
The Contribution of the Urokinase Plasminogen Activator and the Urokinase Receptor to Pleural and Parenchymal Lung Injury and Repair: A Narrative Review

Abstract
:1. Introduction
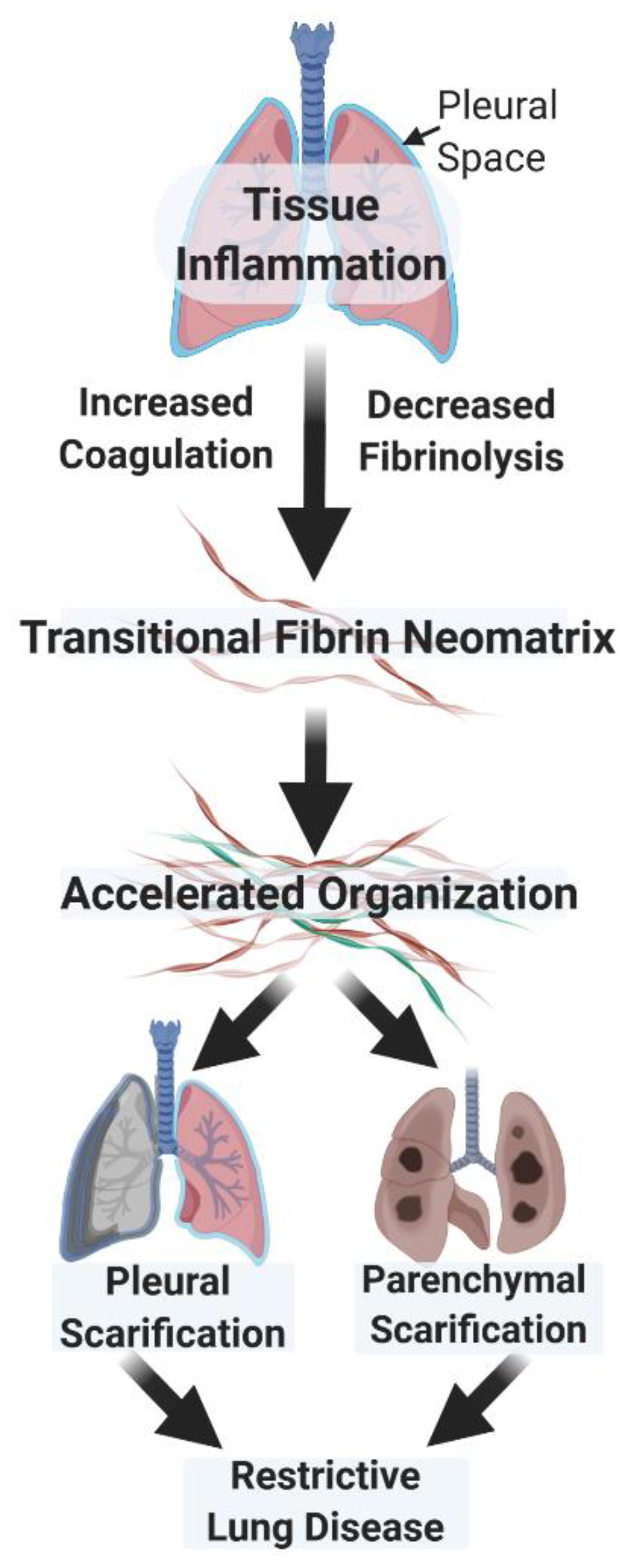
2. Similar Modes of Regulation Govern the Organization of the Fibrinous Transitional Neomatrix in the Settings of Lung and Pleural Injury
