
Distinctive HBV Replication Capacity and Susceptibility to Tenofovir Induced by a Polymerase Point Mutation in Hepatoma Cell Lines and Primary Human Hepatocytes

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
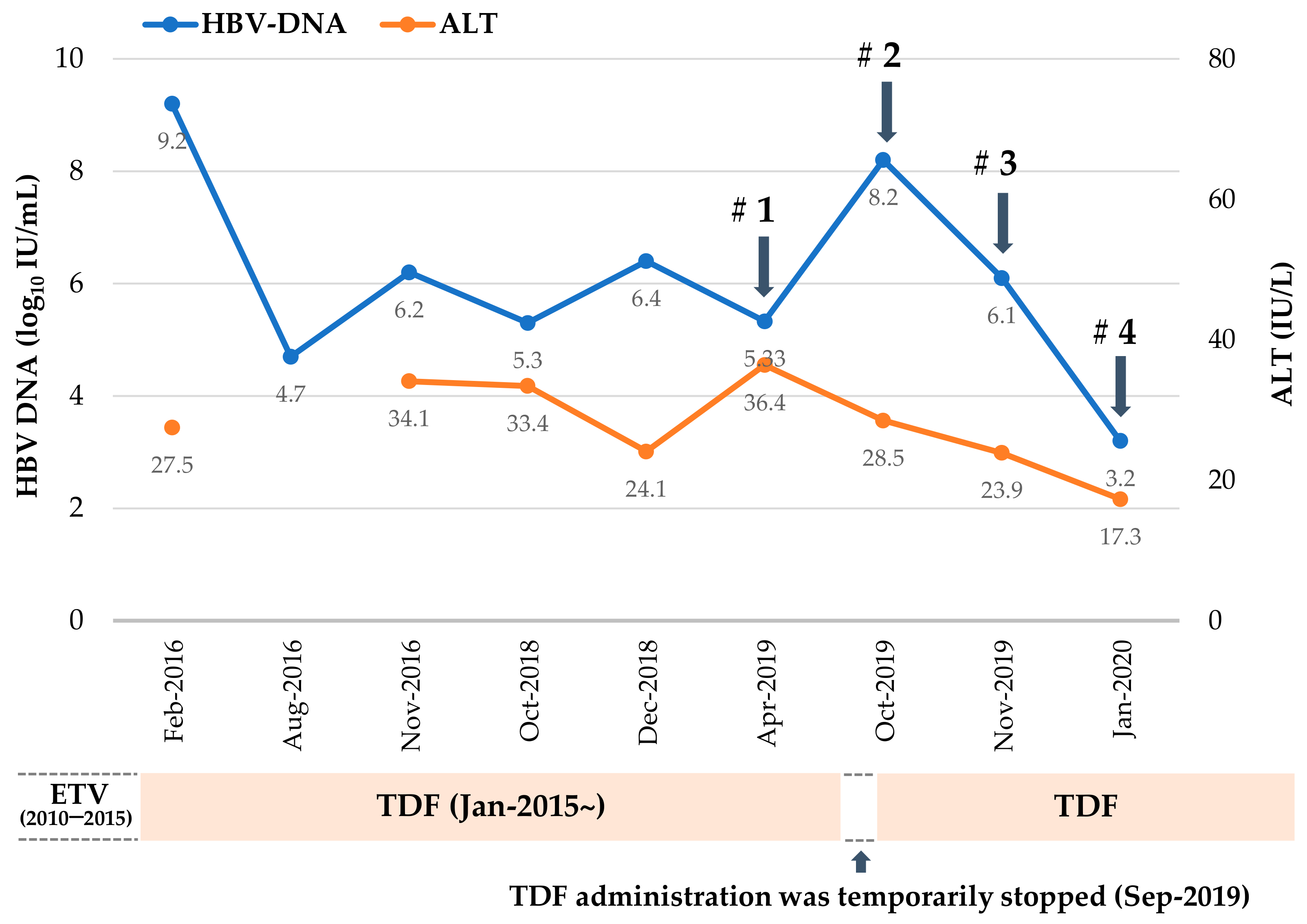
2.1. Mutation Profile of HBV RT Domain Cloned from a TDF-Treated Patient
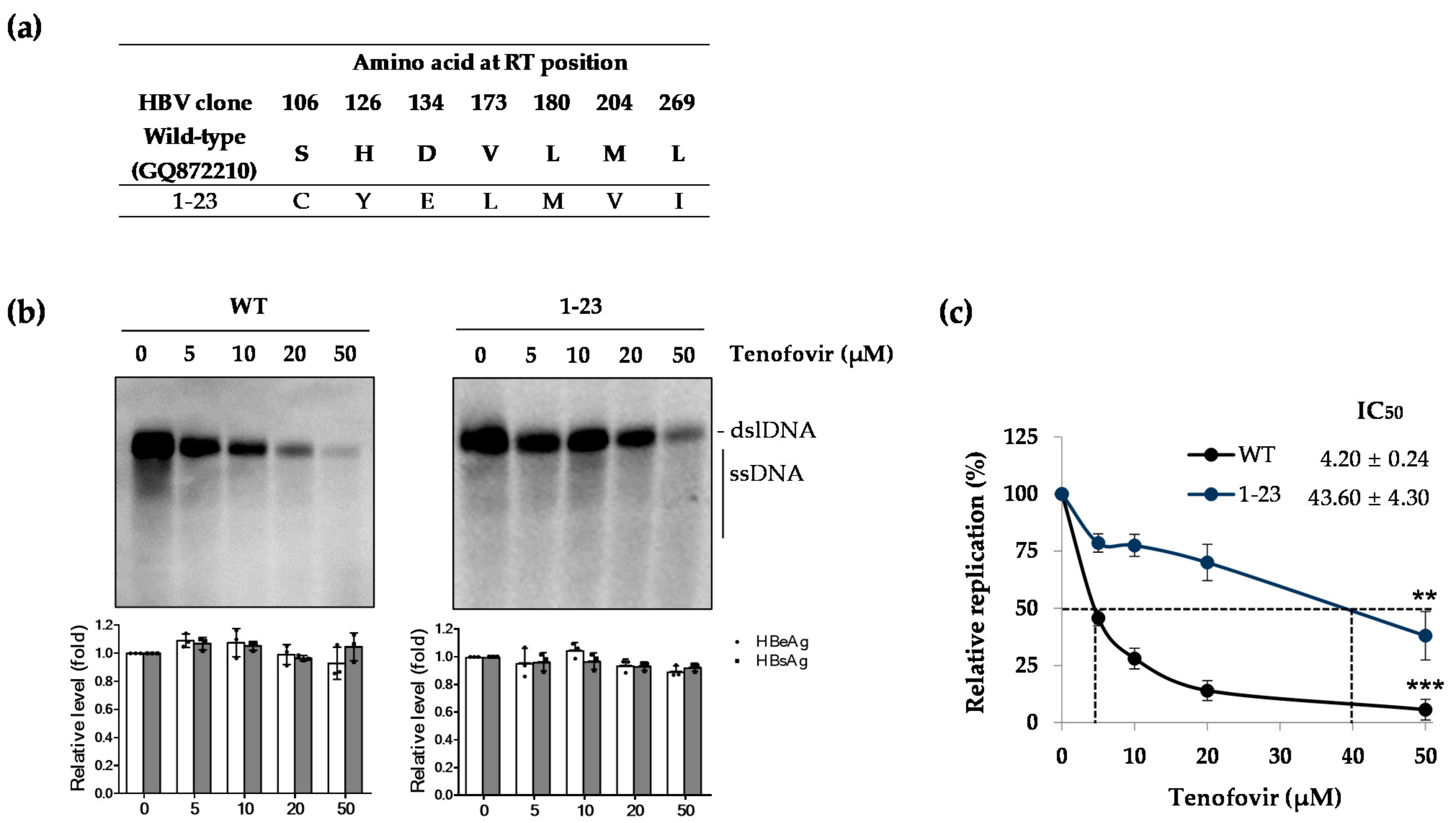
2.2. Patient-Derived HBV Mutant Harboring CYELMVI Mutation Is Resistant to Tenofovir Treatment
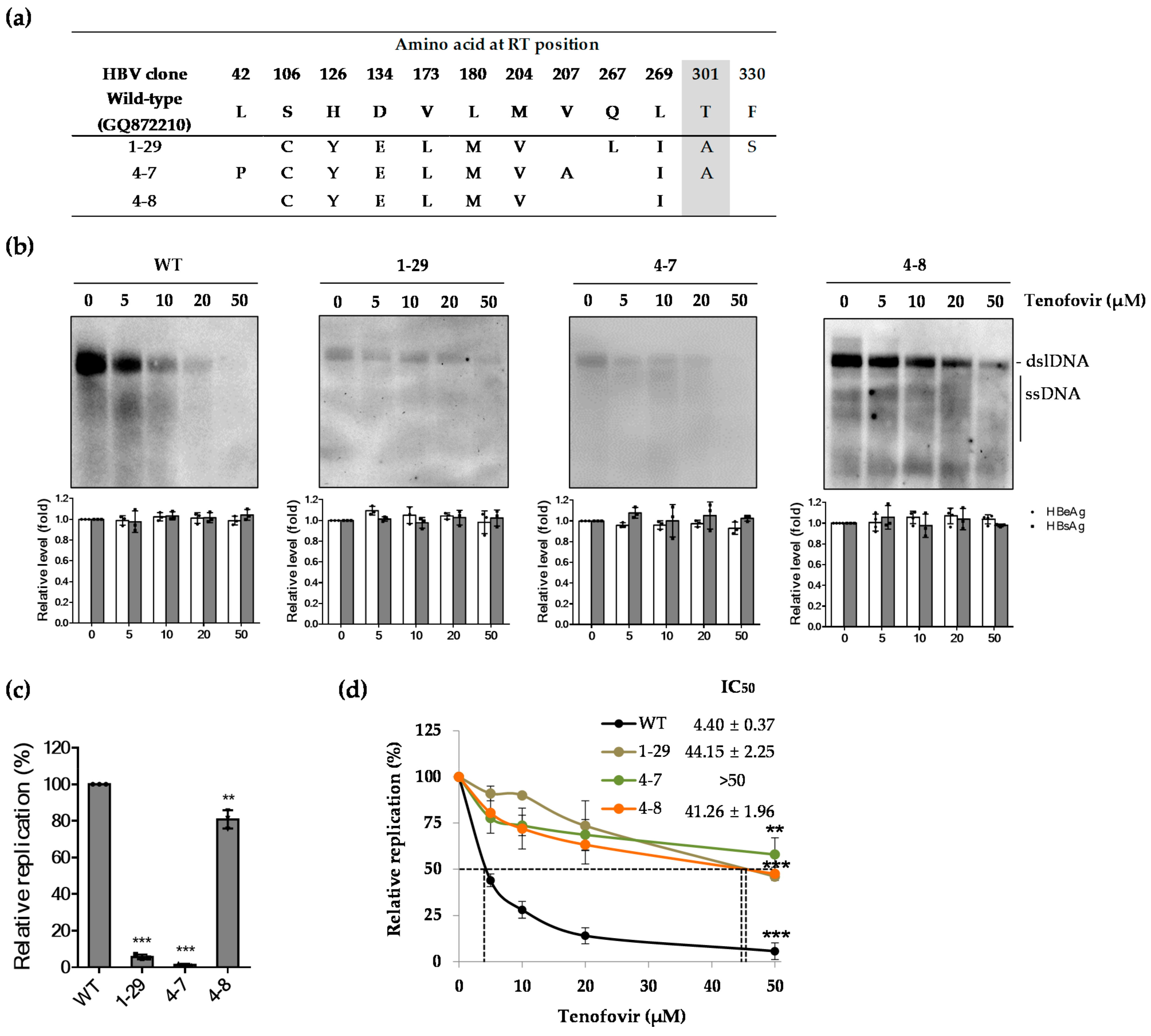
2.3. Effect of Patient-Derived HBV RT Mutants Harboring the rtT301A Mutation on Replication Capacity and Tenofovir Resistance in Hepatoma Cell Lines
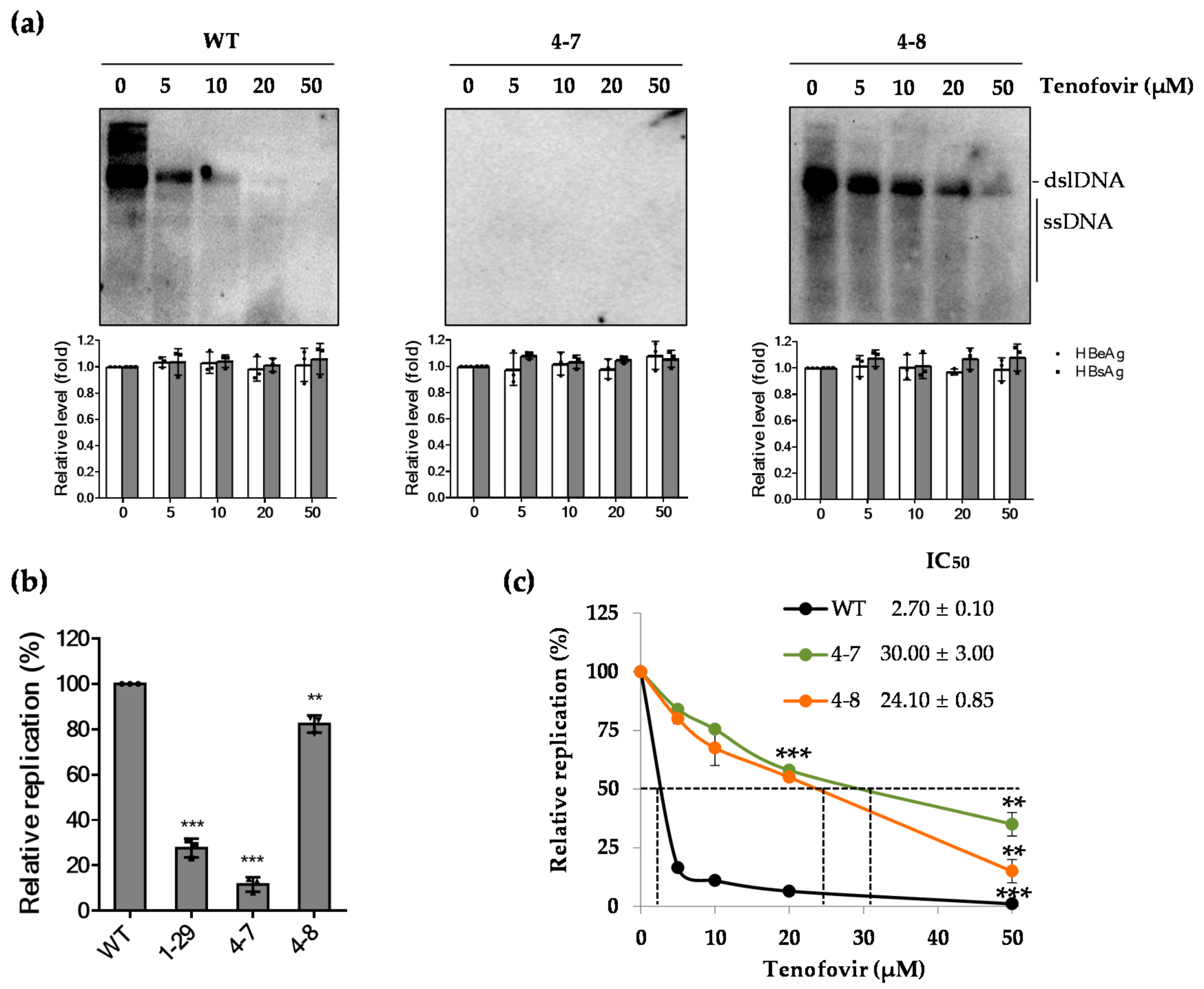
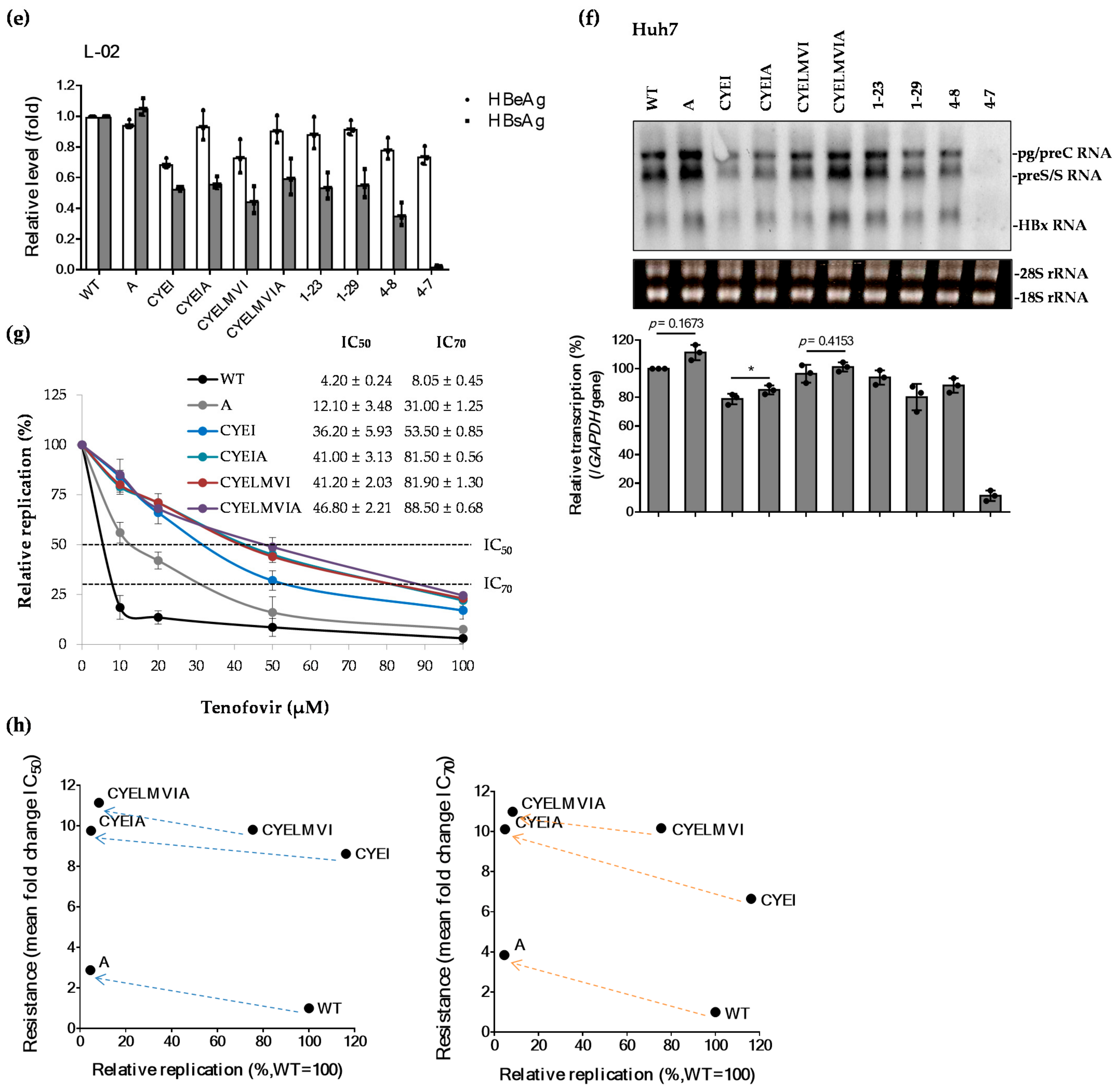
2.4. The rtT301A Mutation Decreases Replication Capacity and Increases Tenofovir Resistance in Hepatoma Cell Lines
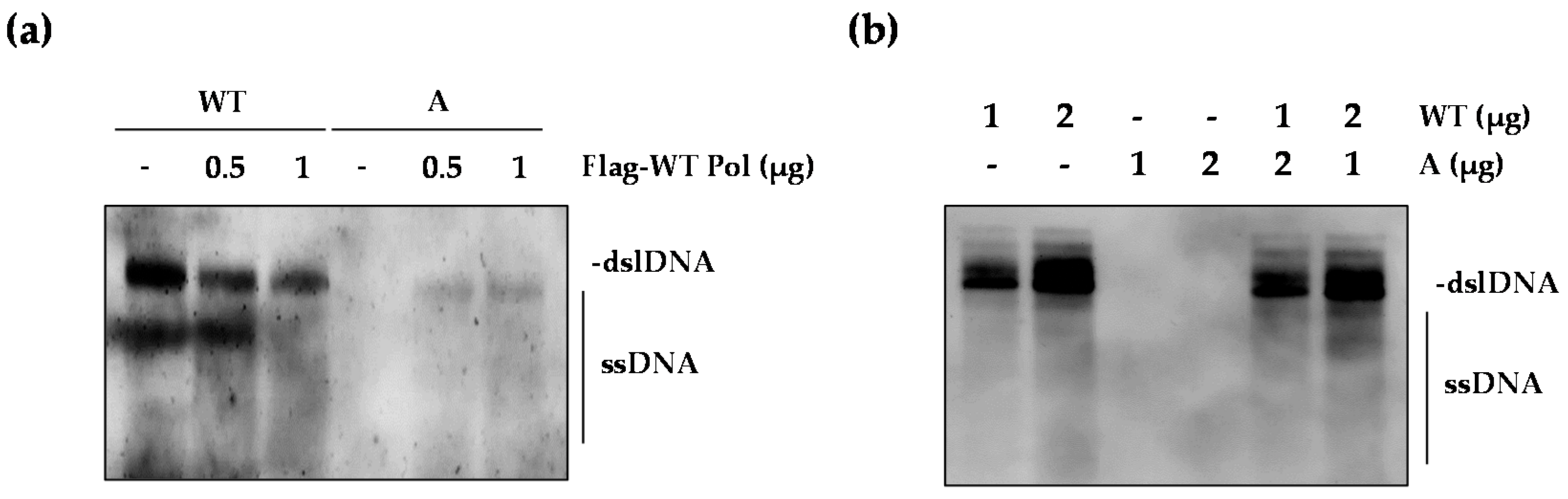
2.5. The WT Polymerase (WT Pol) Partially Rescued the Replication Defect of the rtT301A Mutant
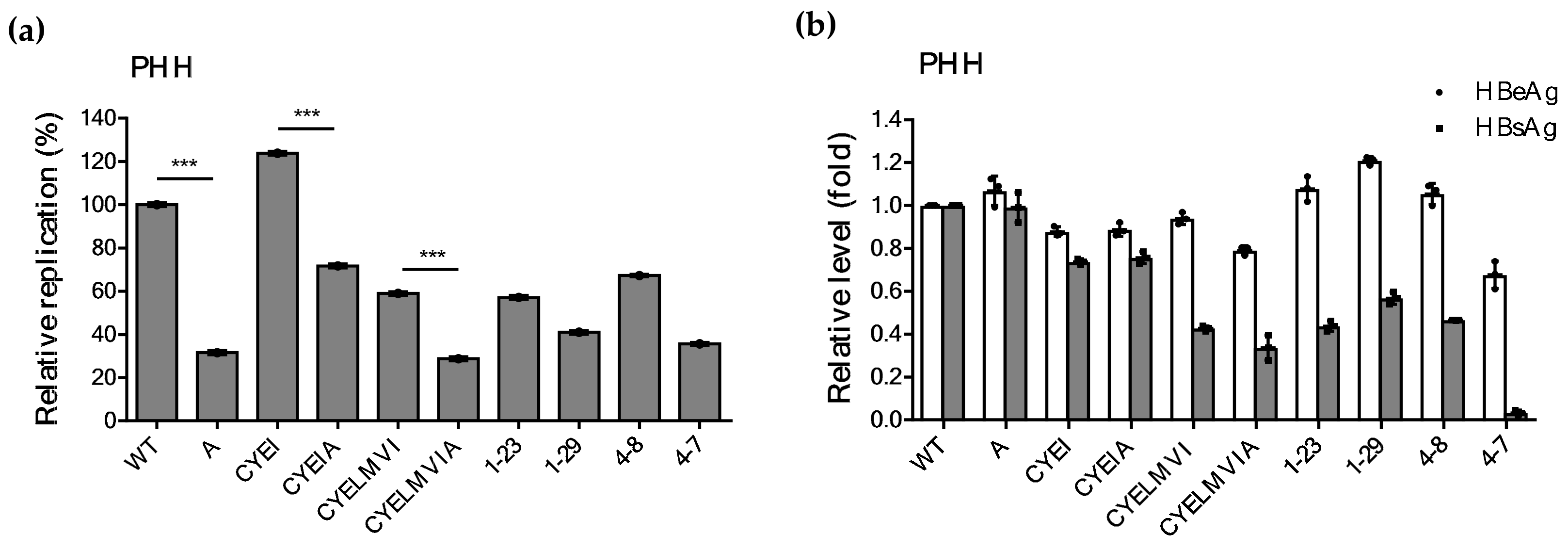
2.6. Impaired Replication Ability of rtT301A Mutants in Hepatoma Cell Lines Is Restored in PHHs
3. Discussion
4. Materials and Methods
4.1. Patient
4.2. HBV RT Sequence Analysis
4.3. Construction of HBV Reverse Transcriptase (RT) Mutant Replicons Harboring Artificially Substituted or Patient-Derived RT Domains by Site-Directed Mutagenesis
4.4. Isolation of Primary Human Hepatocytes (PHHs)
4.5. Cell Culture, Transfection, and Drug Treatment
4.6. Southern and Northern Blot Analysis
4.7. Quantitative Real-Time PCR
4.8. ELISA
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trepo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, N.D.; Seong, B.L. Discovery and development of anti-HBV agents and their resistance. Molecules 2010, 15, 5878–5908. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Aguilar Schall, R.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. 2018 Korean Association for the Study of the Liver (KASL) Clinical Practice Guidelines of Chronic Hepatitis B: What’s Different? Korean J. Gastroenterol. 2019, 73, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J.; Chan, H.L.Y. Tenofovir Alafenamide: A Review in Chronic Hepatitis B. Drugs 2017, 77, 1017–1028. [Google Scholar] [CrossRef]
