IEEE Trans. Control. Syst. Technol. 2020, 28, 183–195. [Google Scholar] [CrossRef]
- Montefusco, F.; Tagliavini, A.; Ferrante, M.; Pedersen, M.G. Concise Whole-Cell Modeling of BKCa-CaV Activity Controlled by Local Coupling and Stoichiometry. Biophys. J. 2017, 112, 2387–2396. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, M.; Tagliavini, A.; Cortese, G.; Riz, M.; Montefusco, F. Recent Advances in Mathematical Modeling and Statistical Analysis of Exocytosis in Endocrine Cells. Math. Biosci. 2016, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montefusco, F.; Cosentino, C.; Amato, F. CORE-Net: Exploiting Prior Knowledge and Preferential Attachment to Infer Biological Interaction Networks. IET Syst. Biol. 2010, 4, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Curatola, W.; Montefusco, F.; Bansal, M.; di Bernardo, D.; Amato, F. Linear Matrix Inequalities Approach to Reconstruction of Biological Networks. IET Syst. Biol. 2007, 1, 164–173. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Di Domenico, M.; Casadonte, R.; Ricci, P.; Santini, M.; Frati, G.; Rizzo, A.; Carratelli, C.R.; Lamberti, M.; Parrotta, E.; Quaresima, B.; et al. Cardiac and Skeletal Muscle Expression of Mutant β-Myosin Heavy Chains, Degree of Functional Impairment and Phenotypic Heterogeneity in Hypertrophic Cardiomyopathy. J. Cell Physiol. 2012, 227, 3471–3476. [Google Scholar] [CrossRef]
- Marcus, F.I.; Edson, S.; Towbin, J.A. Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy: A Practical Guide for Physicians. J. Am. Coll. Cardiol. 2013, 61, 1945–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, M.M.; Calkins, H.; Judge, D.P. Mechanisms of Disease: Molecular Genetics of Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Marotta, P.; Cianflone, E.; Aquila, I.; Vicinanza, C.; Scalise, M.; Marino, F.; Mancuso, T.; Torella, M.; Indolfi, C.; Torella, D. Combining Cell and Gene Therapy to Advance Cardiac Regeneration. Expert Opin. Biol. Ther. 2018, 18, 409–423. [Google Scholar] [CrossRef]
- Cianflone, E.; Aquila, I.; Scalise, M.; Marotta, P.; Torella, M.; Nadal-Ginard, B.; Torella, D. Molecular Basis of Functional Myogenic Specification of Bona Fide Multipotent Adult Cardiac Stem Cells. Cell Cycle 2018, 17, 927–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, T.; Barone, A.; Salatino, A.; Molinaro, C.; Marino, F.; Scalise, M.; Torella, M.; De Angelis, A.; Urbanek, K.; Torella, D.; et al. Unravelling the Biology of Adult Cardiac Stem Cell-Derived Exosomes to Foster Endogenous Cardiac Regeneration and Repair. Int. J. Mol. Sci. 2020, 21, 3725. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/Beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of Adipogenesis by Wnt Signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of Canonical Wnt/Beta-Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, O.; Usami, N.; Kondo, M.; Takahashi, M.; Goto, H.; Shimokata, K.; Kusugami, K.; Sekido, Y. Plakoglobin (Gamma-Catenin) Has TCF/LEF Family-Dependent Transcriptional Activity in Beta-Catenin-Deficient Cell Line. Oncogene 2004, 23, 964–972. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear Plakoglobin Is Essential for Differentiation of Cardiac Progenitor Cells to Adipocytes in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Cristancho, A.G.; Lazar, M.A. Forming Functional Fat: A Growing Understanding of Adipocyte Differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt Signaling Pathways Meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.; Fukata, Y.; Kaibuchi, K. Regulation and Functions of Rho-Associated Kinase. Exp. Cell Res. 2000, 261, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ellawindy, A.; Satoh, K.; Sunamura, S.; Kikuchi, N.; Suzuki, K.; Minami, T.; Ikeda, S.; Tanaka, S.; Shimizu, T.; Enkhjargal, B.; et al. Rho-Kinase Inhibition During Early Cardiac Development Causes Arrhythmogenic Right Ventricular Cardiomyopathy in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2172–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobusue, H.; Onishi, N.; Shimizu, T.; Sugihara, E.; Oki, Y.; Sumikawa, Y.; Chiyoda, T.; Akashi, K.; Saya, H.; Kano, K. Regulation of MKL1 via Actin Cytoskeleton Dynamics Drives Adipocyte Differentiation. Nat. Commun. 2014, 5, 3368. [Google Scholar] [CrossRef] [Green Version]
- Orban, M.; Goedel, A.; Haas, J.; Sandrock-Lang, K.; Gärtner, F.; Jung, C.B.; Zieger, B.; Parrotta, E.; Kurnik, K.; Sinnecker, D.; et al. Functional Comparison of Induced Pluripotent Stem Cell- and Blood-Derived GPIIbIIIa Deficient Platelets. PLoS ONE 2015, 10, e0115978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, M.T.; Santamaria, G.; Parrotta, E.I.; Scalise, S.; Lo Conte, M.; Gasparini, S.; Ferlazzo, E.; Aguglia, U.; Ciampi, C.; Sgura, A.; et al. Establishment and Characterization of Induced Pluripotent Stem Cells (IPSCs) from Central Nervous System Lupus Erythematosus. J. Cell Mol. Med. 2019, 23, 7382–7394. [Google Scholar] [CrossRef] [PubMed]
- Scalise, S.; Scaramuzzino, L.; Lucchino, V.; Esposito, C.; Malatesta, P.; Grillone, K.; Perrotti, N.; Cuda, G.; Parrotta, E.I. Generation of IPSC Lines from Two Patients Affected by Febrile Seizure Due to Inherited Missense Mutation in SCN1A Gene. Stem Cell Res. 2020, 49, 102083. [Google Scholar] [CrossRef]
- Parrotta, E.; De Angelis, M.T.; Scalise, S.; Candeloro, P.; Santamaria, G.; Paonessa, M.; Coluccio, M.L.; Perozziello, G.; De Vitis, S.; Sgura, A.; et al. Two Sides of the Same Coin? Unraveling Subtle Differences between Human Embryonic and Induced Pluripotent Stem Cells by Raman Spectroscopy. Stem Cell Res. Ther. 2017, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Parrotta, E.I.; Scalise, S.; Taverna, D.; De Angelis, M.T.; Sarro, G.; Gaspari, M.; Santamaria, G.; Cuda, G. Comprehensive Proteogenomic Analysis of Human Embryonic and Induced Pluripotent Stem Cells. J. Cell Mol. Med. 2019, 23, 5440–5453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, E.I.; Scalise, S.; Scaramuzzino, L.; Cuda, G. Stem Cells: The Game Changers of Human Cardiac Disease Modelling and Regenerative Medicine. Int. J. Mol. Sci. 2019, 20, 5760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of Cell-Cell Contacts and RhoA/MRTF-A Signaling Regulates Cardiomyocyte Identity. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Salic, A.; Krüger, R.; Heinrich, R.; Kirschner, M.W. The Roles of APC and Axin Derived from Experimental and Theoretical Analysis of the Wnt Pathway. PLoS Biol. 2003, 1, E10. [Google Scholar] [CrossRef] [Green Version]
- Simcha, I.; Shtutman, M.; Salomon, D.; Zhurinsky, J.; Sadot, E.; Geiger, B.; Ben-Ze’ev, A. Differential Nuclear Translocation and Transactivation Potential of Beta-Catenin and Plakoglobin. J. Cell Biol. 1998, 141, 1433–1448. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, A.; Schwientek, P.; Ellinghaus, P.; Summer, H.; Golz, S.; Kassner, A.; Schulz, U.; Gummert, J.; Milting, H. Myocardial Transcriptome Analysis of Human Arrhythmogenic Right Ventricular Cardiomyopathy. Physiol. Genom. 2012, 44, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Benary, U.; Kofahl, B.; Hecht, A.; Wolf, J. Modeling Wnt/β-Catenin Target Gene Expression in APC and Wnt Gradients Under Wild Type and Mutant Conditions. Front. Physiol. 2013, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Rath, O.; Kolch, W.; Cho, K.-H. A Hidden Oncogenic Positive Feedback Loop Caused by Crosstalk between Wnt and ERK Pathways. Oncogene 2007, 26, 4571–4579. [Google Scholar] [CrossRef] [Green Version]
- Sauro, H.M. Enzyme Kinetics for Systems Biology, 2nd ed.; Ambrosius Publishing: New York, NY, USA, 2013; ISBN 978-0-9824773-3-5. [Google Scholar]
- Rossol-Allison, J.; Stemmle, L.N.; Swenson-Fields, K.I.; Kelly, P.; Fields, P.E.; McCall, S.J.; Casey, P.J.; Fields, T.A. Rho GTPase Activity Modulates Wnt3a/Beta-Catenin Signaling. Cell Signal. 2009, 21, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Esufali, S.; Bapat, B. Cross-Talk between Rac1 GTPase and Dysregulated Wnt Signaling Pathway Leads to Cellular Redistribution of Beta-Catenin and TCF/LEF-Mediated Transcriptional Activation. Oncogene 2004, 23, 8260–8271. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ung, C.Y.; Ma, X.H.; Li, B.W.; Low, B.C.; Cao, Z.W.; Chen, Y.Z. Simulation of Crosstalk between Small GTPase RhoA and EGFR-ERK Signaling Pathway via MEKK1. Bioinformatics 2009, 25, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.K.; Kolch, W.; Kholodenko, B.N. When Ubiquitination Meets Phosphorylation: A Systems Biology Perspective of EGFR/MAPK Signalling. Cell Commun. Signal. 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Dassow, G.; Meir, E.; Munro, E.M.; Odell, G.M. The Segment Polarity Network Is a Robust Developmental Module. Nature 2000, 406, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Mayer, C.; Miller, J.; Monier, A.; Salamov, A.; et al. Pan Genome of the Phytoplankton Emiliania Underpins Its Global Distribution. Nature 2013, 499, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.G.; Cosentino, C. Validation and Invalidation of Systems Biology Models Using Robustness Analysis. IET Syst. Biol. 2011, 5, 229–244. [Google Scholar] [CrossRef]
- Salerno, L.; Cosentino, C.; Merola, A.; Bates, D.G.; Amato, F. Validation of a Model of the GAL Regulatory System via Robustness Analysis of Its Bistability Characteristics. BMC Syst. Biol. 2013, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno, L.; Cosentino, C.; Morrone, G.; Amato, F. Computational Modeling of a Transcriptional Switch Underlying B-Lymphocyte Lineage Commitment of Hematopoietic Multipotent Cells. PLoS ONE 2015, 10, e0132208. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Set Name [# Genes (K)] | Description | Genes in Overlap (k) |
|---|---|---|
| GO_PLASMA_MEMBRANE_PROTEIN-COMPLEX [510] | Any protein complex that is part of the plasma membrane | 51 |
| GO_MEMBRANE_PROTEIN_COMPLEX [1020] | Any protein complex that is part of a membrane | 53 |
| GO_ANCHORING_JUNCTION [489] | A cell junction that mechanically attaches a cell (and its cytoskeleton) to neighboring cells or to the extracellular matrix | 32 |
| GO_CELL_JUNCTION [510] | A cellular component that forms a specialized region of connection between two or more cells or between a cell and the extracellular matrix. At a cell junction, anchoring proteins in one cell to cytoskeleton proteins in one cell to cytoskeleton proteins in neighboring cells or to proteins in the extracellular matrix | 39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrotta, E.I.; Procopio, A.; Scalise, S.; Esposito, C.; Nicoletta, G.; Santamaria, G.; De Angelis, M.T.; Dorn, T.; Moretti, A.; Laugwitz, K.-L.; et al. Deciphering the Role of Wnt and Rho Signaling Pathway in iPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling. Int. J. Mol. Sci. 2021, 22, 2004. https://doi.org/10.3390/ijms22042004
Parrotta EI, Procopio A, Scalise S, Esposito C, Nicoletta G, Santamaria G, De Angelis MT, Dorn T, Moretti A, Laugwitz K-L, et al. Deciphering the Role of Wnt and Rho Signaling Pathway in iPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling. International Journal of Molecular Sciences. 2021; 22(4):2004. https://doi.org/10.3390/ijms22042004
Chicago/Turabian StyleParrotta, Elvira Immacolata, Anna Procopio, Stefania Scalise, Claudia Esposito, Giovanni Nicoletta, Gianluca Santamaria, Maria Teresa De Angelis, Tatjana Dorn, Alessandra Moretti, Karl-Ludwig Laugwitz, and et al. 2021. "Deciphering the Role of Wnt and Rho Signaling Pathway in iPSC-Derived ARVC Cardiomyocytes by In Silico Mathematical Modeling" International Journal of Molecular Sciences 22, no. 4: 2004. https://doi.org/10.3390/ijms22042004