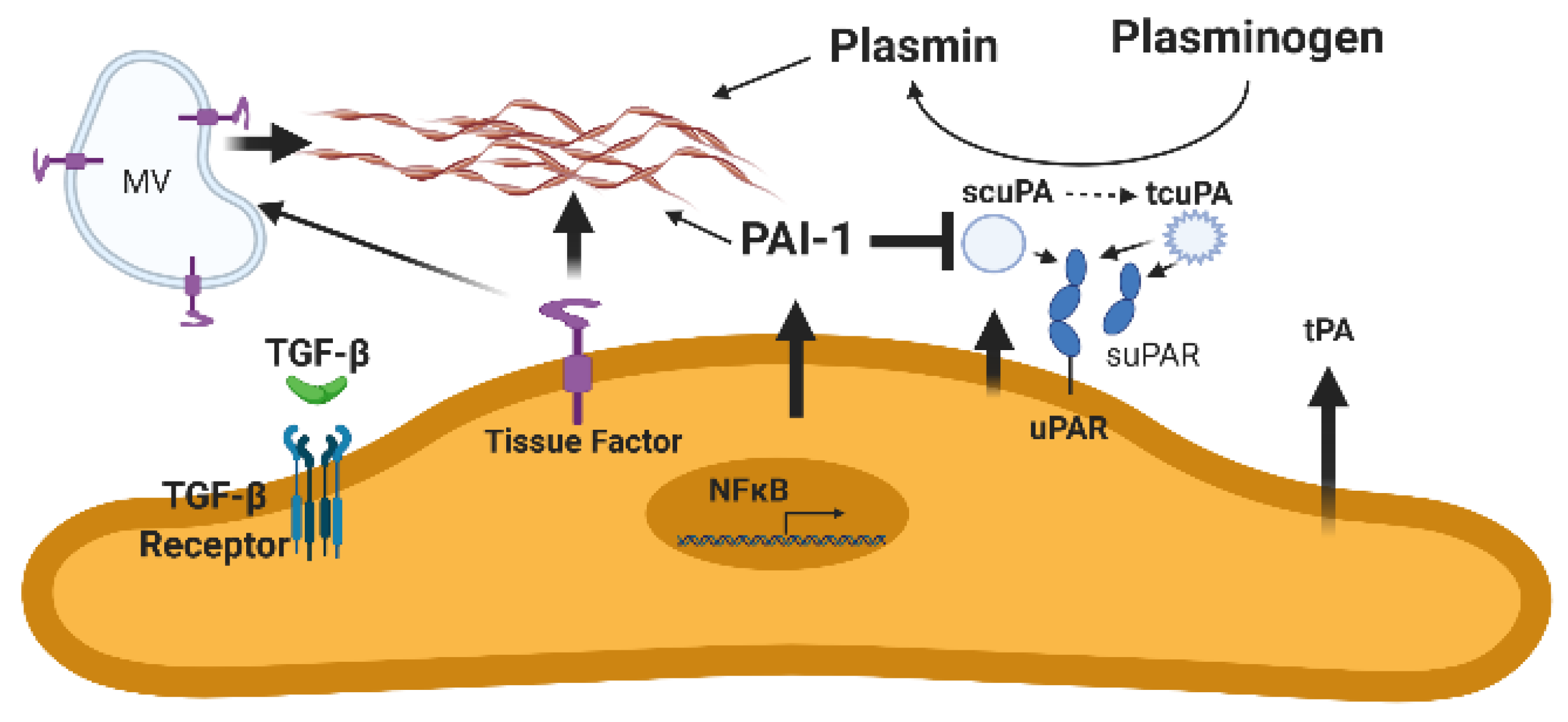
3. The Urokinase/Urokinase Receptor Interaction and Derangements Associated with Lung or Pleural Injury
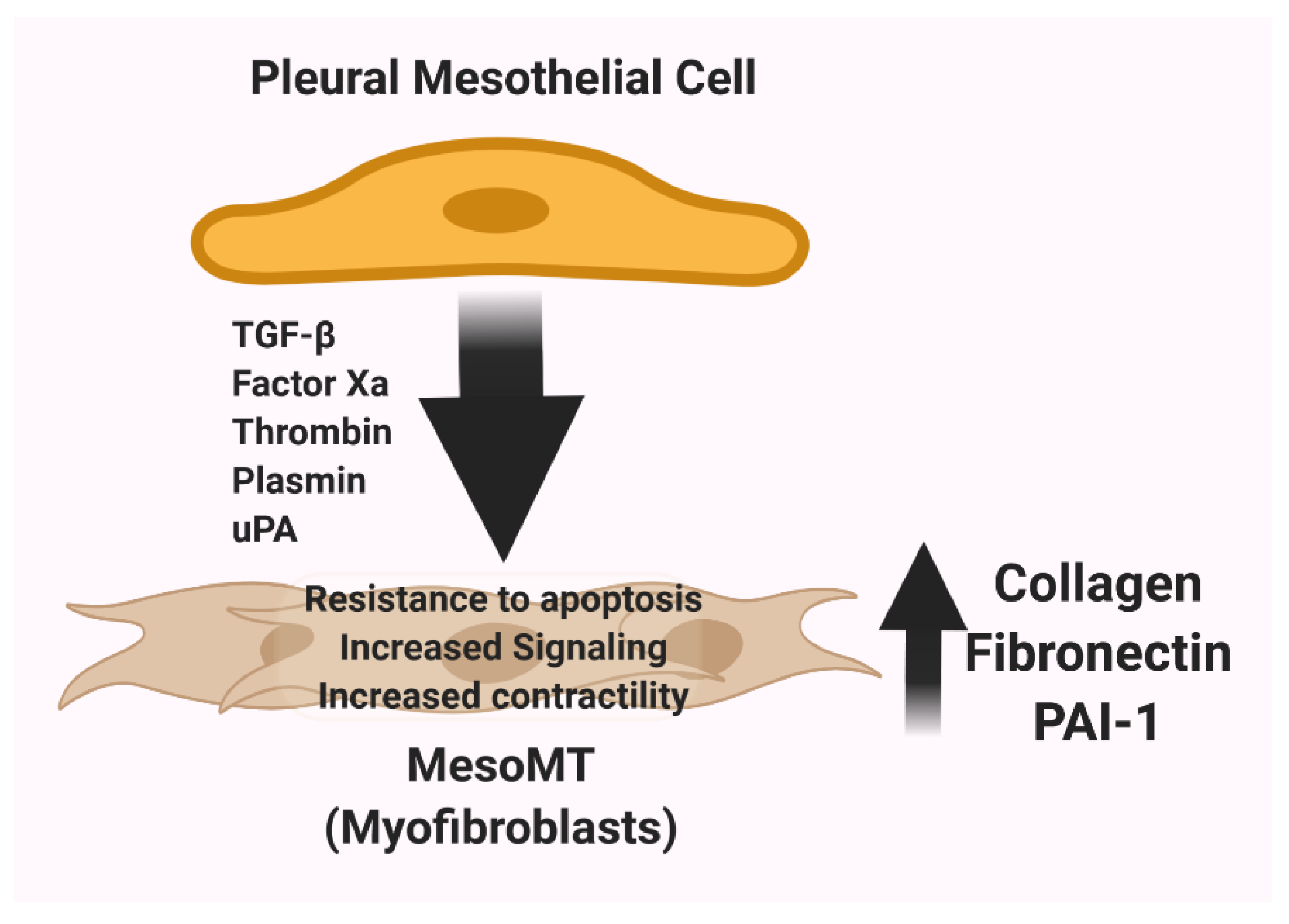
4. The uPA/uPAR System and the Contribution of Mesenchymal Differentiation of Pleural Mesothelial Cells to Pleural Thickening and Scarification
5. The Role of Soluble uPAR in Pleural Injury: Biomarker, Effector, or Both?
6. The Use of scuPA for Treatment of Pleural Loculation
7. New Strategies to Limit Pleural Organization: Clinical Trial Testing of scuPA for Treatment of Empyema or Complicated Parapneumonic Pleural Effusions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tucker, T.; Idell, S. Plasminogen-Plasmin System in the Pathogenesis and Treatment of Lung and Pleural Injury. Semin. Thromb. Hemost. 2013, 39, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.A.; Rahman, N.M.; Lee, Y.C.G.; Florova, G.; Shetty, S.; Idell, R.; Ikebe, M.; Das, K.; Tucker, T.A.; Idell, S. Fibrin turnover and pleural organization: Bench to bedside. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L757–L768. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Gonzalez, K.K.; MacArthur, C.K.; Gillies, C.; Walsh, P.N.; McLarty, J.; Thrall, R.S. Bronchoalveolar Lavage Procoagulant Activity in Bleomycin-Induced Lung Injury in Marmosets: Characterization and Relationship to Fibrin Deposition and Fibrosis. Am. Rev. Respir. Dis. 1987, 136, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; James, K.K.; Gillies, C.; Fair, D.S.; Thrall, R.S. Abnormalities of pathways of fibrin turnover in lung lavage of rats with oleic acid and bleomycin-induced lung injury support alveolar fibrin deposition. Am. J. Pathol. 1989, 135, 387–399. [Google Scholar] [PubMed]
- Idell, S.; James, K.K.; Levin, E.G.; Schwartz, B.S.; Manchanda, N.; Maunder, R.J.; Martin, T.R.; McLarty, J.; Fair, D.S. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J. Clin. Investig. 1989, 84, 695–705. [Google Scholar] [CrossRef]
- Idell, S.; Peters, J.; James, K.K.; Fair, D.S.; Coalson, J.J. Local abnormalities of coagulation and fibrinolytic pathways that promote alveolar fibrin deposition in the lungs of baboons with diffuse alveolar damage. J. Clin. Investig. 1989, 84, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M. Fibrin as a component of the tumor stroma: Origins and biological significance. Cancer Metastasis Rev. 1983, 2, 41–73. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M.; Harvey, V.S.; McDonagh, J. Regulation of extravascular coagulation by microvascular permeability. Science 1985, 227, 1059–1061. [Google Scholar] [CrossRef]
- Brown, L.F.; Dvorak, A.M.; Dvorak, H.F. Leaky Vessels, Fibrin Deposition, and Fibrosis: A Sequence of Events Common to Solid Tumrs and Many Other Types of Disease. Am. Rev. Respir. Dis. 1989, 140, 1104–1107. [Google Scholar] [CrossRef]
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Quax, P.H.; Grimbergen, J.M.; Lansink, M.; Bakker, A.H.; Blatter, M.C.; Belin, D.; van Hinsbergh, V.W.; Verheijen, J.H. Binding of Human Urokinase-Type Plasminogen Activator to Its Receptor: Residues Involved in Species Specificity and Binding. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitrin, R.G. Plasminogen Activation in the Injured Lung: Pulmonology Does Not Recapitulate Hematology. Am. J. Respir. Cell Mol. Biol. 1992, 6, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Gyetko, M.R.; Shollenberger, S.B.; Sitrin, R.G. Urokinase expression in mononuclear phagocytes: Cytokine-specific modulation by interferon-gamma and tumor necrosis factor-alpha. J. Leukoc. Biol. 1992, 51, 256–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakaran, P.; Ware, L.B.; White, K.E.; Cross, M.T.; Matthay, M.A.; Olman, M.A. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L20–L28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karandashova, S.; Florova, G.; Azghani, A.O.; Komissarov, A.A.; Koenig, K.; Tucker, T.A.; Allen, T.C.; Stewart, K.; Tvinnereim, A.; Idell, S. Intrapleural Adenoviral Delivery of Human Plasminogen Activator Inhibitor–1 Exacerbates Tetracycline-Induced Pleural Injury in Rabbits. Am. J. Respir. Cell Mol. Biol. 2013, 48, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Gyetko, M.R.; Idell, S.; Gharaee-Kermani, M.; Shetty, R.S.; Starcher, B.C.; Shetty, S. Regulation of alveolar epithelial cell apoptosis and pulmonary fibrosis by coordinate expression of components of the fibrinolytic system. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L463–L473. [Google Scholar] [CrossRef] [Green Version]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Ji, H.-L.; Neuenschwander, P.F.; Boggaram, V.; Morris, G.F.; Fu, J.; Idell, S.; Shetty, S. Regulation of Lung Injury and Fibrosis by p53-Mediated Changes in Urokinase and Plasminogen Activator Inhibitor-1. Am. J. Pathol. 2013, 183, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Idell, S.; Girard, W.; Koenig, K.B.; McLarty, J.; Fair, D.S. Abnormalities of Pathways of Fibrin Turnover in the Human Pleural Space. Am. Rev. Respir. Dis. 1991, 144, 187–194. [Google Scholar] [CrossRef]