- Park, E.S.; Lee, A.R.; Kim, D.H.; Lee, J.H.; Yoo, J.J.; Ahn, S.H.; Sim, H.; Park, S.; Kang, H.S.; Won, J.; et al. Identification of a quadruple mutation that confers tenofovir resistance in chronic hepatitis B patients. J. Hepatol. 2019, 70, 1093–1102. [Google Scholar] [CrossRef]
- Echevarria, J.M.; Avellon, A. Hepatitis B virus genetic diversity. J. Med. Virol. 2006, 78 (Suppl. S1), S36–S42. [Google Scholar] [CrossRef]
- Park, S.; Park, E.S.; Koo, J.E.; Park, Y.K.; Lee, A.R.; Dezhbord, M.; Cho, E.S.; Ahn, S.H.; Kim, D.H.; Lee, J.H.; et al. Entecavir-resistant hepatitis B virus decreases surface antigenicity: A full genome and functional characterization. Liver Int. 2020, 40, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Guerra, B.; Lee, H.; Averett, D.R.; Pfeiffer, B.; Chavez, D.; Notvall, L.; Bigger, C. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(i)-poly(c), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic replicons. J. Virol. 2003, 77, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- McCormick, C.J.; Challinor, L.; Macdonald, A.; Rowlands, D.J.; Harris, M. Introduction of replication-competent hepatitis C virus transcripts using a tetracycline-regulable baculovirus delivery system. J. Gen. Virol. 2004, 85, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Dastgerdi, E.; Winer, B.Y.; Celia-Terrassa, T.; Kang, Y.; Tabernero, D.; Yagmur, E.; Rodriguez-Frias, F.; Gregori, J.; Luedde, T.; Trautwein, C.; et al. Selection of the highly replicative and partially multidrug resistant rtS78T HBV polymerase mutation during TDF-ETV combination therapy. J. Hepatol. 2017, 67, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Wang, J.; Zhao, X.; Li, Y.; Zhang, Q.; Song, C.; Zeng, H.; Wang, X. Entecavir resistance mutations rtL180M/T184L/M204V combined with rtA200V lead to tenofovir resistance. Liver Int. 2020, 40, 83–91. [Google Scholar] [CrossRef]
- Liu, Y.; Corsa, A.C.; Buti, M.; Cathcart, A.L.; Flaherty, J.F.; Miller, M.D.; Kitrinos, K.M.; Marcellin, P.; Gane, E.J. No detectable resistance to tenofovir disoproxil fumarate in HBeAg+ and HBeAg- patients with chronic hepatitis B after 8 years of treatment. J. Viral Hepat. 2017, 24, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Corsa, A.C.; Liu, Y.; Flaherty, J.F.; Mitchell, B.; Fung, S.K.; Gane, E.; Miller, M.D.; Kitrinos, K.M. No resistance to tenofovir disoproxil fumarate through 96 weeks of treatment in patients with lamivudine-resistant chronic hepatitis B. Clin. Gastroenterol. Hepatol. 2014, 12, 2106–2112.e1. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Jung, E.U.; Kim, B.H.; Lee, J.H.; Cho, H.; Ahn, H.; Choi, W.M.; Cho, Y.Y.; Lee, M.; Yoo, J.J.; et al. Tenofovir monotherapy versus tenofovir plus lamivudine or telbivudine combination therapy in treatment of lamivudine-resistant chronic hepatitis B. Antimicrob. Agents Chemother. 2015, 59, 972–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.S.; Byun, K.S.; Yoo, B.C.; Kwon, S.Y.; Kim, Y.J.; An, J.; Lee, H.C.; Lee, Y.S. Tenofovir monotherapy versus tenofovir and entecavir combination therapy in patients with entecavir-resistant chronic hepatitis B with multiple drug failure: Results of a randomised trial. Gut 2016, 65, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.S.; Yoo, B.C.; Byun, K.S.; Kwon, S.Y.; Kim, Y.J.; An, J.; Lee, H.C.; Lee, Y.S. Tenofovir monotherapy versus tenofovir and entecavir combination therapy in adefovir-resistant chronic hepatitis B patients with multiple drug failure: Results of a randomised trial. Gut 2016, 65, 1042–1051. [Google Scholar] [CrossRef]
- Ghany, M.G.; Doo, E.C. Antiviral resistance and hepatitis B therapy. Hepatology 2009, 49, S174–S184. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Yuan, Z.H.; Wu, L.; Ding, J.P.; Wen, Y.M. A single amino acid in the reverse transcriptase domain of hepatitis B virus affects virus replication efficiency. J. Virol. 2001, 75, 11827–11833. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.H.; Kim, D.H.; Lee, A.R.; Kim, B.K.; Park, Y.K.; Park, E.S.; Ahn, S.H.; Shin, G.C.; Park, S.; Kang, H.S.; et al. Substitution at rt269 in Hepatitis B Virus Polymerase Is a Compensatory Mutation Associated with Multi-Drug Resistance. PLoS ONE 2015, 10, e0136728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, W.E.t.; Yang, H.; Westland, C.E.; Das, K.; Arnold, E.; Gibbs, C.S.; Miller, M.D.; Xiong, S. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J. Virol. 2003, 77, 11833–11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, H.; Zhang, J.; Zhang, J.; Guo, H. Naturally occurring core protein mutations compensate for the reduced replication fitness of a lamivudine-resistant HBV isolate. Antiviral Res. 2019, 165, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Warner, N.; Locarnini, S.; Kuiper, M.; Bartholomeusz, A.; Ayres, A.; Yuen, L.; Shaw, T. The L80I substitution in the reverse transcriptase domain of the hepatitis B virus polymerase is associated with lamivudine resistance and enhanced viral replication in vitro. Antimicrob. Agents Chemother. 2007, 51, 2285–2292. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xin, S.; Ye, X.; Chen, R.; Xu, Z.; Li, X.; Ye, H.; Cheng, S.; Xu, D. Increased occurrence of mutant rtI233V of HBV in patients receiving adefovir therapy. Antivir. Ther. 2016, 21, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Blum, H.E.; Galun, E.; Liang, T.J.; von Weizsacker, F.; Wands, J.R. Naturally occurring missense mutation in the polymerase gene terminating hepatitis B virus replication. J. Virol. 1991, 65, 1836–1842. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.C.; Krastev, Z.; Horban, A.; Petersen, J.; Sperl, J.; Dinh, P.; Martins, E.B.; Yee, L.J.; Flaherty, J.F.; Kitrinos, K.M.; et al. Efficacy of tenofovir disoproxil fumarate at 240 weeks in patients with chronic hepatitis B with high baseline viral load. Hepatology 2013, 58, 505–513. [Google Scholar] [CrossRef]
- Clark, D.N.; Hu, J. Unveiling the roles of HBV polymerase for new antiviral strategies. Future Virol. 2015, 10, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Shaw, T.; Bartholomeusz, A.; Locarnini, S. HBV drug resistance: Mechanisms, detection and interpretation. J. Hepatol. 2006, 44, 593–606. [Google Scholar] [CrossRef]
