
Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-cient lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut

 ,
, 
Abstract
:1. Introduction
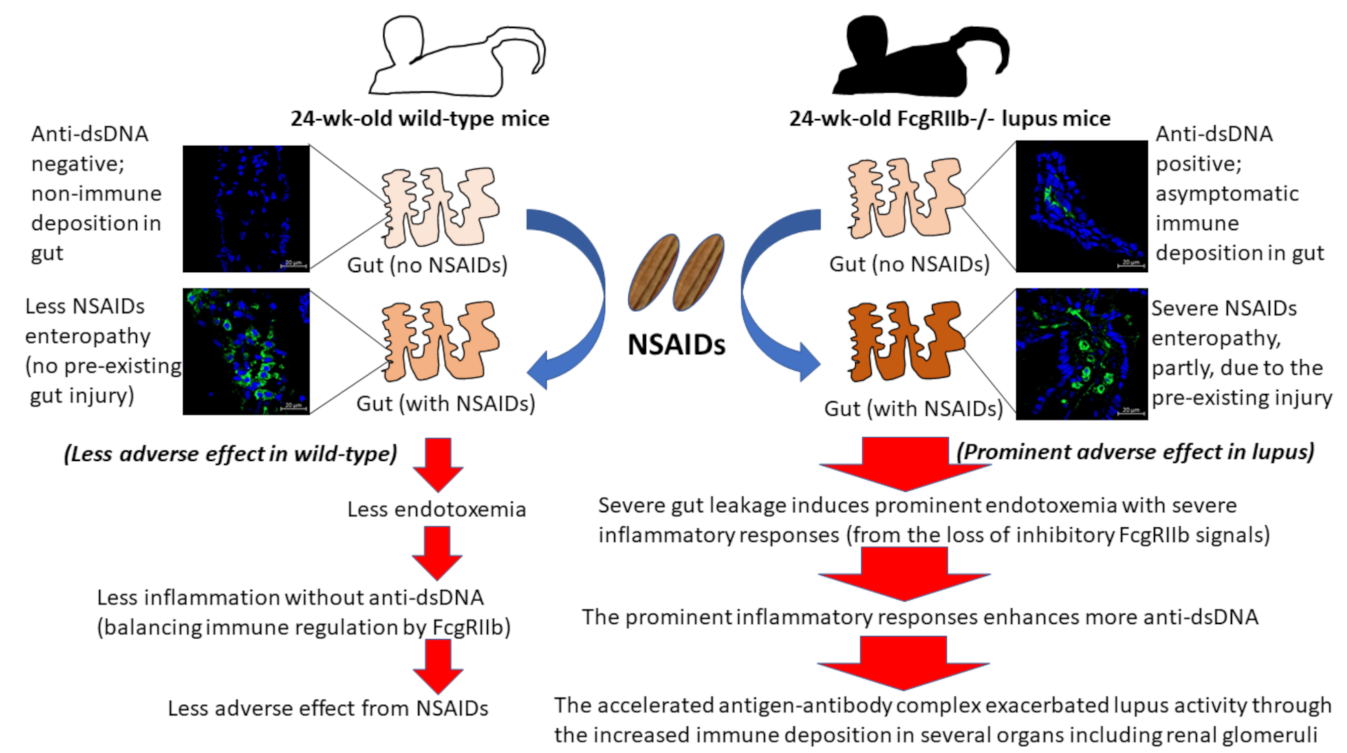