- Idell, S.; Kumar, A.; Zwieb, C.; Holiday, D.; Koenig, K.B.; Johnson, A.R. Effects of TGF-beta and TNF-alpha on procoagulant and fibrinolytic pathways of human tracheal epithelial cells. Am. J. Physiol. Content 1994, 267, 693. [Google Scholar] [CrossRef]
- Idell, S.; Koenig, K.B.; Fair, D.S.; Martin, T.R.; McLarty, J.; Maunder, R.J. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am. J. Physiol. Cell. Mol. Physiol. 1991, 261, L240–L248. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Gonzalez, K.; Bradford, H.; MacArthur, C.K.; Fein, A.M.; Maunder, R.J.; Garcia, J.G.; Griffith, D.E.; Weiland, J.; Martin, T.R. Procoagulant Activity in Bronchoalveolar Lavage in the Adult Respiratory Distress Syndrome. Contribution of Tissue Factor Associated with Factor Vii. Am. Rev. Respir. Dis. 1987, 136, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Zwieb, C.; Kumar, A.; Koenig, K.B.; Johnson, A.R. Pathways of Fibrin Turnover of Human Pleural Mesothelial CellsIn Vitro. Am. J. Respir. Cell Mol. Biol. 1992, 7, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Zwieb, C.; Boggaram, J.; Holiday, D.; Johnson, A.R.; Raghu, G. Mechanisms of fibrin formation and lysis by human lung fibroblasts: Influence of TGF-beta and TNF-alpha. Am. J. Physiol. Content 1992, 263, L487–L494. [Google Scholar] [CrossRef]
- Bertozzi, P.; Astedt, B.; Zenzius, L.; Lynch, K.; LeMaire, F.; Zapol, W.; Chapman, H.A. Depressed Bronchoalveolar Urokinase Activity in Patients with Adult Respiratory Distress Syndrome. N. Engl. J. Med. 1990, 322, 890–897. [Google Scholar] [CrossRef]
- Chapman, H.A. Disorders of lung matrix remodeling. J. Clin. Investig. 2004, 113, 148–157. [Google Scholar] [CrossRef]
- Chapman, H.A. Plasminogen activators, integrins, and the coordinated regulation of cell adhesion and migration. Curr. Opin. Cell Biol. 1997, 9, 714–724. [Google Scholar] [CrossRef]
- Chapman, H.A.; Stahl, M.; Allen, C.L.; Yee, R.; Fair, D.S. Regulation of the Procoagulant Activity within the Bronchoalveolar Compartment of Normal Human Lung. Am. Rev. Respir. Dis. 1988, 137, 1417–1425. [Google Scholar] [CrossRef]
- Gross, T.J.; Simon, R.H.; Kelly, C.J.; Sitrin, R.G. Rat alveolar epithelial cells concomitantly express plasminogen activator inhibitor-1 and urokinase. Am. J. Physiol. Cell. Mol. Physiol. 1991, 260, L286–L295. [Google Scholar] [CrossRef]
- Gross, T.J.; Simon, R.H.; Sitrin, R.G. Expression of Urokinase-type Plasminogen Activator by Rat Pulmonary Alveolar Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1990, 3, 449–456. [Google Scholar] [CrossRef]
- Gross, T.J.; Simon, R.H.; Sitrin, R.G. Tissue Factor Procoagulant Expression by Rat Alveolar Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1992, 6, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Sitrin, R.G.; Pan, P.; Blackwood, R.A.; Huang, J.; Petty, H.R. Cutting edge: Evidence for a signaling partnership between urokinase receptors (CD87) and L-selectin (CD62L) in human polymorphonuclear neutrophils. J. Immunol. 2001, 166, 4822–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitrin, R.G.; Pan, P.M.; Harper, H.A.; Todd, R.F.; Harsh, D.M.; Blackwood, R.A. Clustering of Urokinase Receptors (uPAR.; CD87) Induces Proinflammatory Signaling in Human Polymorphonuclear Neutrophils. J. Immunol. 2000, 165, 3341–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitrin, R.G.; Todd, R.F.; Albrecht, E.; Gyetko, M.R. The urokinase receptor (CD87) facilitates CD11b/CD18-mediated adhesion of human monocytes. J. Clin. Investig. 1996, 97, 1942–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitrin, R.G.; Todd, R.F.; Mizukami, I.F.; Gross, T.J.; Shollenberger, S.B.; Gyetko, M.R. Cytokine-specific regulation of urokinase receptor (CD87) expression by U937 mononuclear phagocytes. Blood 1994, 84, 1268–1275. [Google Scholar]
- Bastarache, J.A. The complex role of fibrin in acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L275–L276. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Wang, L.; Geiser, T.; Wang, Z.; Albertine, K.H.; Matthay, M.A.; Ware, L.B. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax 2007, 62, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Van der Poll, T.; Levi, M.; Nick, J.A.; Abraham, E. Activated Protein C Inhibits Local Coagulation after Intrapulmonary Delivery of Endotoxin in Humans. Am. J. Respir. Crit. Care Med. 2005, 171, 1125–1128. [Google Scholar] [CrossRef]
- Bdeir, K.; Murciano, J.C.; Tomaszewski, J.; Koniaris, L.; Martinez, J.; Cines, D.B.; Muzykantov, V.R.; Higazi, A.A. Urokinase Mediates Fibrinolysis in the Pulmonary Microvasculature. Blood 2000, 96, 1820–1826. [Google Scholar] [CrossRef]
- Higazi, A.A.; Bdeir, K.; Hiss, E.; Arad, S.; Kuo, A.; Barghouti, I.; Cines, D.B. Lysis of Plasma Clots by Urokinase-Soluble Urokinase Receptor Complexes. Blood 1998, 92, 2075–2083. [Google Scholar] [CrossRef]
- Sisson, T.H.; Hanson, K.E.; Subbotina, N.; Patwardhan, A.; Hattori, N.; Simon, R.H. Inducible lung-specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L1023–L1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaisgood, C.M.; French, E.L.; Noga, C.; Simon, R.H.; Ploplis, V.A. The Development of Bleomycin-Induced Pulmonary Fibrosis in Mice Deficient for Components of the Fibrinolytic System. Am. J. Pathol. 2000, 157, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Hattori, N.; Degen, J.L.; Sisson, T.H.; Liu, H.; Moore, B.B.; Pandrangi, R.G.; Simon, R.H.; Drew, A.F. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Investig. 2000, 106, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horowitz, J.C.; Tschumperlin, D.J.; Kim, K.K.; Osterholzer, J.J.; Subbotina, N.; Ajayi, I.O.; Teitz-Tennenbaum, S.; Virk, A.; Dotson, M.; Liu, F.; et al. Urokinase Plasminogen Activator Overexpression Reverses Established Lung Fibrosis. Thromb. Haemost. 2019, 119, 1968–1980. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Scotton, C.J. Coagulation Cascade Proteinases in Lung Injury and Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Lübke, N.; Ermert, M.; Schermuly, R.T.; Weissmann, N.; Breithecker, A.; Markart, P.; Ruppert, C.; Quanz, K.; Ermert, L.; et al. Prevention of Bleomycin-induced Lung Fibrosis by Aerosolization of Heparin or Urokinase in Rabbits. Am. J. Respir. Crit. Care Med. 2003, 168, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Günther, A.; Mosavi, P.; Heinemann, S.; Ruppert, C.; Muth, H.; Markart, P.; Grimminger, F.; Walmrath, D.; Temmesfeld-Wollbrück, B.; Seeger, W. Alveolar Fibrin Formation Caused by Enhanced Procoagulant and Depressed Fibrinolytic Capacities in Severe Pneumonia. Am. J. Respir. Crit. Care Med. 2000, 161, 454–462. [Google Scholar] [CrossRef]
- Idell, S.; Peterson, B.T.; Gonzalez, K.K.; Gray, L.D.; Bach, R.; McLarty, J.; Fair, D.S. Local Abnormalities of Coagulation and Fibrinolysis and Alveolar Fibrin Deposition in Sheep with Oleic Acid-induced Lung Injury. Am. Rev. Respir. Dis. 1988, 138, 1282–1294. [Google Scholar] [CrossRef]
- Idell, S.; James, K.K.; Coalson, J.J. Fibrinolytic Activity in Bronchoalveolar Lavage of Baboons with Diffuse Alveolar Damage: Trends in Two Forms of Lung Injury. Crit. Care Med. 1992, 20, 1431–1440. [Google Scholar] [CrossRef]
- Idell, S.; Mazar, A.; Cines, D.; Kuo, A.; Parry, G.; Gawlak, S.; Juarez, J.; Koenig, K.; Azghani, A.; Hadden, W.; et al. Single-Chain Urokinase Alone or Complexed to Its Receptor in Tetracycline-induced Pleuritis in Rabbits. Am. J. Respir. Crit. Care Med. 2002, 166, 920–926. [Google Scholar] [CrossRef]
- Burnham, E.L.; Hyzy, R.C.; Paine, R., III; Kelly, A.M.; Quint, L.E.; Lynch, D.; Curran-Everett, D.; Moss, M.; Standiford, T.J. Detection of Fibroproliferation by Chest High-Resolution Ct Scan in Resolving Ards. Chest 2014, 146, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsaers, S.E.; Prele, C.M.; Brody, A.R.; Idell, S. Pathogenesis of pleural fibrosis. Respirology 2004, 9, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Murao, S.; Yamakawa, K. A Systematic Summary of Systematic Reviews on Anticoagulant Therapy in Sepsis. J. Clin. Med. 2019, 8, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechowicz, K.; Drozdzal, S.; Machaj, F.; Rosik, J.; Szostak, B.; Zegan-Baranska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. Covid-19: The Potential Treatment of Pulmonary Fibrosis Associated with Sars-Cov-2 Infection. J. Clin. Med. 2020, 9, 1917. [Google Scholar] [CrossRef]
- Ji, H.J.; Su, Z.; Zhao, R.; Komissarov, A.A.; Yi, G.; Liu, S.L.; Idell, S.; Matthay, M.A. Insufficient Hyperfibrinolysis in Covid-19: A Systematic Review of Thrombolysis Based on Meta-Analysis and Meta-Regression. medRxiv 2020. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of Cell Signalling by Upar. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Ellis, V.; Behrendt, N.; Danø, K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem. 1991, 266, 12752–12758. [Google Scholar] [CrossRef]
- Tucker, T.A.; Dean, C.; Komissarov, A.A.; Koenig, K.; Mazar, A.P.; Pendurthi, U.; Allen, T.; Idell, S. The Urokinase Receptor Supports Tumorigenesis of Human Malignant Pleural Mesothelioma Cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 685–696. [Google Scholar] [CrossRef]
- Shetty, S.; Padijnayayveetil, J.; Tucker, T.; Stankowska, D.; Idell, S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L967–L975. [Google Scholar] [CrossRef]
- Shetty, S.; Kumar, A.; Johnson, A.R.; Pueblitz, S.; Holiday, D.; Raghu, G.; Idell, S. Differential expression of the urokinase receptor in fibroblasts from normal and fibrotic human lungs. Am. J. Respir. Cell Mol. Biol. 1996, 15, 78–87. [Google Scholar] [CrossRef]
- Shetty, S.; Idell, S. A urokinase receptor mRNA binding protein from rabbit lung fibroblasts and mesothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 1998, 274, L871–L882. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Hattori, N.; Xu, Y.; Simon, R.H. Treatment of Bleomycin-Induced Pulmonary Fibrosis by Transfer of Urokinase-Type Plasminogen Activator Genes. Hum. Gene Ther. 1999, 10, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Eitzman, D.T.; McCoy, R.D.; Zheng, X.; Fay, W.P.; Shen, T.; Ginsburg, D.; Simon, R.H. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J. Clin. Investig. 1996, 97, 232–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zoelen, M.A.; Florquin, S.; de Beer, R.; Pater, J.M.; Verstege, M.I.; Meijers, J.C.; van der Poll, T. Urokinase Plasminogen Activator Receptor-Deficient Mice Demonstrate Reduced Hyperoxia-Induced Lung Injury. Am. J. Pathol. 2009, 174, 2182–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijneveld, A.W.; Levi, M.; Florquin, S.; Speelman, P.; Carmeliet, P.; Van Der Poll, T. Urokinase Receptor Is Necessary for Adequate Host Defense Against Pneumococcal Pneumonia. J. Immunol. 2002, 168, 3507–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip-Joët, F.; Alessi, M.-C.; Aillaud, M.; Barriere, J.-R.; Arnaud, A.; Juhan-Vague, I. Fibrinolytic and inflammatory processes in pleural effusions. Eur. Respir. J. 1995, 8, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.-L.; Chen, Y.-C.; Chang, S.-C. Effect of Repeated Thoracenteses on Fluid Characteristics, Cytokines, and Fibrinolytic Activity in Malignant Pleural Effusion. Chest 2003, 123, 1188–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckert, L.; Brockway, B.; Simpson, G.; Southcott, A.M.; Lee, Y.C.G.; Rahman, N.; Light, R.W.; Shoemaker, S.; Gillies, J.; Komissarov, A.A.; et al. Phase 1 Trial of Intrapleural Lti-01; Single Chain Urokinase in Complicated Parapneumonic Effusions or Empyema. JCI Insight 2019, 5, e127470. [Google Scholar] [CrossRef]
- Idell, S. The pathogenesis of pleural space loculation and fibrosis. Curr. Opin. Pulm. Med. 2008, 14, 310–315. [Google Scholar] [CrossRef]
- Tucker, T.A.; Jeffers, A.; Alvarez, A.; Owens, S.; Koenig, K.; Quaid, B.; Komissarov, A.A.; Florova, G.; Kothari, H.; Pendurthi, U.; et al. Plasminogen Activator Inhibitor-1 Deficiency Augments Visceral Mesothelial Organization, Intrapleural Coagulation and Lung Restriction in Mice with Carbon Black/Bleomycin-Induced Pleural Injury. Am. J. Respir. Cell Mol. Biol. 2013, 50, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Florova, G.; Azghani, A.; Karandashova, S.; Schaefer, C.; Koenig, K.; Stewart-Evans, K.; Declerck, P.J.; Idell, S.; Komissarov, A.A. Targeting of Plasminogen Activator Inhibitor 1 Improves Fibrinolytic Therapy for Tetracycline-Induced Pleural Injury in Rabbits. Am. J. Respir. Cell Mol. Biol. 2015, 52, 429–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idell, S.; Allen, T.; Chen, S.; Koenig, K.; Mazar, A.; Azghani, A. Intrapleural activation, processing, efficacy, and duration of protection of single-chain urokinase in evolving tetracycline-induced pleural injury in rabbits. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L25–L32. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Azghani, A.; Chen, S.; Koenig, K.; Mazar, A.; Kodandapani, L.; Bdeir, K.; Cines, D.; Kulikovskaya, I.; Allen, T. Intrapleural Low-Molecular-Weight Urokinase or Tissue Plasminogen Activator Versus Single-Chain Urokinase in Tetracycline-Induced Pleural Loculation in Rabbits. Exp. Lung Res. 2007, 33, 419–440. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.A.; Florova, G.; Azghani, A.O.; Buchanan, A.; Boren, J.; Allen, T.; Rahman, N.M.; Koenig, K.; Chamiso, M.; Karandashova, S.; et al. Dose dependency of outcomes of intrapleural fibrinolytic therapy in new rabbit empyema models. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L389–L399. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.A.; Florova, G.; Azghani, A.O.; Buchanan, A.; Bradley, W.M.; Schaefer, C.; Koenig, K.; Idell, S. The Time Course of Resolution of Adhesions During Fibrinolyitic Therapy in Tetracycline-Induced Pleural Injury in Rabbits. Am. J. Physiol. Lung. Cell Mol. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, J.P.; Hallifax, R.; Rahman, N.M. New therapeutic approaches to pleural infection. Curr. Opin. Infect. Dis. 2013, 26, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Idell, S.; Rahman, N.M. Intrapleural Fibrinolytic Therapy for Empyema and Pleural Loculation: Knowns and Unknowns. Ann. Am. Thorac. Soc. 2018, 15, 515–517. [Google Scholar] [CrossRef]
- Idell, S. Update on the Use of Fibrinolysins in Pleural Disease. Clin. Pulm. Med. 2005, 12, 184–190. [Google Scholar] [CrossRef]
- Mazar, A.P.; Ahn, R.W.; O’Halloran, T.V. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr. Pharm. Des. 2011, 17, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Tucker, T.A.; Williams, L.; Koenig, K.; Kothari, H.; Komissarov, A.A.; Florova, G.; Mazar, A.P.; Allen, T.C.; Bdeir, K.; Mohan Rao, L.V.; et al. Lipoprotein Receptor-Related Protein 1 Regulates Collagen 1 Expression, Proteolysis, and Migration in Human Pleural Mesothelial Cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Tucker, T.A.; Jeffers, A.; Boren, J.; Quaid, B.; Owens, S.; Koenig, K.B.; Tsukasaki, Y.; Florova, G.; Komissarov, A.A.; Ikebe, M.; et al. Organizing empyema induced in mice by Streptococcus pneumoniae: Effects of plasminogen activator inhibitor-1 deficiency. Clin. Transl. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Decologne, N.; Wettstein, G.; Kolb, M.; Margetts, P.; Garrido, C.; Camus, P.; Bonniaud, P. Bleomycin induces pleural and subpleural fibrosis in the presence of carbon particles. Eur. Respir. J. 2009, 35, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Decologne, N.; Kolb, M.; Margetts, P.J.; Menetrier, F.; Artur, Y.; Garrido, C.; Gauldie, J.; Camus, P.; Bonniaud, P. Tgf-Beta1 Induces Progressive Pleural Scarring and Subpleural Fibrosis. J. Immunol. 2007, 179, 6043–6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, S.; Jeffers, A.; Boren, J.; Tsukasaki, Y.; Koenig, K.; Ikebe, M.; Idell, S.; Tucker, T.A. Mesomesenchymal Transition of Pleural Mesothelial Cells Is Pi3k and Nf-Kappab Dependent. Am. J. Physiol. Lung. Cell Mol. Physiol. 2015, 308, L1265–L1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boren, J.; Shryock, G.; Fergis, A.; Jeffers, A.; Owens, S.; Qin, W.; Koenig, K.B.; Tsukasaki, Y.; Komatsu, S.; Ikebe, M.; et al. Inhibition of Glycogen Synthase Kinase 3beta Blocks Mesomesenchymal Transition and Attenuates Streptococcus Pneumonia-Mediated Pleural Injury in Mice. Am. J. Pathol. 2017, 187, 2461–2472. [Google Scholar] [CrossRef] [Green Version]
- Ploug, M.; Gardsvoll, H.; Jorgensen, T.J.; Lonborg, H.L.; Dano, K. Structural Analysis of the Interaction between Urokinase-Type Plasminogen Activator and Its Receptor: A Potential Target for Anti-Invasive Cancer Therapy. Biochem. Soc. Trans. 2002, 30, 177–183. [Google Scholar] [CrossRef]
- Pyke, C.; Eriksen, J.; Solberg, H.; Nielsen, B.; Kristensen, P.; Lund, L.R.; Dano, K. An alternatively spliced variant of mRNA for the human receptor for urokinase plasminogen activator. FEBS Lett. 1993, 326, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The Molecular Crystal Ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Arnold, D.T.; Hamilton, F.W.; Elvers, K.T.; Frankland, S.W.; Zahan-Evans, N.; Patole, S.; Medford, A.; Bhatnagar, R.; Maskell, N.A. Pleural Fluid suPAR Levels Predict the Need for Invasive Management in Parapneumonic Effusions. Am. J. Respir. Crit. Care Med. 2020, 201, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Van Veen, M.; Matas-Rico, E.; van de Wetering, K.; Leyton-Puig, D.; Kedziora, K.M.; De Lorenzi, V.; Stijf-Bultsma, Y.; van den Broek, B.; Jalink, K.; Sidenius, N.; et al. Negative Regulation of Urokinase Receptor Activity by a Gpi-Specific Phospholipase C in Breast Cancer Cells. eLife 2017, 6, e23649. [Google Scholar] [CrossRef]
- Masucci, M.; Pedersen, N.; Blasi, F. A soluble, ligand binding mutant of the human urokinase plasminogen activator receptor. J. Biol. Chem. 1991, 266, 8655–8658. [Google Scholar] [CrossRef]
- Higazi, A.A.-R.; Cohen, R.L.; Henkin, J.; Kniss, D.; Schwartz, B.S.; Cines, D.B. Enhancement of the Enzymatic Activity of Single-chain Urokinase Plasminogen Activator by Soluble Urokinase Receptor. J. Biol. Chem. 1995, 270, 17375–17380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, N.; Danø, K. Effect of purified, soluble urokinase receptor on the plasminogen-prourokinase activation system. FEBS Lett. 1996, 393, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.M.; Maskell, N.A.; West, A.; Teoh, R.; Arnold, A.; Mackinlay, C.; Peckham, D.; Davies, C.W.; Ali, N.; Kinnear, W.; et al. Intrapleural Use of Tissue Plasminogen Activator and DNase in Pleural Infection. N. Engl. J. Med. 2011, 365, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Bouros, D.; Schiza, S.; Tzanakis, N.; Chalkiadakis, G.; Drositis, J.; Siafakas, N. Intrapleural Urokinase versus Normal Saline in the Treatment of Complicated Parapneumonic Effusions and Empyema. Am. J. Respir. Crit. Care Med. 1999, 159, 37–42. [Google Scholar] [CrossRef]
- Alemán, C.; Porcel, J.M.; Alegre-Martin, J.; Ruiz, E.; Bielsa, S.; Andreu, J.; Deu, M.; Suñé, P.; Deu-Martín, M.; López, I.; et al. Intrapleural Fibrinolysis with Urokinase Versus Alteplase in Complicated Parapneumonic Pleural Effusions and Empyemas: A Prospective Randomized Study. Lung 2015, 193, 993–1000. [Google Scholar] [CrossRef]
- Bédat, B.; Plojoux, J.; Noel, J.; Morel, A.; Worley, J.; Triponez, F.; Karenovics, W. Comparison of intrapleural use of urokinase and tissue plasminogen activator/DNAse in pleural infection. ERJ Open Res. 2019, 5, 00084–02019. [Google Scholar] [CrossRef] [Green Version]
- Komissarov, A.A.; Mazar, A.P.; Koenig, K.; Kurdowska, A.K.; Idell, S. Regulation of Intrapleural Fibrinolysis by Urokinase-Alpha-Macroglobulin Complexes in Tetracycline-Induced Pleural Injury in Rabbits. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L568–L577. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Single Chain uPA; scuPA | A Proenzyme that Can Bind uPAR and Localize PA Activity to the Cell Surface |
| Two-chain uPA; tcuPA | Conversion of scuPA to this two-chain form generates a much more active PA, can likewise bind uPAR and is mainly involved in pericellular proteolysis. |
| Tissue type plasminogen activator; tPA | Has a greater affinity for fibrin than tcuPA, relatively more involved in intravascular fibrinolysis, and binding to fibrin increases its PA activity. |
| Plasminogen activator inhibitor 1; PAI-1 | Main PA inhibitor in extravascular fluids in lung and pleural injury, where it can exist in active, cleaved, and inactivated or latent forms. Active PAI-1 can inhibit both tcuPA and tPA. |
| uPA receptor; uPAR | Multidomain surface glycoprotein responsible for cellular localization of uPA and can be cleaved by uPA. Capable of mediating signaling through interactions with other surface receptors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucker, T.A.; Idell, S. The Contribution of the Urokinase Plasminogen Activator and the Urokinase Receptor to Pleural and Parenchymal Lung Injury and Repair: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 1437. https://doi.org/10.3390/ijms22031437
Tucker TA, Idell S. The Contribution of the Urokinase Plasminogen Activator and the Urokinase Receptor to Pleural and Parenchymal Lung Injury and Repair: A Narrative Review. International Journal of Molecular Sciences. 2021; 22(3):1437. https://doi.org/10.3390/ijms22031437
Chicago/Turabian StyleTucker, Torry A., and Steven Idell. 2021. "The Contribution of the Urokinase Plasminogen Activator and the Urokinase Receptor to Pleural and Parenchymal Lung Injury and Repair: A Narrative Review" International Journal of Molecular Sciences 22, no. 3: 1437. https://doi.org/10.3390/ijms22031437