- Melegari, M.; Scaglioni, P.P.; Wands, J.R. Hepatitis B virus mutants associated with 3TC and famciclovir administration are replication defective. Hepatology 1998, 27, 628–633. [Google Scholar] [CrossRef]
- Hu, J.; Toft, D.O.; Seeger, C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 1997, 16, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kim, S.; Ryu, W.S. DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J. Virol. 2009, 83, 5815–5824. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Hu, J. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 2008, 82, 6852–6861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seglen, P.O. Preparation of rat liver cells. 3. Enzymatic requirements for tissue dispersion. Exp. Cell Res. 1973, 82, 391–398. [Google Scholar] [CrossRef]
- Ahn, S.H.; Park, Y.K.; Park, E.S.; Kim, J.H.; Kim, D.H.; Lim, K.H.; Jang, M.S.; Choe, W.H.; Ko, S.Y.; Sung, I.K.; et al. The impact of the hepatitis B virus polymerase rtA181T mutation on replication and drug resistance is potentially affected by overlapping changes in surface gene. J. Virol. 2014, 88, 6805–6818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucifora, J.; Salvetti, A.; Marniquet, X.; Mailly, L.; Testoni, B.; Fusil, F.; Inchauspe, A.; Michelet, M.; Michel, M.L.; Levrero, M.; et al. Detection of the hepatitis B virus (HBV) covalently-closed-circular DNA (cccDNA) in mice transduced with a recombinant AAV-HBV vector. Antiviral Res. 2017, 145, 14–19. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood Sample | Amino Acid at RT Position | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HBV Clone | 8 | 12 | 38 | 42 | 55 | 106 | 110 | 117 | 126 | 128 | 134 | 148 | 164 | 167 | 173 | 180 | 181 | 190 | 191 | 199 | 204 | 207 | 214 | 223 | 226 | 226 | 238 | 241 | 247 | 266 | 267 | 269 | 270 | 274 | 275 | 292 | 293 | 295 | 300 | 301 | 313 | 317 | 320 | 323 | 324 | 329 | 330 | 332 | 336 | |
| Wild-Type (GQ872210) | E | H | T | L | H | S | R | S | H | T | D | Y | L | R | V | L | A | V | V | L | M | V | V | S | N | N | N | K | L | V | Q | L | K | R | K | G | L | G | F | T | A | S | A | F | S | A | F | C | L | |
| #1 | 1-1 | G | ||||||||||||||||||||||||||||||||||||||||||||||||
| 1-4 | P | C | Y | E | L | L | M | V | A | L | I | A | ||||||||||||||||||||||||||||||||||||||
| 1-5 | A | I | A | H | I | A | ||||||||||||||||||||||||||||||||||||||||||||
| 1-6,7,8 | M | V | ||||||||||||||||||||||||||||||||||||||||||||||||
| 1-9 | P | L | A | M | ||||||||||||||||||||||||||||||||||||||||||||||
| 1-10 | P | |||||||||||||||||||||||||||||||||||||||||||||||||
| 1-17 | C | Y | E | L | M | V | I | Q | R | C | ||||||||||||||||||||||||||||||||||||||||
| 1-19 | C | Y | E | H | L | M | V | I | A | W | ||||||||||||||||||||||||||||||||||||||||
| 1-23,28 | C | Y | E | L | M | V | I | |||||||||||||||||||||||||||||||||||||||||||
| 1-29,30,31,38 | C | Y | E | L | M | V | L | I | A | S | ||||||||||||||||||||||||||||||||||||||||
| 1-32 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 1-33 | P | |||||||||||||||||||||||||||||||||||||||||||||||||
| 1-34 | C | Y | E | L | M | V | L | I | A | G | ||||||||||||||||||||||||||||||||||||||||
| 1-35,37 | A | R | I | H | I | A | ||||||||||||||||||||||||||||||||||||||||||||
| 1-36 | R | A | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| #2 | 2-1 | A | R | I | H | I | L | A | T | |||||||||||||||||||||||||||||||||||||||||
| 2-2 | A | C | N | H | I | L | A | |||||||||||||||||||||||||||||||||||||||||||
| 2-3 | A | L | V | H | I | L | A | L | ||||||||||||||||||||||||||||||||||||||||||
| 2-4 | A | R | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 2-6 | A | C | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 2-7 | A | I | H | P | I | L | A | T | ||||||||||||||||||||||||||||||||||||||||||
| 2-8 | A | C | I | H | I | L | A | T | ||||||||||||||||||||||||||||||||||||||||||
| 2-9 | A | C | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 2-11 | A | R | C | H | I | L | A | |||||||||||||||||||||||||||||||||||||||||||
| 2-12 | A | F | I | H | I | L | A | |||||||||||||||||||||||||||||||||||||||||||
| #3 | 3-1 | A | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 3-2 | A | H | I | L | A | T | ||||||||||||||||||||||||||||||||||||||||||||