2. Results
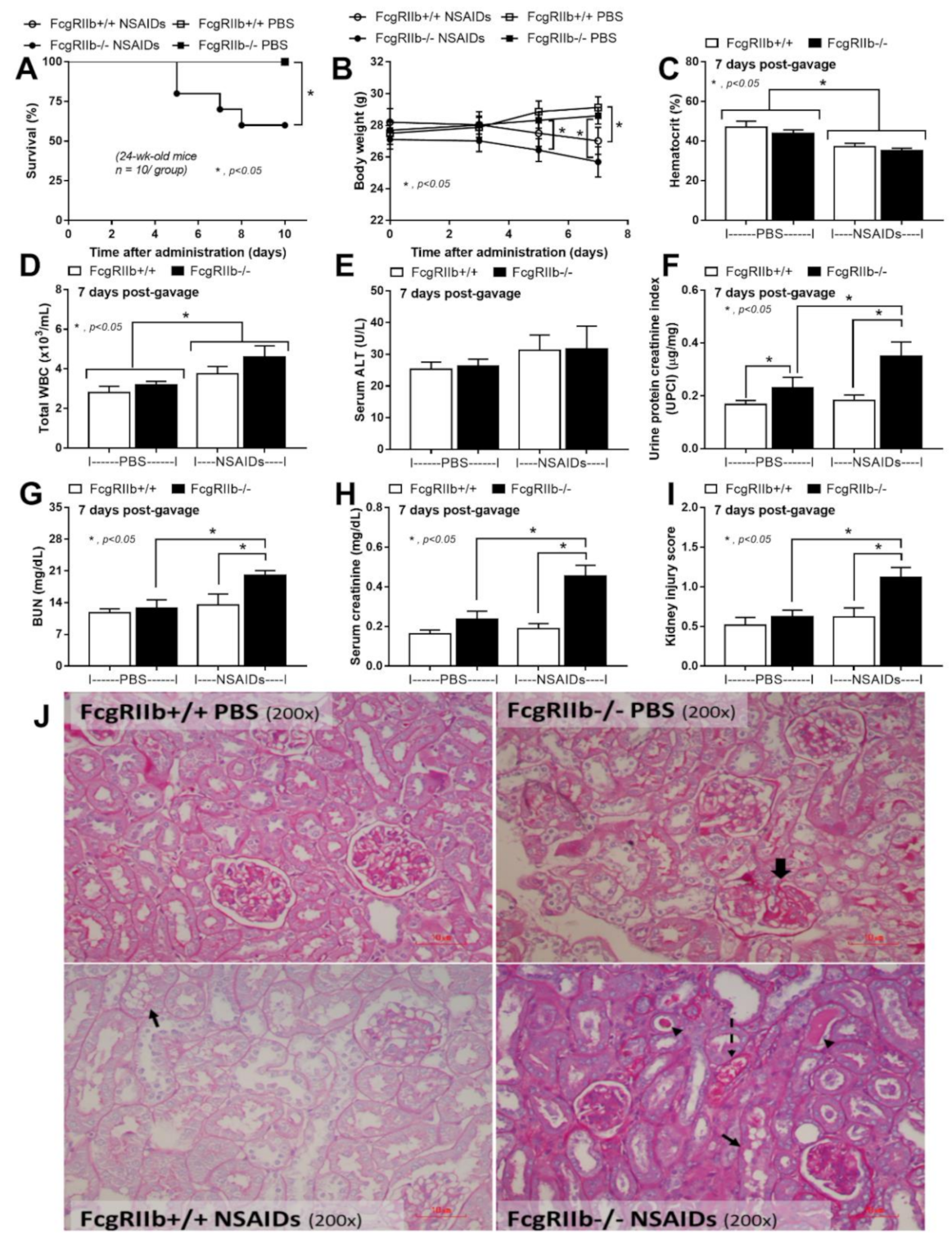
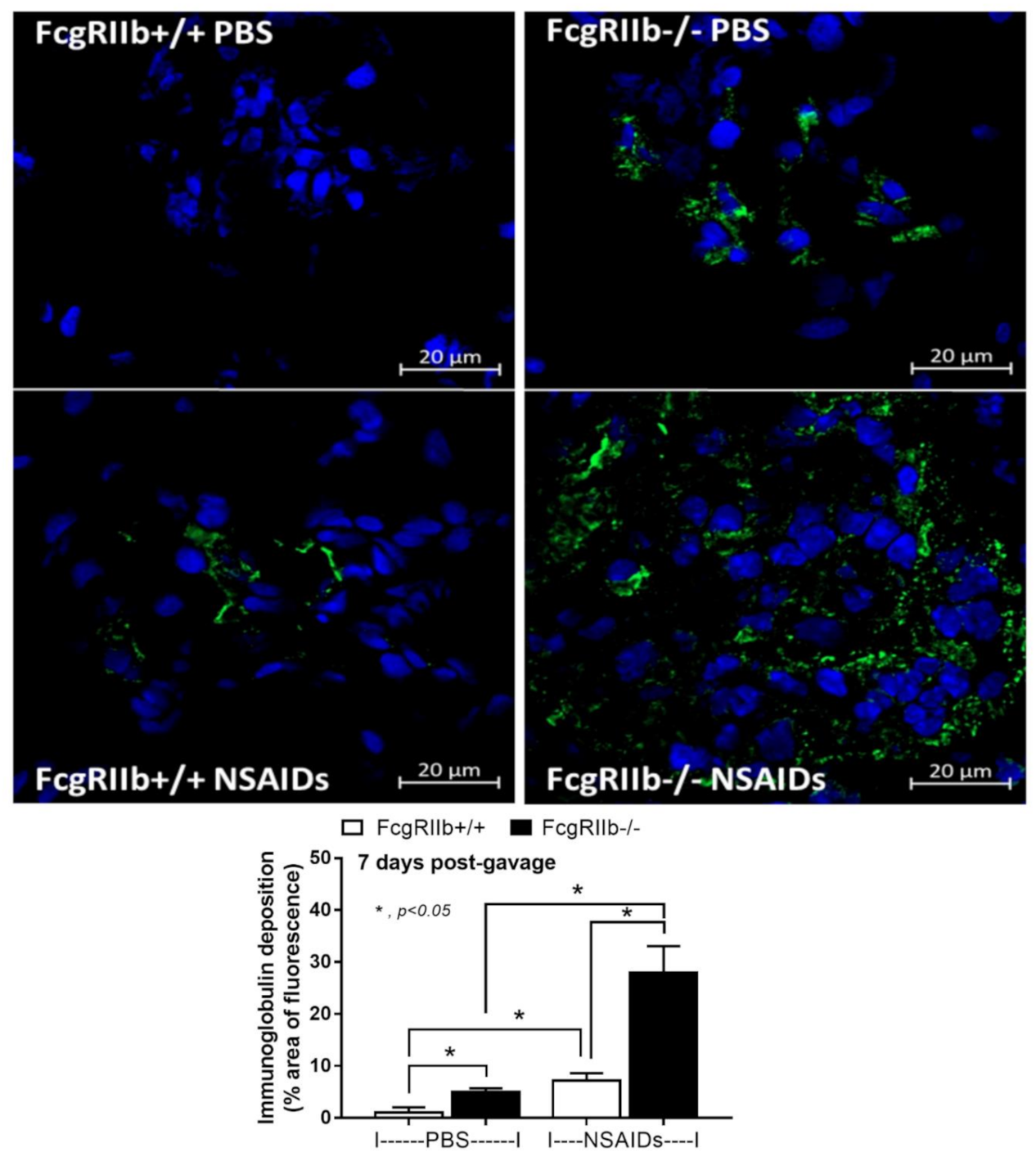
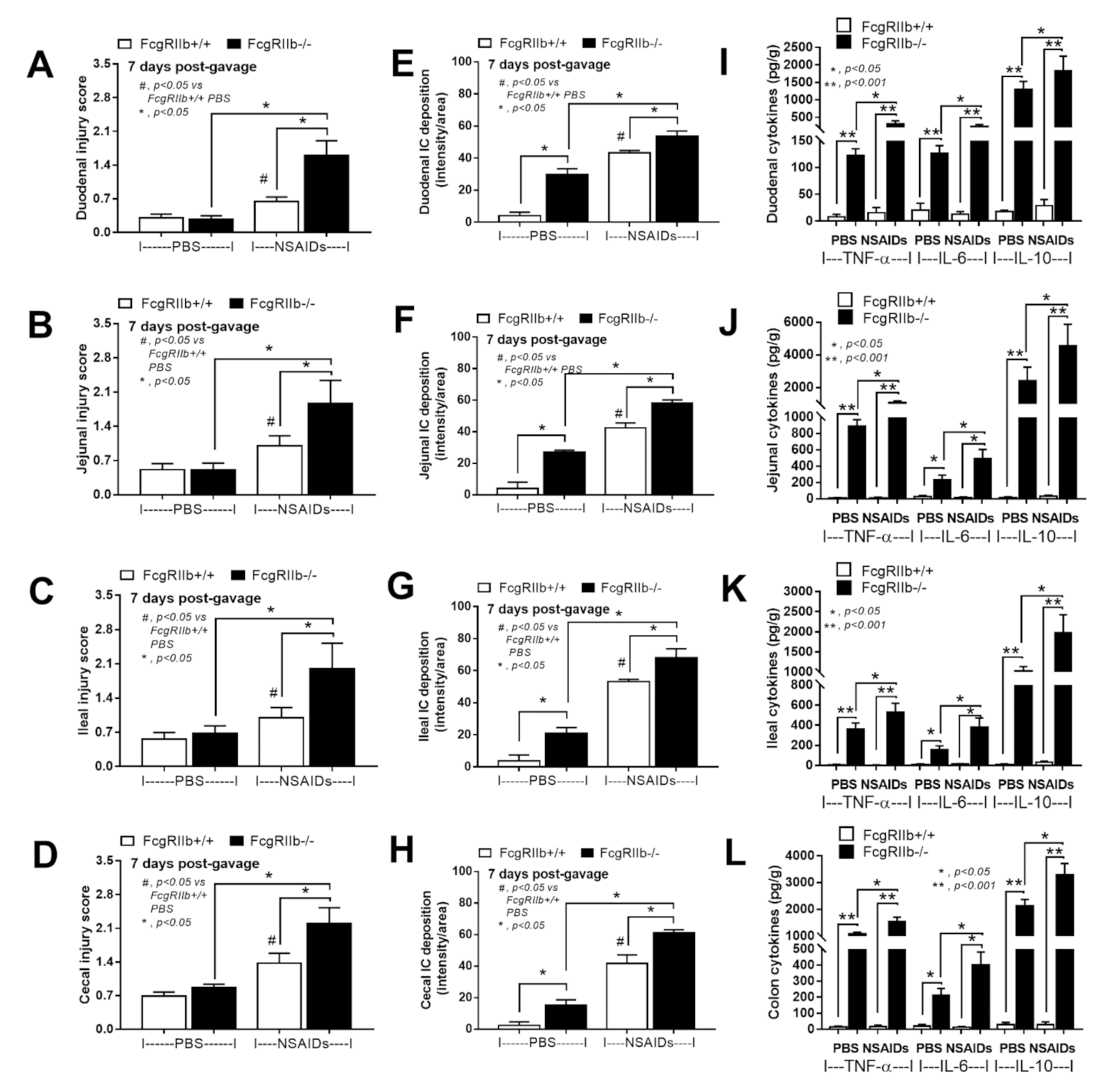
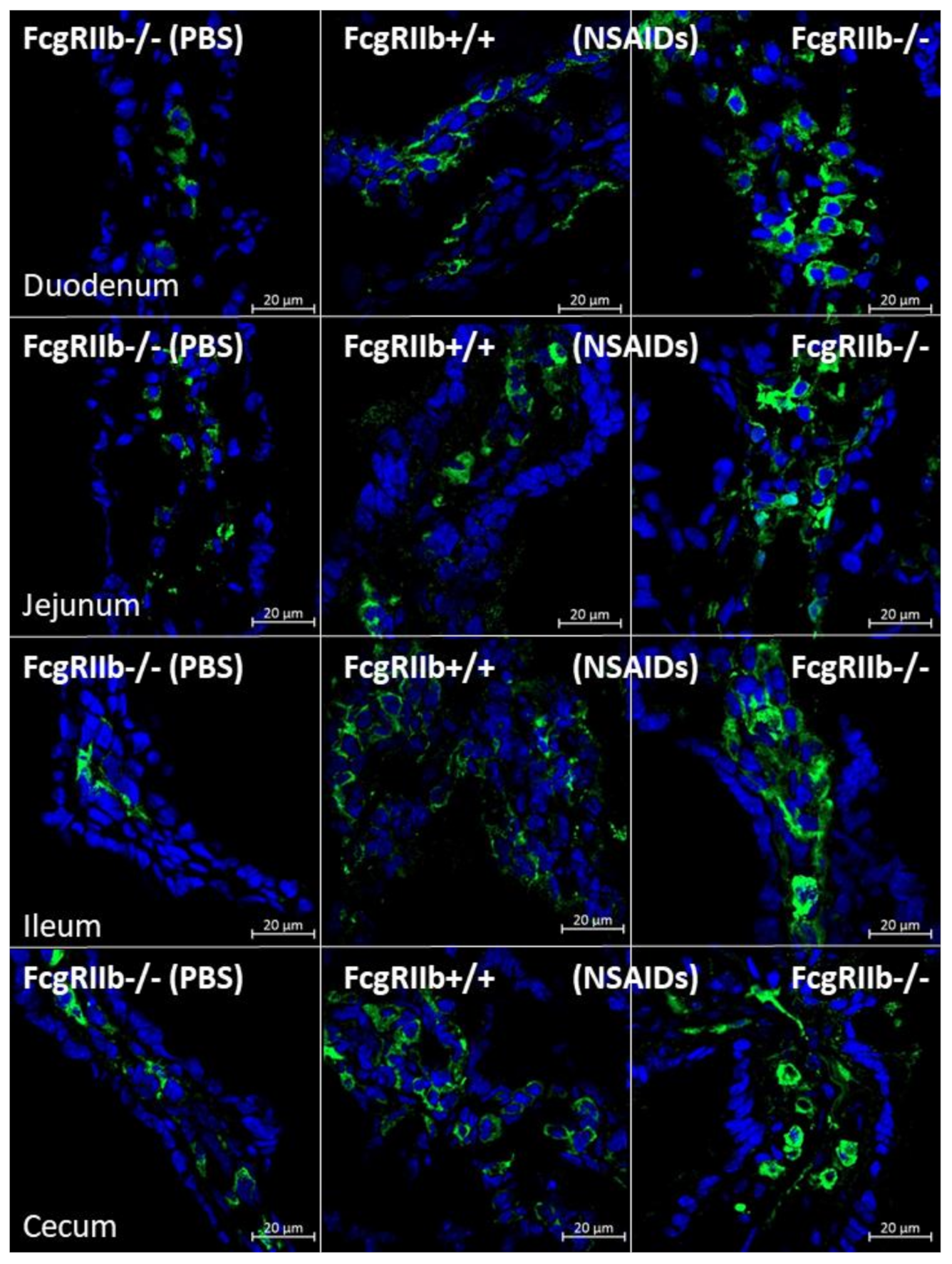
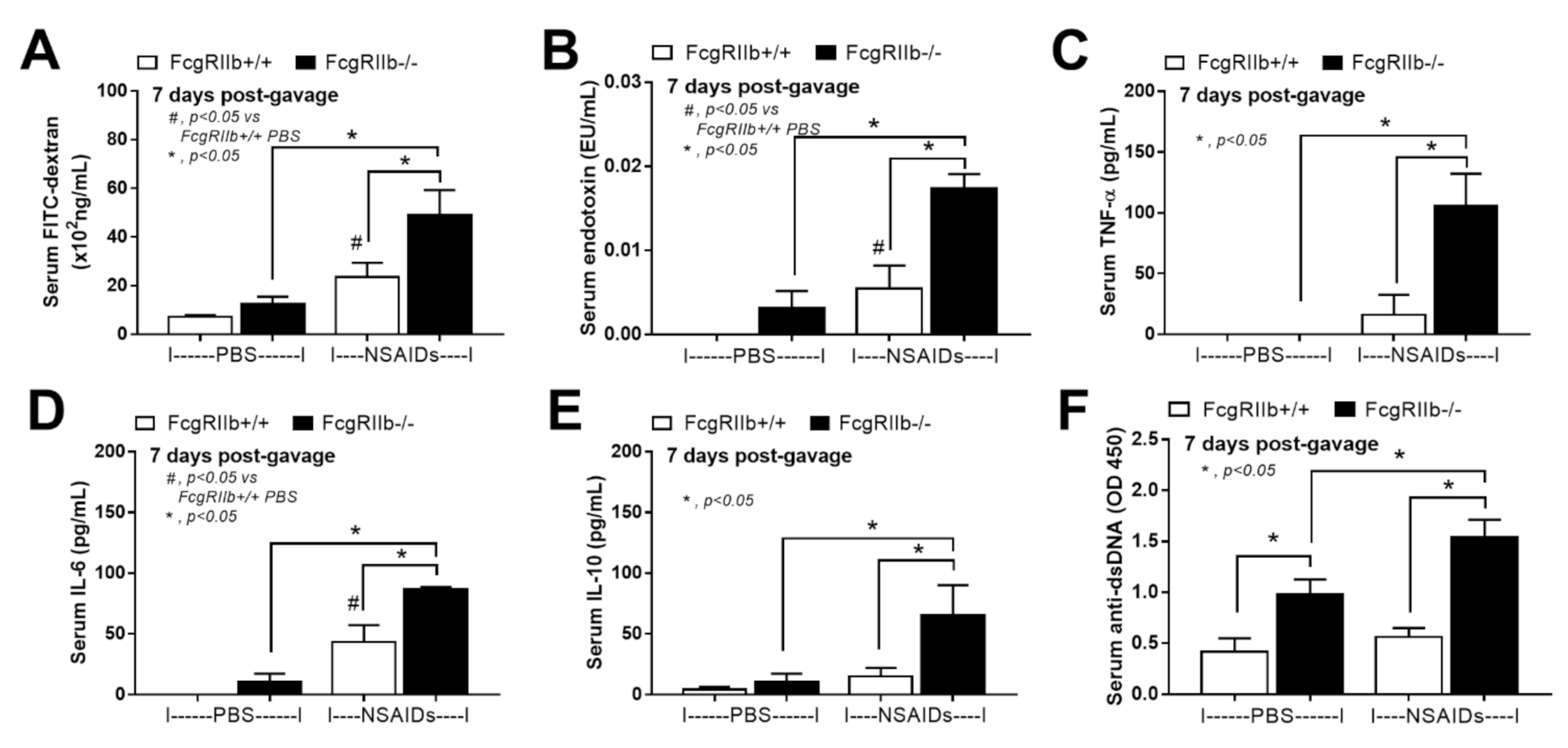
2.1. Prominent Indomethacin-Induced Renal Injury, Enterocolitis, and Endotoxemia in FcgRIIb-/-Mice Compared with Wild-Type Mice
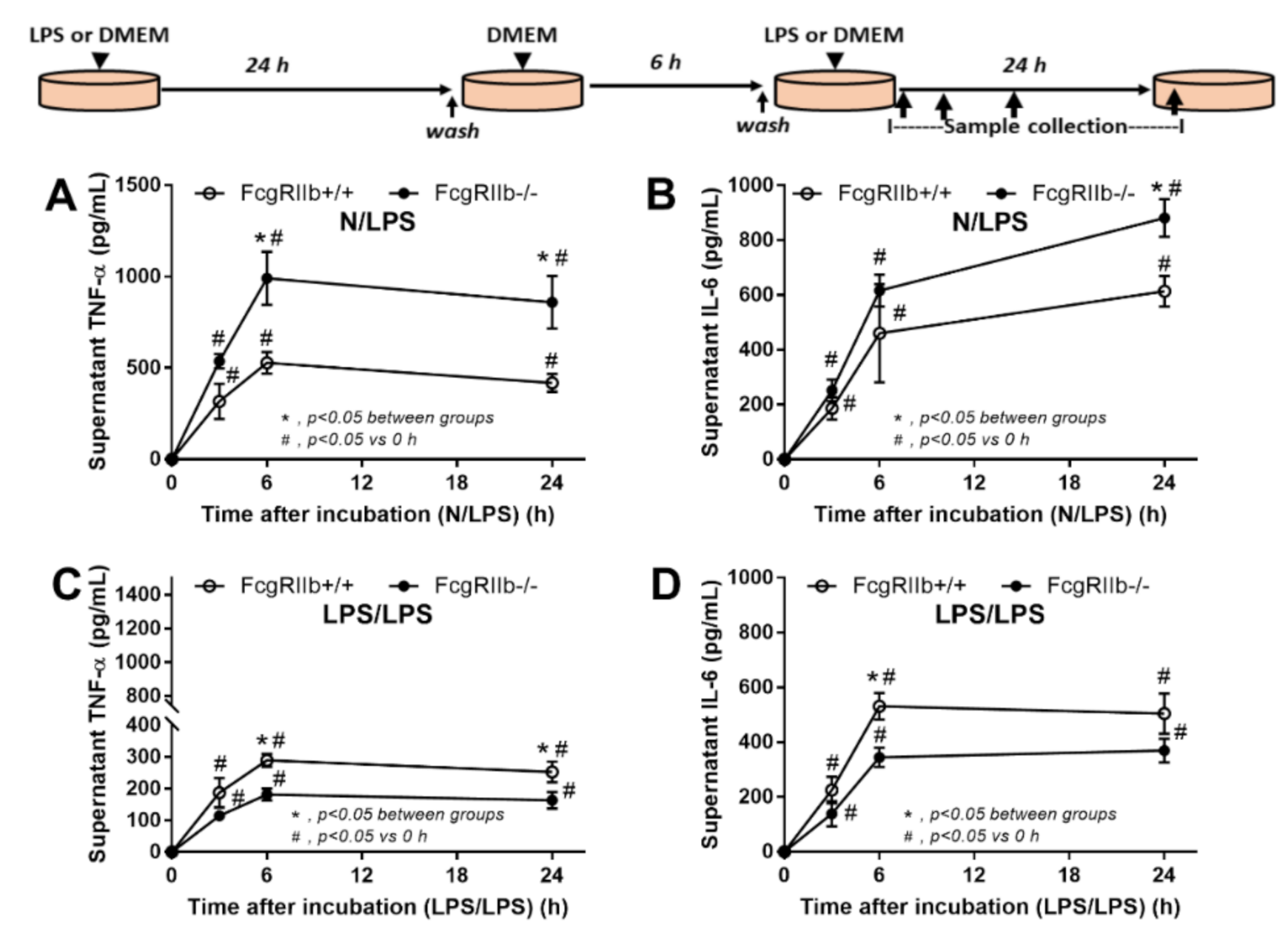
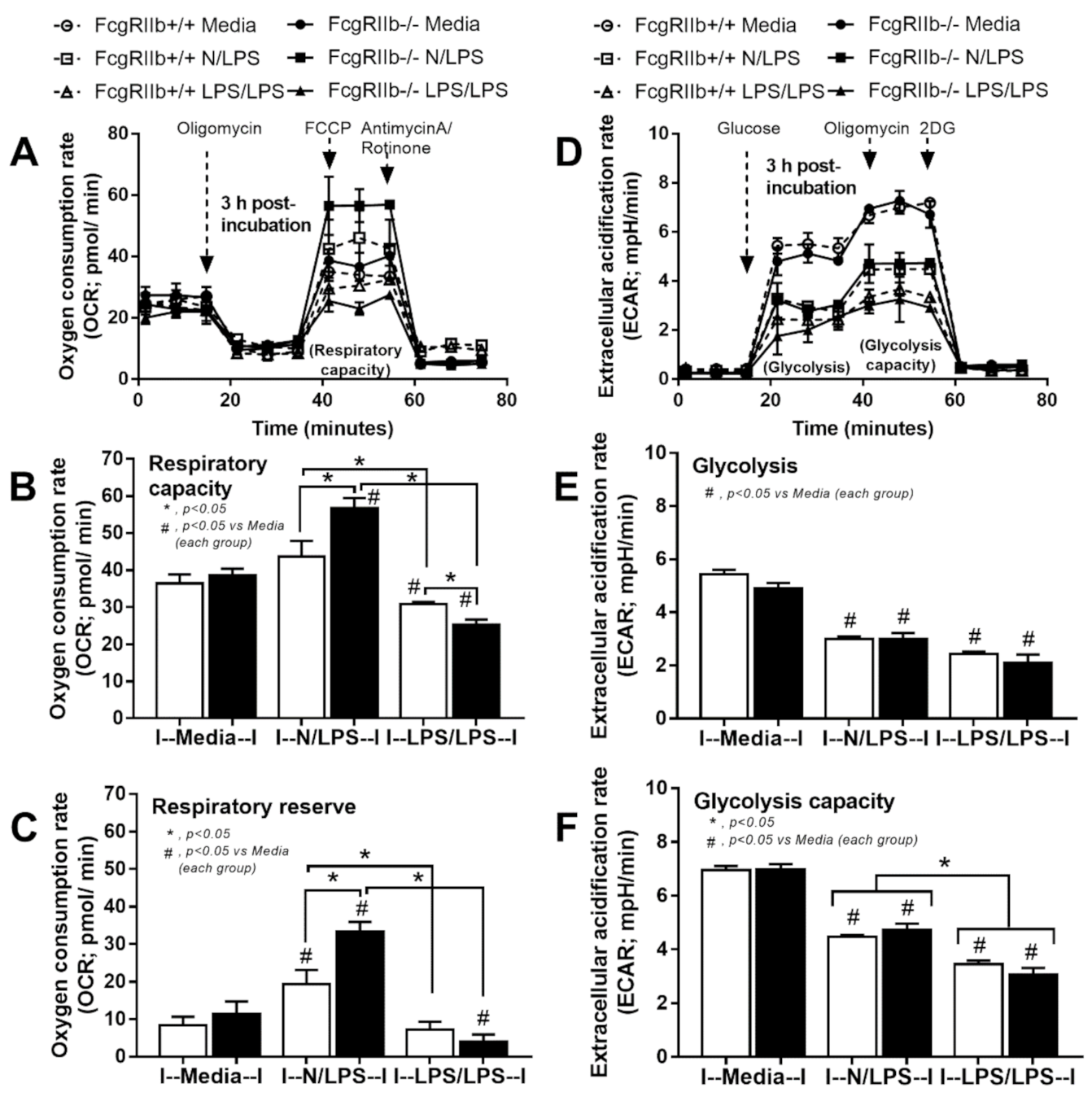
2.2. Prominent Responses against Endotoxin in FcgRIIb-/- Macrophages Compared to Wild-Type Macrophages In Vitro Implied Less Impact of LPS Tolerant Macrophages in NSAIDs-Administered Mice
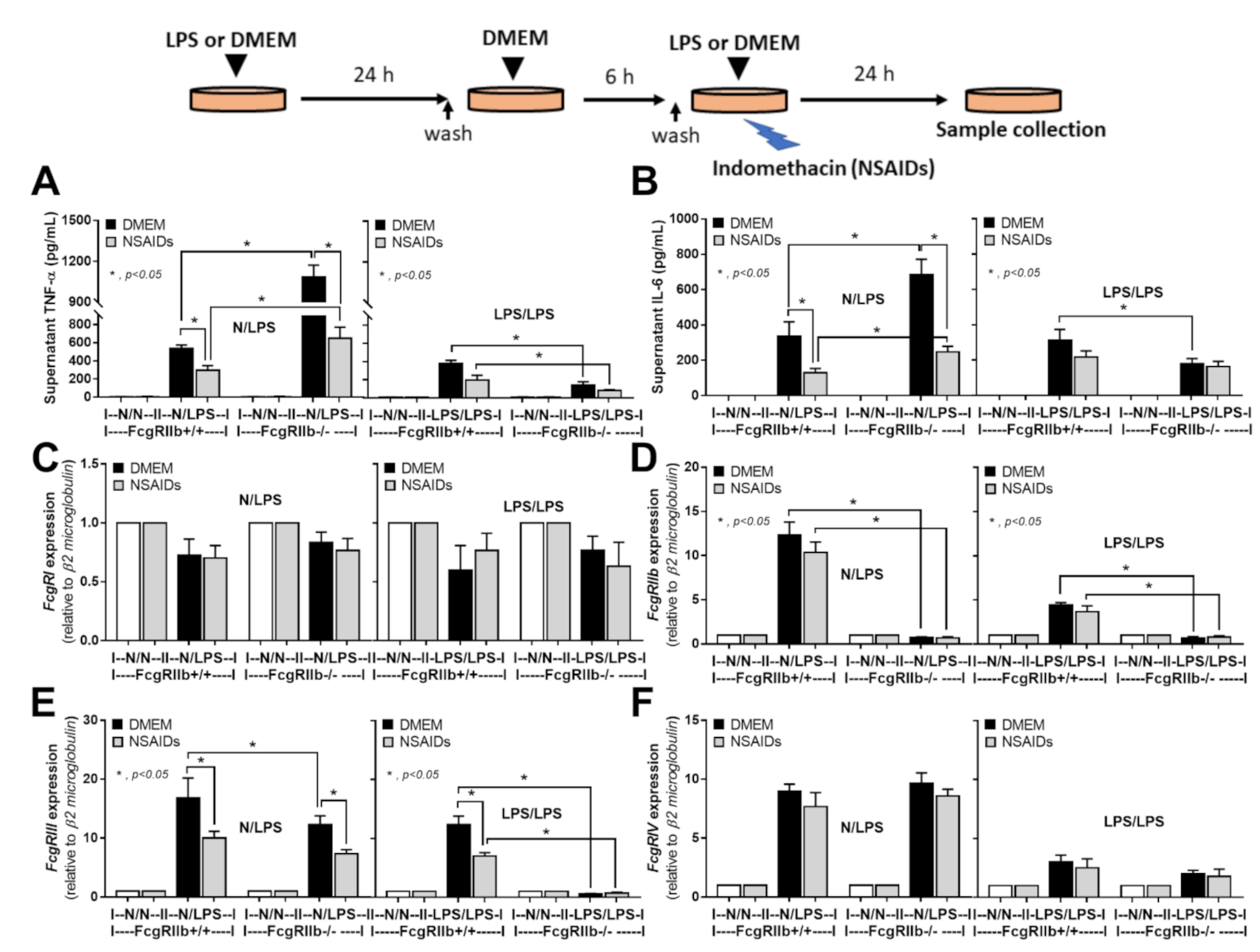
2.3. An Anti-Inflammatory Effect of NSAIDs in LPS-Activated Macrophages
3. Discussion
3.1. Prominent Indomethacin-Induced Nephropathy and Enteropathy in FcgRIIb-/- Mice Compared to Wild-Type Mice: An Impact of Immune Deposition in Asymptomatic Lupus
3.2. Prominent Inflammatory Responses against Endotoxin in FcgRIIb-/- Mice over Wild-Type Mice, an Inhibitory Effect of FcgRIIb.
4. Materials and Methods
4.1. Animals and Animal Model
4.2. Gut Permeability Determination
4.3. Histology Analysis and Immunofluorescent Imaging
4.4. Bone Marrow Derived Macrophages and the In Vitro Experiments
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolland, S.; Ravetch, J.V. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Clatworthy, M.R.; Willcocks, L.; Urban, B.; Langhorne, J.; Williams, T.N.; Peshu, N.; Watkins, N.A.; Floto, R.A.; Smith, K.G. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc. Natl. Acad. Sci. USA 2007, 104, 7169–7174. [Google Scholar] [CrossRef] [Green Version]
- Crispín, J.C.; Hedrich, C.M.; Tsokos, G.C. Gene-function studies in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 476–484. [Google Scholar] [CrossRef]
- Chu, Z.T.; Tsuchiya, N.; Kyogoku, C.; Ohashi, J.; Qian, Y.P.; Xu, S.B.; Mao, C.Z.; Chu, J.Y.; Tokunaga, K. Association of Fcgamma receptor IIb polymorphism with susceptibility to systemic lupus erythematosus in Chinese: A common susceptibility gene in the Asian populations. Tissue Antigens 2004, 63, 21–27. [Google Scholar] [CrossRef]
- Issara-Amphorn, J.; Surawut, S.; Worasilchai, N.; Thim-Uam, A.; Finkelman, M.; Chindamporn, A.; Palaga, T.; Hirankarn, N.; Pisitkun, P.; Leelahavanichkul, A. The Synergy of Endotoxin and (1→3)-β-D-Glucan, from Gut Translocation, Worsens Sepsis Severity in a Lupus Model of Fc Gamma Receptor IIb-Deficient Mice. J. Innate Immun. 2018, 10, 189–201. [Google Scholar] [CrossRef]
- Surawut, S.; Makjaroen, J.; Thim-Uam, A.; Wongphoom, J.; Palaga, T.; Pisitkun, P.; Chindamporn, A.; Leelahavanichkul, A. Increased susceptibility against Cryptococcus neoformans of lupus mouse models (pristane-induction and FcGRIIb deficiency) is associated with activated macrophage, regardless of genetic background. J. Microbiol. 2019, 57, 45–53. [Google Scholar] [CrossRef]
- Surawut, S.; Ondee, T.; Taratummarat, S.; Palaga, T.; Pisitkun, P.; Chindamporn, A.; Leelahavanichkul, A. The role of macrophages in the susceptibility of Fc gamma receptor IIb deficient mice to Cryptococcus neoformans. Sci. Rep. 2017, 7, 40006. [Google Scholar] [CrossRef] [Green Version]
- Ondee, T.; Gillen, J.; Visitchanakun, P.; Somparn, P.; Issara-Amphorn, J.; Dang Phi, C.; Chancharoenthana, W.; Gurusamy, D.; Nita-Lazar, A.; Leelahavanichkul, A. Lipocalin-2 (Lcn-2) Attenuates Polymicrobial Sepsis with LPS Preconditioning (LPS Tolerance) in FcGRIIb Deficient Lupus Mice. Cells 2019, 8, 1064. [Google Scholar] [CrossRef] [Green Version]
- Ondee, T.; Jaroonwitchawan, T.; Pisitkun, T.; Gillen, J.; Nita-Lazar, A.; Leelahavanichkul, A.; Somparn, P. Decreased Protein Kinase C-β Type II Associated with the Prominent Endotoxin Exhaustion in the Macrophage of FcGRIIb−/− Lupus Prone Mice is Revealed by Phosphoproteomic Analysis. Int. J. Mol. Sci. 2019, 20, 1354. [Google Scholar] [CrossRef] [Green Version]
- Ondee, T.; Surawut, S.; Taratummarat, S.; