
Signatures of Dermal Fibroblasts from RDEB Pediatric Patients

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Description of the Experimental Material
2.2. State of Health and Genetics of Patients d1–d4
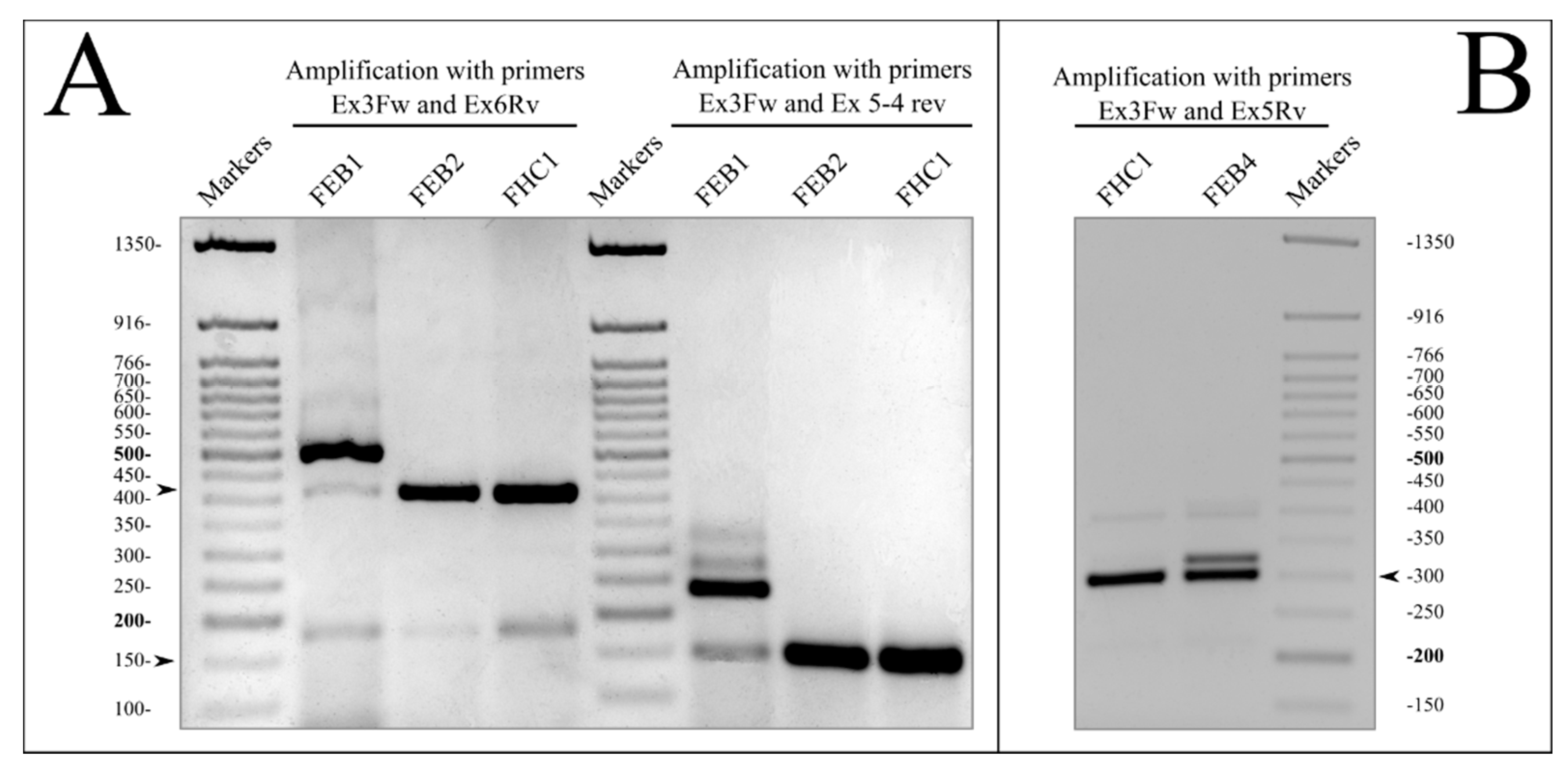
2.3. Mutations in the COL7A1 Gene
RT-PCR Analysis for Verification of COL7A1 Splice Defects
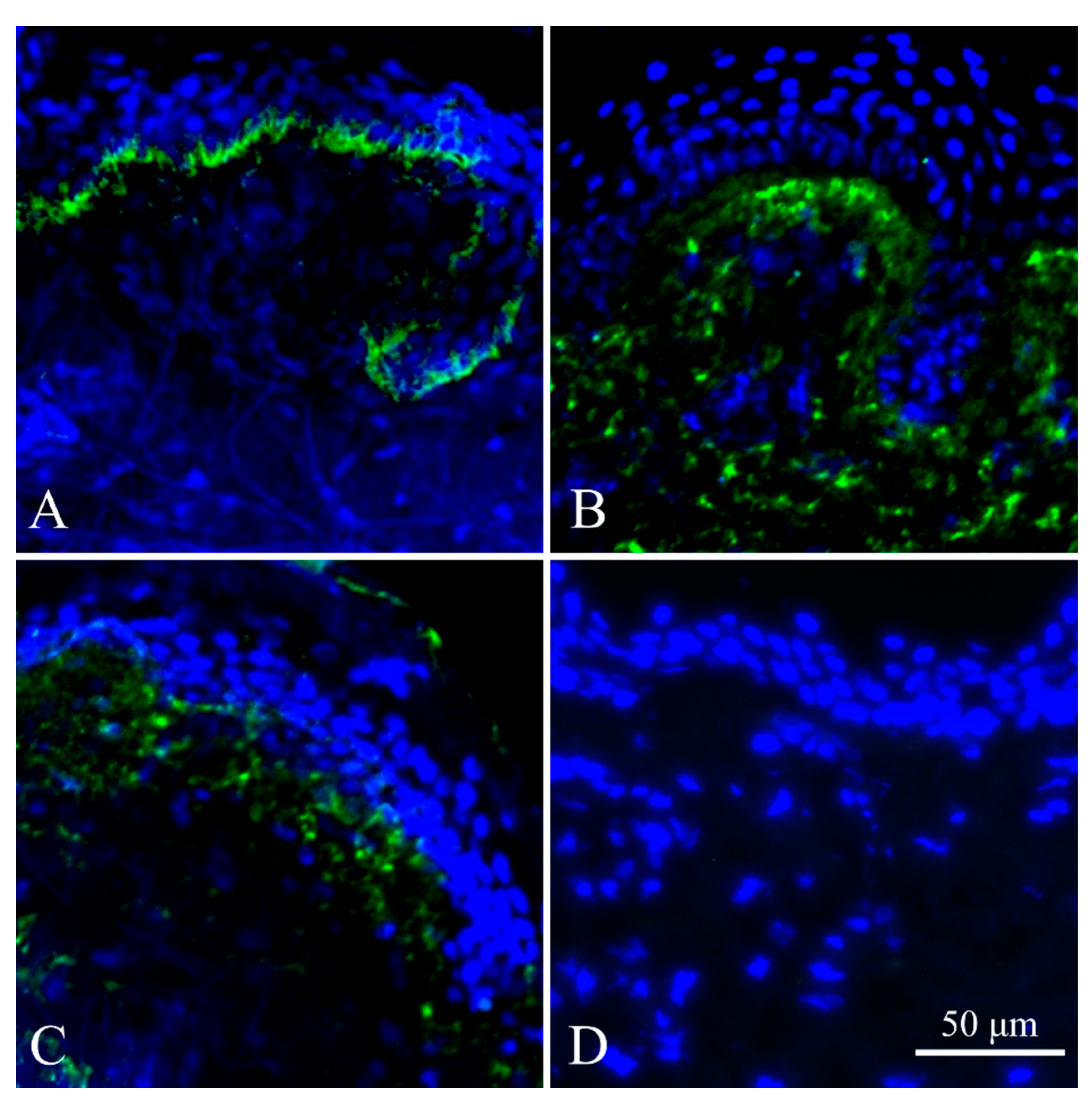
2.4. Immunohistochemistry of Skin Biopsy (IHC)
2.5. Characterization of Fibroblast Cell Lines
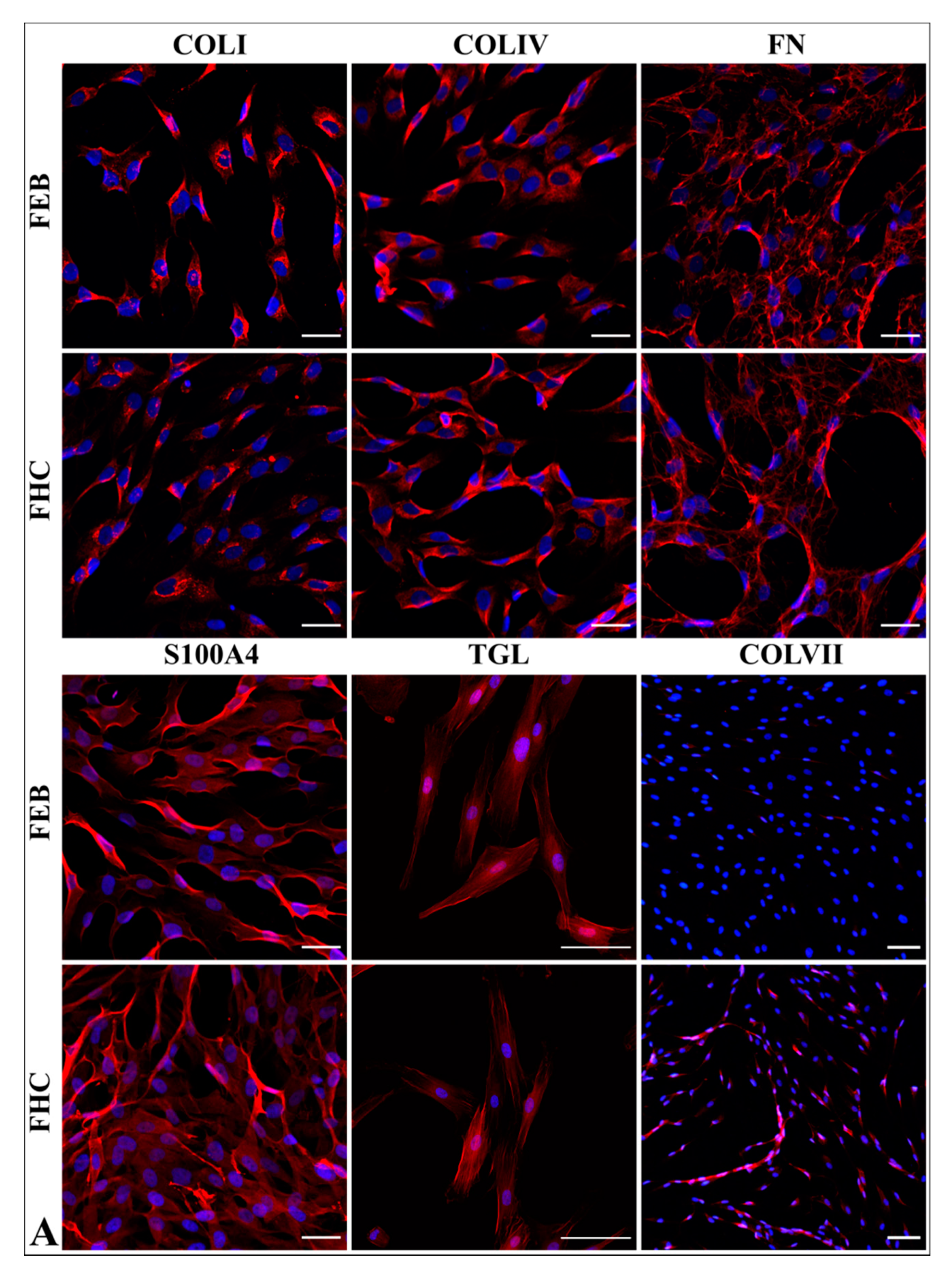
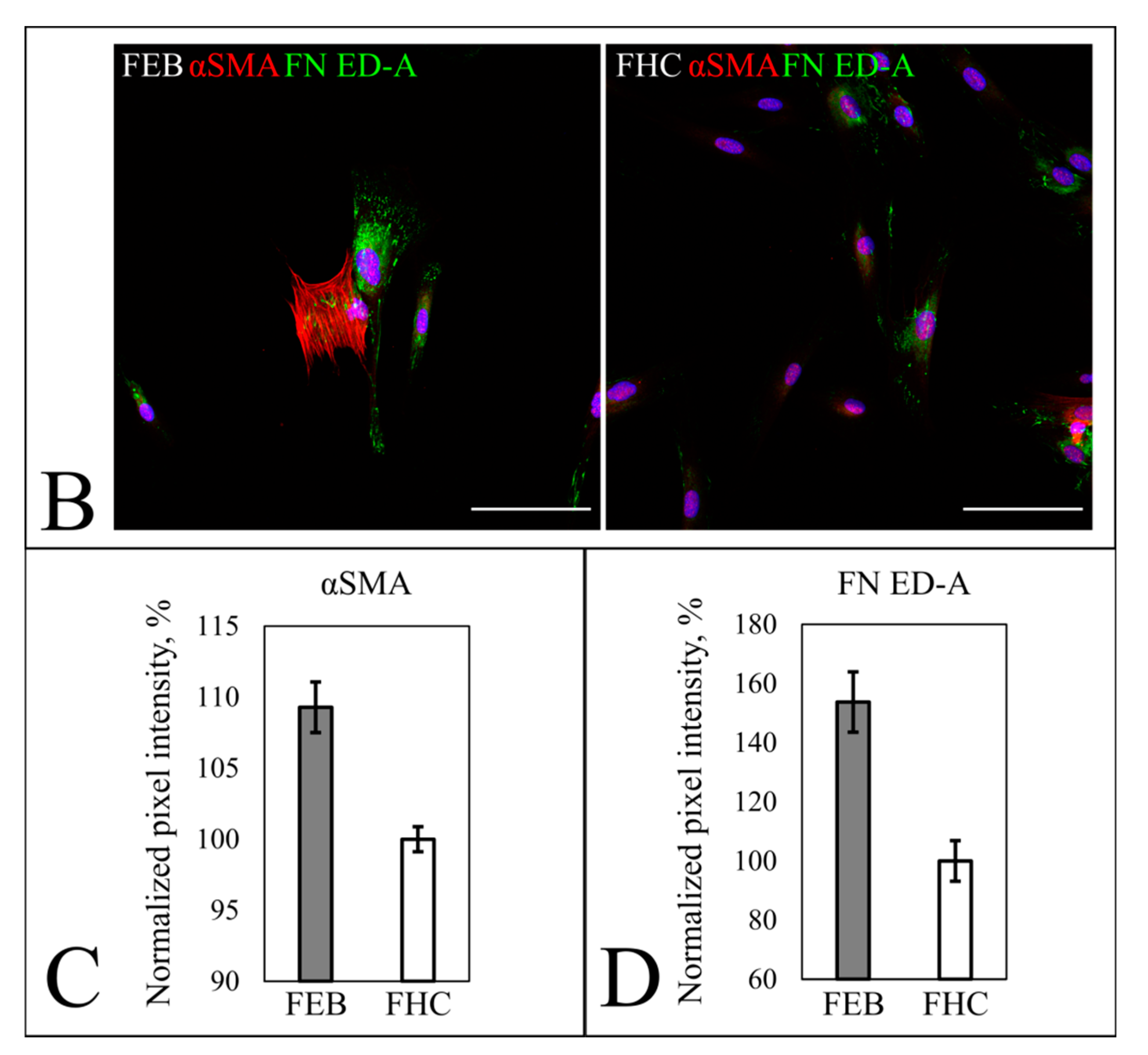
2.5.1. Fibroblast Markers’ Expression
2.5.2. Morphology Analysis
2.5.3. Contraction of Collagen Gel
2.5.4. Western Blot of Collagen VII
2.6. Transcriptome Analysis
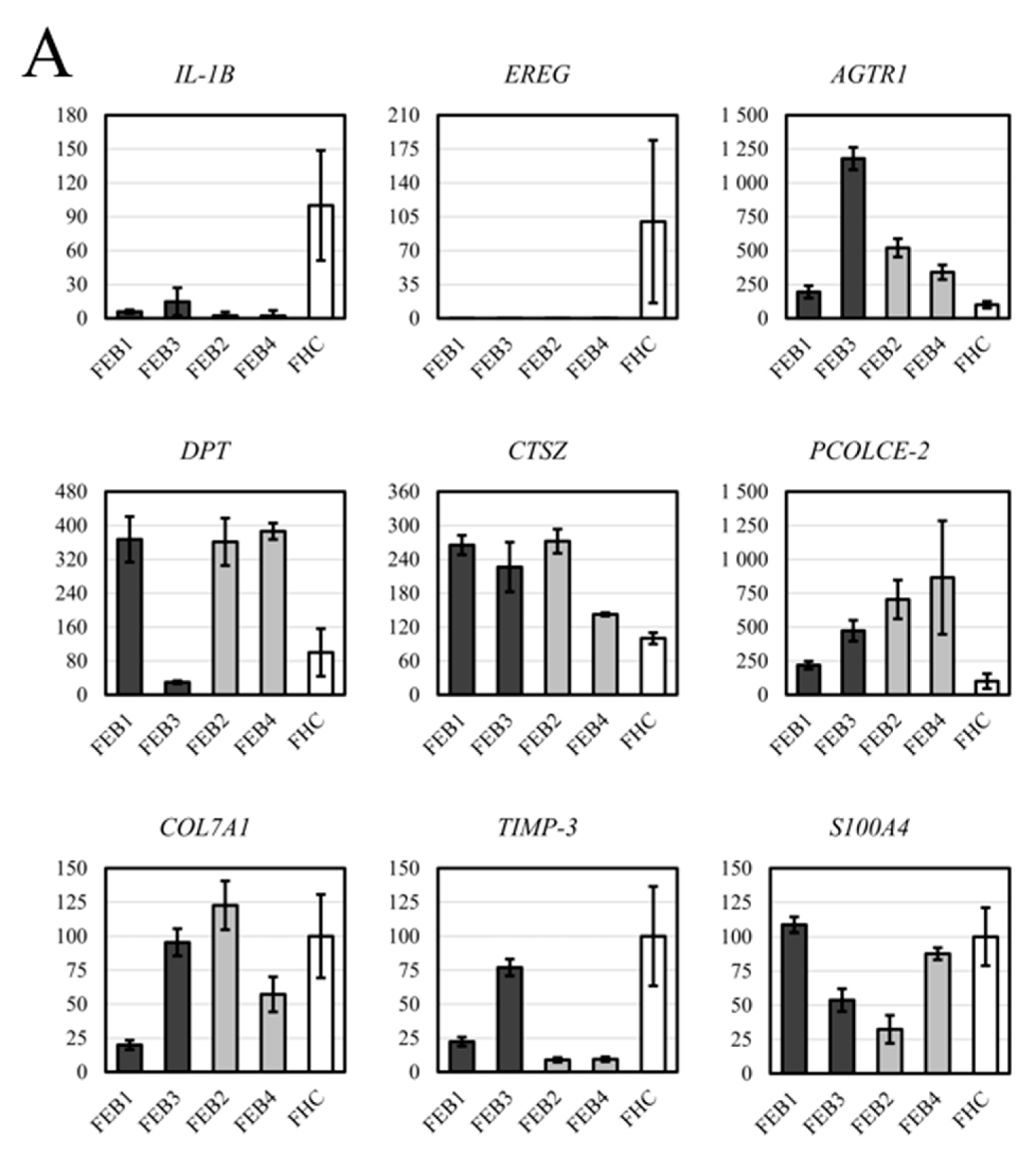
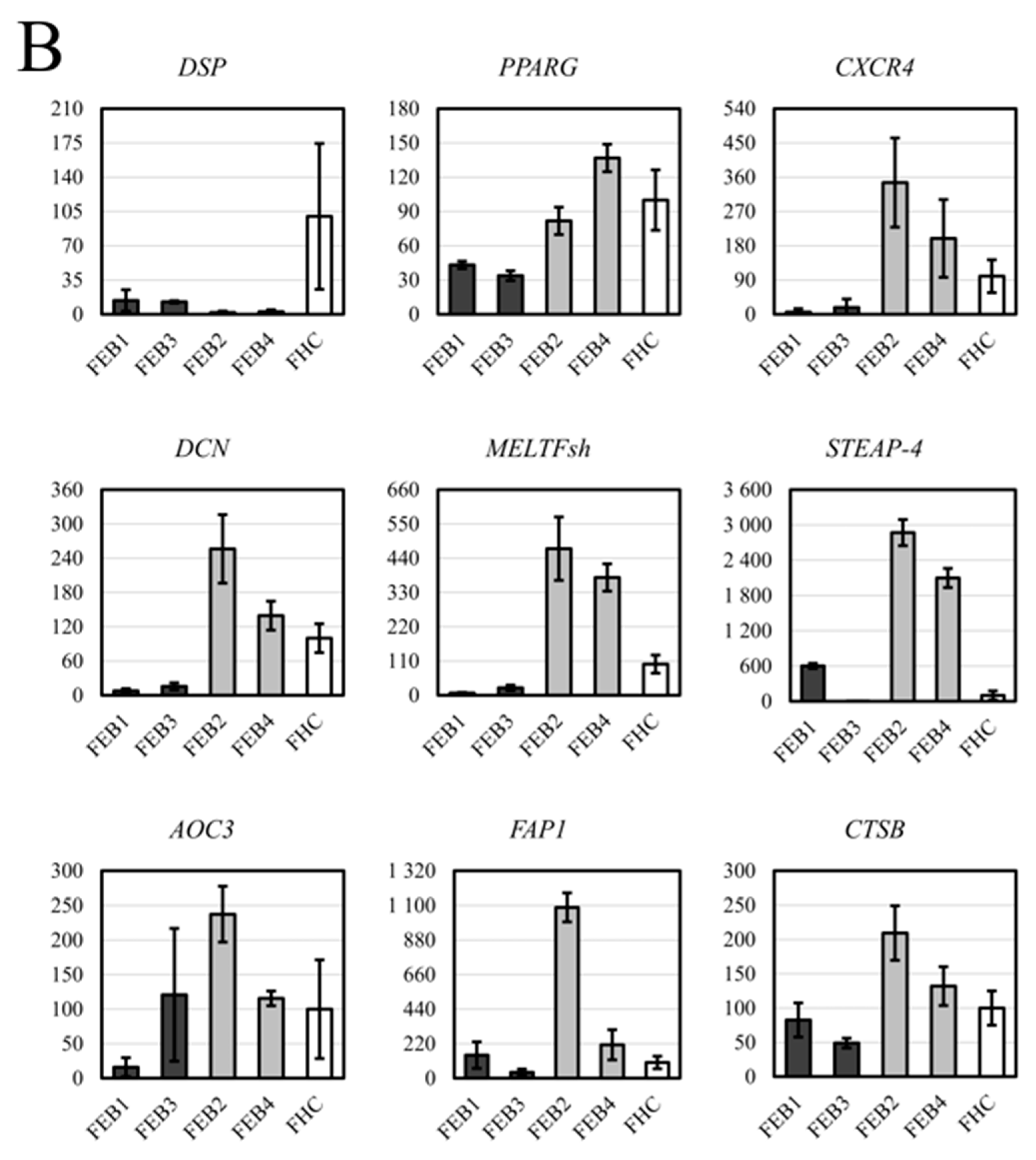
2.7. RT-qPCR Analysis for DEG of FEB and FHC
3. Discussion
4. Materials and Methods
4.1. Obtaining Skin Biopsy and Blood Samples
4.2. Immunohistochemistry of Collagen VII in Skin
4.3. Primary Cell Isolation and Culture
4.4. GenBank Accession Number and Clinical Resources
4.5. Preparation of DNA, cDNA and Reverse Transcriptase Polymerase Chain Reaction (RT-PCR), One-Tube PCR
4.6. RNA-Seq Analysis
4.7. RT-qPCR
4.8. Genetic Constructs
4.9. Cell Morphology Analysis
4.10. Immunocytochemical Fluorescence Analysis
- -
- Primary anti-collagen I antibodies (RAH C11, Imtek, Moscow, Russia)
- -
- Primary anti-collagen IV antibodies (ab6586, Abcam, Cambridge, UK)
- -
- Primary anti-fibronectin antibodies (ab2413, Abcam, Cambridge, UK)
- -
- Primary anti-S100A4 antibodies (ab27957, Abcam, Cambridge, UK)
- -
- Primary anti-α-SMA antibodies (ab5694, Abcam, Cambridge, UK)
- -
- Primary anti-FN ED-A antibodies (ab6328, Abcam, Cambridge, UK)
- -
- Primary anti-SM22ɑ antibodies (ab10135, Abcam, Cambridge, UK)
- -
- Primary anti-collagen VII antibodies (C6805, Merck, Kenilworth, NJ, USA)
- -
- Secondary anti-mouse Alexa-594 (A21201, Invitrogen, Carlsbad, CA, USA)
- -
- Secondary anti-mouse Alexa-488 (A11029, Invitrogen, Carlsbad, CA, USA)
- -
- Secondary anti-rabbit Alexa-594 (A21442, Invitrogen, Carlsbad, CA, USA)
- -
- Secondary anti-goat Alexa-488 (A21467, Invitrogen, Carlsbad, CA, USA)
4.11. Confocal Imaging
4.12. Semiquantitative Immunocytochemical Collagen VII, α-SMA and FN ED-A Expression Assay
4.13. Collagen Gel Contraction
4.14. Western Blot Analysis
4.15. RNA-Seq Data Analysis
4.16. Gene Ontology Analysis
4.17. Software
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chung, H.J.; Uitto, J. Type VII Collagen: The Anchoring Fibril Protein at Fault in Dystrophic Epidermolysis Bullosa. Dermatol. Clin. 2010, 28, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, J.-D.; Johnson, L.B.; Weiner, M.; Li, K.-P.; Suchindran, C. Epidermolysis Bullosa and the Risk of Life-Threatening Cancers: The National EB Registry Experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic Twins Discordant for Recessive Dystrophic Epidermolysis Bullosa Phenotype Highlight the Role of TGF-β Signalling in Modifying Disease Severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, A.G.; Dellambra, E.; Logli, E.; Zambruno, G.; Castiglia, D. Epidermolysis Bullosa-Associated Squamous Cell Carcinoma: From Pathogenesis to Therapeutic Perspectives. Int. J. Mol. Sci. 2019, 20, 5707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March, O.P.; Kocher, T.; Koller, U. Context-Dependent Strategies for Enhanced Genome Editing of Genodermatoses. Cells 2020, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, S.S.; Yan, W.; Frew, J.W.; Cohn, H.I.; Rhodes, L.M.; Tran, K.; Melbourne, W.; Nelson, J.A.; Sturm, M.; Fogarty, J.; et al. A Phase II Randomized Vehicle-Controlled Trial of Intradermal Allogeneic Fibroblasts for Recessive Dystrophic Epidermolysis Bullosa. J. Am. Acad. Dermatol. 2013, 69, 898–908.e7. [Google Scholar] [CrossRef]
- Atanasova, V.S.; Russell, R.J.; Webster, T.G.; Cao, Q.; Agarwal, P.; Lim, Y.Z.; Krishnan, S.; Fuentes, I.; Guttmann-Gruber, C.; McGrath, J.A.; et al. Thrombospondin-1 Is a Major Activator of TGF-β Signaling in Recessive Dystrophic Epidermolysis Bullosa Fibroblasts. J. Investig. Dermatol. 2019, 139, 1497–1505.e5. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, F.; De Domenico, E.; Nyström, A.; Mastroeni, S.; Abeni, D.; Baldini, E.; Ulisse, S.; Uva, P.; Bruckner-Tuderman, L.; Zambruno, G.; et al. Decorin Counteracts Disease Progression in Mice with Recessive Dystrophic Epidermolysis Bullosa. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 81, 3–16. [Google Scholar] [CrossRef]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan Ameliorates Dystrophic Epidermolysis Bullosa and Uncovers New Disease Mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef]
- Walker, E.J.; Heydet, D.; Veldre, T.; Ghildyal, R. Transcriptomic Changes during TGF-β-Mediated Differentiation of Airway Fibroblasts to Myofibroblasts. Sci. Rep. 2019, 9, 20377. [Google Scholar] [CrossRef] [Green Version]
- Pfendner, E.G.; Lucky, A.W. Dystrophic Epidermolysis Bullosa. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- van den Akker, P.C.; Mellerio, J.E.; Martinez, A.E.; Liu, L.; Meijer, R.; Dopping-Hepenstal, P.J.C.; van Essen, A.J.; Scheffer, H.; Hofstra, R.M.W.; McGrath, J.A.; et al. The Inversa Type of Recessive Dystrophic Epidermolysis Bullosa Is Caused by Specific Arginine and Glycine Substitutions in Type VII Collagen. J. Med. Genet. 2011, 48, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csikós, M.; Szocs, H.I.; Lászik, A.; Mecklenbeck, S.; Horváth, A.; Kárpáti, S.; Bruckner-Tuderman, L. High Frequency of the 425A-->G Splice-Site Mutation and Novel Mutations of the COL7A1 Gene in Central Europe: Significance for Future Mutation Detection Strategies in Dystrophic Epidermolysis Bullosa. Br. J. Dermatol. 2005, 152, 879–886. [Google Scholar] [CrossRef]
- Gorell, E.S.; Nguyen, N.; Siprashvili, Z.; Marinkovich, M.P.; Lane, A.T. Characterization of Patients with Dystrophic Epidermolysis Bullosa for Collagen VII Therapy. Br. J. Dermatol. 2015, 173, 821–823. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.S.; Grüninger, G.; Imsak, R.; Müller, M.L.; Schumann, H.; Kiritsi, D.; Emmert, S.; Borozdin, W.; Kohlhase, J.; Bruckner-Tuderman, L.; et al. Forty-Two Novel COL7A1 Mutations and the Role of a Frequent Single Nucleotide Polymorphism in the MMP1 Promoter in Modulation of Disease Severity in a Large European Dystrophic Epidermolysis Bullosa Cohort. Br. J. Dermatol. 2009, 161, 1089–1097. [Google Scholar] [CrossRef]
- Jeřábková, B.; Kopečková, L.; Bučková, H.; Veselý, K.; Valíčková, J.; Fajkusová, L. Analysis of the COL7A1 Gene in Czech Patients with Dystrophic Epidermolysis Bullosa Reveals Novel and Recurrent Mutations. J. Dermatol. Sci. 2010, 59, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kahofer, P.; Bruckner-Tuderman, L.; Metze, D.; Lemmink, H.; Scheffer, H.; Smolle, J. Dystrophic Epidermolysis Bullosa Inversa with COL7A1 Mutations and Absence of GDA-J/F3 Protein. Pediatr. Dermatol. 2003, 20, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, K.; Sawamura, D.; Goto, M.; Nakamura, H.; Jung, S.-Y.; Kim, S.-C.; Shimizu, H. Epidermolysis Bullosa Simplex in Japanese and Korean Patients: Genetic Studies in 19 Cases. Br. J. Dermatol. 2006, 155, 313–317. [Google Scholar] [CrossRef]
- Almaani, N.; Liu, L.; Dopping-Hepenstal, P.J.C.; Lai-Cheong, J.E.; Wong, A.; Nanda, A.; Moss, C.; Martinéz, A.E.; Mellerio, J.E.; McGrath, J.A. Identical Glycine Substitution Mutations in Type VII Collagen May Underlie Both Dominant and Recessive Forms of Dystrophic Epidermolysis Bullosa. Acta Derm. Venereol. 2011, 91, 262–266. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, K.; Furukawa, F.; Imamura, S. The Epitope for Anti-Type VII Collagen Monoclonal Antibody (LH7:2) Locates at the Central Region of the N-Terminal Non-Collagenous Domain of Type VII Collagen. Br. J. Dermatol. 1994, 131, 472–476. [Google Scholar] [CrossRef]
- Woodley, D.T.; Remington, J.; Chen, M. Autoimmunity to Type VII Collagen: Epidermolysis Bullosa Acquisita. Clin. Rev. Allergy Immunol. 2007, 33, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Escámez, M.J.; García, M.; Cuadrado-Corrales, N.; Llames, S.G.; Charlesworth, A.; De Luca, N.; Illera, N.; Sánchez-Jimeno, C.; Holguín, A.; Duarte, B.; et al. The First COL7A1 Mutation Survey in a Large Spanish Dystrophic Epidermolysis Bullosa Cohort: C.6527insC Disclosed as an Unusually Recurrent Mutation. Br. J. Dermatol. 2010, 163, 155–161. [Google Scholar] [CrossRef]
- Cuadrado-Corrales, N.; Sánchez-Jimeno, C.; García, M.; Escámez, M.-J.; Illera, N.; Hernández-Martín, Á.; Trujillo-Tiebas, M.-J.; Ayuso, C.; Del Rio, M. A Prevalent Mutation with Founder Effect in Spanish Recessive Dystrophic Epidermolysis Bullosa Families. BMC Med. Genet. 2010, 11, 139. [Google Scholar] [CrossRef] [Green Version]
- Chacón-Solano, E.; León, C.; Díaz, F.; García-García, F.; García, M.; Escámez, M.J.; Guerrero-Aspizua, S.; Conti, C.J.; Mencía, Á.; Martínez-Santamaría, L.; et al. Fibroblast Activation and Abnormal Extracellular Matrix Remodelling as Common Hallmarks in Three Cancer-Prone Genodermatoses. Br. J. Dermatol. 2019, 181, 512–522. [Google Scholar] [CrossRef] [PubMed]
- GEO Accession Viewer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119501 (accessed on 22 December 2020).
- Nyström, A.; Velati, D.; Mittapalli, V.R.; Fritsch, A.; Kern, J.S.; Bruckner-Tuderman, L. Collagen VII Plays a Dual Role in Wound Healing. J. Clin. Investig. 2013, 123, 3498–3509. [Google Scholar] [CrossRef] [Green Version]
- Küttner, V.; Mack, C.; Rigbolt, K.T.; Kern, J.S.; Schilling, O.; Busch, H.; Bruckner-Tuderman, L.; Dengjel, J. Global Remodelling of Cellular Microenvironment Due to Loss of Collagen VII. Mol. Syst. Biol. 2013, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Thriene, K.; Grüning, B.A.; Bornert, O.; Erxleben, A.; Leppert, J.; Athanasiou, I.; Weber, E.; Kiritsi, D.; Nyström, A.; Reinheckel, T.; et al. Combinatorial Omics Analysis Reveals Perturbed Lysosomal Homeostasis in Collagen VII-Deficient Keratinocytes. Mol. Cell. Proteom. MCP 2018, 17, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Vendrell, X.; Bautista-Llácer, R.; Alberola, T.M.; García-Mengual, E.; Pardo, M.; Urries, A.; Sánchez, J. Pregnancy after PGD for Recessive Dystrophic Epidermolysis Bullosa Inversa: Genetics and Preimplantation Genetics. J. Assist. Reprod. Genet. 2011, 28, 825–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-Specific Genome Editing for Correction of Induced Pluripotent Stem Cells Derived from Dominant Dystrophic Epidermolysis Bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [Green Version]
- Kuzmiak, H.A.; Maquat, L.E. Applying Nonsense-Mediated MRNA Decay Research to the Clinic: Progress and Challenges. Trends Mol. Med. 2006, 12, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Gardella, R.; Belletti, L.; Zoppi, N.; Marini, D.; Barlati, S.; Colombi, M. Identification of Two Splicing Mutations in the Collagen Type VII Gene (COL7A1) of a Patient Affected by the Localisata Variant of Recessive Dystrophic Epidermolysis Bullosa. Am. J. Hum. Genet. 1996, 59, 292–300. [Google Scholar]
- Hammami-Hauasli, N.; Kalinke, D.U.; Schumann, H.; Kalinke, U.; Pontz, B.F.; Anton-Lamprecht, I.; Pulkkinen, L.; Zimmermann, M.; Uitto, J.; Bruckner-Tuderman, L. A Combination of a Common Splice Site Mutation and a Frameshift Mutation in the COL7A1 Gene: Absence of Functional Collagen VII in Keratinocytes and Skin. J. Investig. Dermatol. 1997, 109, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Woodley, D.T.; Hou, Y.; Martin, S.; Li, W.; Chen, M. Characterization of Molecular Mechanisms Underlying Mutations in Dystrophic Epidermolysis Bullosa Using Site-Directed Mutagenesis. J. Biol. Chem. 2008, 283, 17838–17845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovnanian, A.; Rochat, A.; Bodemer, C.; Petit, E.; Rivers, C.A.; Prost, C.; Fraitag, S.; Christiano, A.M.; Uitto, J.; Lathrop, M.; et al. Characterization of 18 New Mutations in COL7A1 in Recessive Dystrophic Epidermolysis Bullosa Provides Evidence for Distinct Molecular Mechanisms Underlying Defective Anchoring Fibril Formation. Am. J. Hum. Genet. 1997, 61, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titeux, M.; Pendaries, V.; Tonasso, L.; Décha, A.; Bodemer, C.; Hovnanian, A. A Frequent Functional SNP in the MMP1 Promoter Is Associated with Higher Disease Severity in Recessive Dystrophic Epidermolysis Bullosa. Hum. Mutat. 2008, 29, 267–276. [Google Scholar] [CrossRef]
- Breitenbach, J.S.; Rinnerthaler, M.; Trost, A.; Weber, M.; Klausegger, A.; Gruber, C.; Bruckner, D.; Reitsamer, H.A.; Bauer, J.W.; Breitenbach, M. Transcriptome and Ultrastructural Changes in Dystrophic Epidermolysis Bullosa Resemble Skin Aging. Aging 2015, 7, 389–411. [Google Scholar] [CrossRef] [Green Version]
- Rashidghamat, E.; McGrath, J.A. Novel and Emerging Therapies in the Treatment of Recessive Dystrophic Epidermolysis Bullosa. Intractable Rare Dis. Res. 2017, 6, 6–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, E.S.; Yoon, G.; Kang, H.T. A Comparative Analysis of the Cell Biology of Senescence and Aging. Cell. Mol. Life Sci. CMLS 2009, 66, 2503–2524. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Ivarsson, B.; Merrill, C. Production of a Tissue-like Structure by Contraction of Collagen Lattices by Human Fibroblasts of Different Proliferative Potential In Vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 1274–1278. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Nabeshima, K.; Shimao, Y.; Kataoka, H.; Koono, M. Cell Density-Dependent Regulation of Fibronectin Splicing at the EDA Region in Fibroblasts: Cell Density Also Modulates the Responses of Fibroblasts to TGF-β and Cancer Cell-Conditioned Medium. Cancer Lett. 1998, 129, 45–54. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, J.; Zhang, X.; Zhou, X.; Lu, S.; Huang, X.; Shao, J.; Lou, G.; Yang, D.; Geng, Y.-J. Role of MicroRNA-29b in Angiotensin II-Induced Epithelial-Mesenchymal Transition in Renal Tubular Epithelial Cells. Int. J. Mol. Med. 2014, 34, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lago, J.C.; Puzzi, M.B. The Effect of Aging in Primary Human Dermal Fibroblasts. PLoS ONE 2019, 14, e0219165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Dungrawala, H.; Hua, H.; Manukyan, A.; Abraham, L.; Lane, W.; Mead, H.; Wright, J.; Schneider, B.L. Cell Size and Growth Rate Are Major Determinants of Replicative Lifespan. Cell Cycle 2011, 10, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and Characterization of a Fibroblast Marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Marinkovich, M.P.; Veis, A.; Cai, X.; Rao, C.N.; O’Toole, E.A.; Woodley, D.T. Interactions of the Amino-Terminal Noncollagenous (NC1) Domain of Type VII Collagen with Extracellular Matrix Components A POTENTIAL ROLE IN EPIDERMAL-DERMAL ADHERENCE IN HUMAN SKIN. J. Biol. Chem. 1997, 272, 14516–14522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-Associated Stromal Cells as Key Contributors to the Tumor Microenvironment. Breast Cancer Res. BCR 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Hosper, N.A.; van den Berg, P.P.; de Rond, S.; Popa, E.R.; Wilmer, M.J.; Masereeuw, R.; Bank, R.A. Epithelial-to-Mesenchymal Transition in Fibrosis: Collagen Type I Expression Is Highly Upregulated after EMT, but Does Not Contribute to Collagen Deposition. Exp. Cell Res. 2013, 319, 3000–3009. [Google Scholar] [CrossRef]
- Lawson, D.; Harrison, M.; Shapland, C. Fibroblast transgelin and smooth muscle SM22α are the same protein, the expression of which is down-regulated in many cell lines. Cell Motil. 1997, 38, 250–257. [Google Scholar] [CrossRef]
- Breitenbach, J.; Gruber, C.; Klausegger, A.; Trost, A.; Bogner, B.; Reitsamer, H.; Bauer, J.W. Pseudosyndactyly—An Inflammatory and Fibrotic Wound Healing Disorder in Recessive Dystrophic Epidermolysis Bullosa. JDDG J. Dtsch. Dermatol. Ges. 2015, 13, 1257–1266. [Google Scholar] [CrossRef]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In Search of Definitions: Cancer-Associated Fibroblasts and Their Markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Hsia, L.; Ashley, N.; Ouaret, D.; Wang, L.M.; Wilding, J.; Bodmer, W.F. Myofibroblasts Are Distinguished from Activated Skin Fibroblasts by the Expression of AOC3 and Other Associated Markers. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [Green Version]
- Küttner, V.; Mack, C.; Gretzmeier, C.; Bruckner-Tuderman, L.; Dengjel, J. Loss of Collagen VII Is Associated with Reduced Transglutaminase 2 Abundance and Activity. J. Investig. Dermatol. 2014, 134, 2381–2389. [Google Scholar] [CrossRef] [Green Version]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An Essential Role for Fibronectin Extra Type III Domain A in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Steiglitz, B.M.; Keene, D.R.; Greenspan, D.S. PCOLCE2 Encodes a Functional Procollagen C-Proteinase Enhancer (PCPE2) That Is a Collagen-Binding Protein Differing in Distribution of Expression and Post-Translational Modification from the Previously Described PCPE1. J. Biol. Chem. 2002, 277, 49820–49830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.W.; You, F.; Januszyk, M.; Gurtner, G.C.; Kuang, A.A. Transcriptional Profiling of Rapamycin-Treated Fibroblasts From Hypertrophic and Keloid Scars. Ann. Plast. Surg. 2014, 72, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.L.; Kempsell, K.E.; Szakmany, T.; Ball, G. Development of a Bioinformatics Framework for Identification and Validation of Genomic Biomarkers and Key Immunopathology Processes and Controllers in Infectious and Non-Infectious Severe Inflammatory Response Syndrome. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Alexeev, V.; Salas-Alanis, J.C.; Palisson, F.; Mukhtarzada, L.; Fortuna, G.; Uitto, J.; South, A.; Igoucheva, O. Pro-Inflammatory Chemokines and Cytokines Dominate the Blister Fluid Molecular Signature in Patients with Epidermolysis Bullosa and Affect Leukocyte and Stem Cell Migration. J. Investig. Dermatol. 2017, 137, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annicchiarico, G.; Morgese, M.G.; Esposito, S.; Lopalco, G.; Lattarulo, M.; Tampoia, M.; Bonamonte, D.; Brunetti, L.; Vitale, A.; Lapadula, G.; et al. Proinflammatory Cytokines and Antiskin Autoantibodies in Patients with Inherited Epidermolysis Bullosa. Medicine 2015, 94, e1528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jing, Y.; Ding, L.; Zhang, X.; Song, Y.; Chen, S.; Zhao, X.; Huang, X.; Pu, Y.; Wang, Z.; et al. Epiregulin Reprograms Cancer-Associated Fibroblasts and Facilitates Oral Squamous Cell Carcinoma Invasion via JAK2-STAT3 Pathway. J. Exp. Clin. Cancer Res. CR 2019, 38, 274. [Google Scholar] [CrossRef] [Green Version]
- Massip-Copiz, M.; Clauzure, M.; Valdivieso, Á.G.; Santa-Coloma, T.A. Epiregulin (EREG) Is Upregulated through an IL-1β Autocrine Loop in Caco-2 Epithelial Cells with Reduced CFTR Function. J. Cell. Biochem. 2018, 119, 2911–2922. [Google Scholar] [CrossRef]
- Carpenter, B.L.; Liu, J.; Qi, L.; Wang, C.; O’Connor, K.L. Integrin Α6β4 Upregulates Amphiregulin and Epiregulin through Base Excision Repair-Mediated DNA Demethylation and Promotes Genome-Wide DNA Hypomethylation. Sci. Rep. 2017, 7, 6174. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.L.; Chen, M.; Knifley, T.; Davis, K.A.; Harrison, S.M.W.; Stewart, R.L.; O’Connor, K.L. Integrin Α6β4 Promotes Autocrine Epidermal Growth Factor Receptor (EGFR) Signaling to Stimulate Migration and Invasion toward Hepatocyte Growth Factor (HGF). J. Biol. Chem. 2015, 290, 27228–27238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmeier, V.; Etich, J.; Pitzler, L.; Frie, C.; Koch, M.; Fischer, M.; Rappl, G.; Abken, H.; Tomasek, J.J.; Brachvogel, B. Identification of a Myofibroblast-Specific Expression Signature in Skin Wounds. Matrix Biol. 2018, 65, 59–74. [Google Scholar] [CrossRef]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-Transmembrane Epithelial Antigens of the Prostate Comprise a Novel Inflammatory Nexus in Patients with Pustular Skin Disorders. J. Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Grievink, H.W.; Luisman, T.; Kluft, C.; Moerland, M.; Malone, K.E. Comparison of Three Isolation Techniques for Human Peripheral Blood Mononuclear Cells: Cell Recovery and Viability, Population Composition, and Cell Functionality. Biopreserv. Biobank. 2016, 14, 410–415. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilin, A.K.; Rippa, A.L.; Sharobaro, V.I.; Gurskaya, N.G.; Vorotelyak, E.A. The Reconstructed Human Epidermis In Vitro—A Model for Basic and Applied Research of Human Skin. Vestn. Dermatol. Venerol. 2020, 96, 24–34. [Google Scholar] [CrossRef]
- Chermnykh, E.S.; Kiseleva, E.V.; Rogovaya, O.S.; Rippa, A.L.; Vasiliev, A.V.; Vorotelyak, E.A. Tissue-Engineered Biological Dressing Accelerates Skin Wound Healing in Mice via Formation of Provisional Connective Tissue. Histol. Histopathol. 2018, 33, 1189–1199. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 4, 402–408. [Google Scholar] [CrossRef] [PubMed]
- GitHub–Nicjac/PHANTAST-FIJI: Repository for the FIJI/ImageJ Implementation of the Phase Contrast Microscopy Segmentation Toolbox (PHANTAST). Available online: https://github.com/nicjac/PHANTAST-FIJI (accessed on 29 December 2020).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reduce + Visualize GO. Available online: https://ssayols.github.io/rrvgo/ (accessed on 22 December 2020).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Form of RDEB | Gender/Age | Cell Line | C7 Level | Mutation 1 | Mutation 2 |
|---|---|---|---|---|---|---|
| d1 RDEB | generalized severe ORPHA 79408 | M/8 | FEB1 | diminished | c.425A > G (p.K142R) | c.425A > G (p.K142R) |
| d2 RDEB | inversa ORPHA 79409 | M/16 | FEB2 | diminished | c.682 + 1G > A | c.6205 C > T (p.R2069C) |
| d3 * RDEB | generalized severe ORPHA 79408 | M/5 | FEB3 | diminished | c.8245G > A (p.G2749R) | c.8245G > A (p.G2749R) |
| d4 RDEB | generalized intermediate ORPHA 89842 | M/21 | FEB4 | diminished | c.520G > A (p.G174R) | undetermined |
| d5 | - | F/58 | FHC1 | normal | ||
| d6 | - | F/57 | FHC2 | normal | ||
| d7 | - | M/8 | FHC3 | normal | ||
| d8 | - | F/24 | FHC4 | normal | ||
| d9 | - | M/24 | FHC5 | not shown | ||
| d10 | - | F/25 | FHC6 | not shown | ||
| d11 | - | M/35 | FHC7 | not shown | ||
| d12 | - | M/8 | KHC8 ** | not shown |
| Cell Line | Area | Perimeter | Fit Ellipse Minor Axis | Circularity | Solidity | Minimum Caliper Diameter | Contraction of Collagen Gel | FSC | SSC |
|---|---|---|---|---|---|---|---|---|---|
| FEB1 | high | low | low | high | |||||
| FEB2 | high | high | high | low | high | high | high | ||
| FEB3 | low | high | low | low | |||||
| FEB4 | high | high | low | high | high | high |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beilin, A.K.; Evtushenko, N.A.; Lukyanov, D.K.; Murashkin, N.N.; Ambarchian, E.T.; Pushkov, A.A.; Savostyanov, K.V.; Fisenko, A.P.; Rogovaya, O.S.; Vasiliev, A.V.; et al. Signatures of Dermal Fibroblasts from RDEB Pediatric Patients. Int. J. Mol. Sci. 2021, 22, 1792. https://doi.org/10.3390/ijms22041792
Beilin AK, Evtushenko NA, Lukyanov DK, Murashkin NN, Ambarchian ET, Pushkov AA, Savostyanov KV, Fisenko AP, Rogovaya OS, Vasiliev AV, et al. Signatures of Dermal Fibroblasts from RDEB Pediatric Patients. International Journal of Molecular Sciences. 2021; 22(4):1792. https://doi.org/10.3390/ijms22041792
Chicago/Turabian StyleBeilin, Arkadii K., Nadezhda A. Evtushenko, Daniil K. Lukyanov, Nikolay N. Murashkin, Eduard T. Ambarchian, Alexander A. Pushkov, Kirill V. Savostyanov, Andrey P. Fisenko, Olga S. Rogovaya, Andrey V. Vasiliev, and et al. 2021. "Signatures of Dermal Fibroblasts from RDEB Pediatric Patients" International Journal of Molecular Sciences 22, no. 4: 1792. https://doi.org/10.3390/ijms22041792