| 3-3 | A | H | I | L | A | T | ||||||||||||||||||||||||||||||||||||||||||||
| 3-4 | A | R | C | G | F | H | I | L | A | |||||||||||||||||||||||||||||||||||||||||
| 3-5 | A | I | H | I | L | A | T | |||||||||||||||||||||||||||||||||||||||||||
| 3-7 | A | R | H | I | L | S | A | |||||||||||||||||||||||||||||||||||||||||||
| 3-9 | A | T | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 3-10 | A | R | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| #4 | 4-1 | |||||||||||||||||||||||||||||||||||||||||||||||||
| 4-2 | A | C | N | L | A | |||||||||||||||||||||||||||||||||||||||||||||
| 4-3 | E | S | ||||||||||||||||||||||||||||||||||||||||||||||||
| 4-4 | K | V | P | |||||||||||||||||||||||||||||||||||||||||||||||
| 4-7 | P | C | Y | E | L | M | V | A | L | I | A | |||||||||||||||||||||||||||||||||||||||
| 4-8 | C | Y | E | L | M | V | I | |||||||||||||||||||||||||||||||||||||||||||
| 4-11 | A | C | H | I | L | A | ||||||||||||||||||||||||||||||||||||||||||||
| 4-12 | P | I | S | |||||||||||||||||||||||||||||||||||||||||||||||
| 4-14 | A | H | I | L | T | |||||||||||||||||||||||||||||||||||||||||||||
| Clone | Region | Mutations in the Corresponding Gene | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | RT | L42 | S106 | H126 | D134 | V173 | L180 | M204 | V207 | Q267 | L269 | T301 | F330 |
| S | S34 | L98 | T118 | I126 | W165 | W172 | W196 | W199 | - | - | - | - | |
| 1-23 | RT | C | Y | E | L | M | V | I | |||||
| S | V | - | S | - | - | - | |||||||
| 1-29 | RT | C | Y | E | L | M | V | L | I | A | S | ||
| S | V | - | S | - | - | - | |||||||
| 4-8 | RT | C | Y | E | L | M | V | I | |||||
| S | V | - | S | - | - | - | |||||||
| 4-7 | RT | P | C | Y | E | L | M | V | A | I | A | ||
| S | P | V | - | S | - | - | - | R | |||||
| Clone | Huh7 | |||
|---|---|---|---|---|
| IC50 | IC70 | Fold Resistance (/IC50) | Fold Resistance (/IC70) | |
| WT | 4.20 ± 0.24 | 8.05 ± 0.45 | 1 | 1.00 |
| A | 12.10 ± 3.48 | 31.00 ± 1.25 | 2.88 | 3.85 |
| CYEI | 36.20 ± 5.93 | 53.50 ± 0.85 | 8.62 | 6.65 |
| CYEIA | 41.00 ± 3.13 | 81.50 ± 0.56 | 9.76 | 10.12 |
| CYELMVI | 41.20 ± 2.03 | 81.90 ± 1.30 | 9.81 | 10.17 |
| CYELMVIA | 46.80 ± 2.21 | 88.50 ± 0.68 | 11.14 | 10.99 |
| 1-23 | 43.60 ± 4.30 | >50 | 10.38 | >11.90 |
| 1-29 | 44.15 ± 2.25 | >50 | 10.51 | >11.90 |
| 4-8 | 41.26 ± 1.96 | >50 | 9.82 | >11.90 |
| 4-7 | >50 | >50 | >11.90 | >11.90 |
| Clone | Replication Ability (%) | ||
|---|---|---|---|
| Huh7 | HepG2 | PHH | |
| WT | 100 | 100 | 100 ± 0.93 |
| A | 4.51 ± 0.73 | 16.45 ± 1.96 | 31.6 ± 0.97 |
| CYEI | 116.18 ± 2.61 | 125.09 ± 2.14 | 123.84 ± 0.90 |
| CYEIA | 4.81 ± 0.71 | 25.69 ± 9.63 | 71.7 ± 0.93 |
| CYELMVI | 75.50 ± 0.50 | 85.26 ± 5.26 | 58.93 ± 0.85 |
| CYELMVIA | 8.26 ± 2.06 | 23.79 ± 1.79 | 28.76 ± 0.96 |
| 1-23 | 79.00 ± 1.00 | 81.52 ± 1.02 | 57.12 ± 0.58 |
| 1-29 | 5.50 ± 1.50 | 25.62 ± 3.00 | 40.96 ± 0.72 |
| 4-8 | 81.13 ± 2.13 | 84.49 ± 0.75 | 67.26 ± 0.88 |
| 4-7 | 2.50 ± 0.50 | 15.90 ± 2.77 | 35.63 ± 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.R.; Cho, J.-Y.; Kim, J.C.; Dezhbord, M.; Choo, S.Y.; Ahn, C.H.; Kim, N.Y.; Shin, J.J.; Park, S.; Park, E.-S.; et al. Distinctive HBV Replication Capacity and Susceptibility to Tenofovir Induced by a Polymerase Point Mutation in Hepatoma Cell Lines and Primary Human Hepatocytes. Int. J. Mol. Sci. 2021, 22, 1606. https://doi.org/10.3390/ijms22041606
Lee AR, Cho J-Y, Kim JC, Dezhbord M, Choo SY, Ahn CH, Kim NY, Shin JJ, Park S, Park E-S, et al. Distinctive HBV Replication Capacity and Susceptibility to Tenofovir Induced by a Polymerase Point Mutation in Hepatoma Cell Lines and Primary Human Hepatocytes. International Journal of Molecular Sciences. 2021; 22(4):1606. https://doi.org/10.3390/ijms22041606
Chicago/Turabian StyleLee, Ah Ram, Ju-Yeon Cho, Jong Chul Kim, Mehrangiz Dezhbord, Soo Yeun Choo, Chang Hyun Ahn, Na Yeon Kim, Jae Jin Shin, Soree Park, Eun-Sook Park, and et al. 2021. "Distinctive HBV Replication Capacity and Susceptibility to Tenofovir Induced by a Polymerase Point Mutation in Hepatoma Cell Lines and Primary Human Hepatocytes" International Journal of Molecular Sciences 22, no. 4: 1606. https://doi.org/10.3390/ijms22041606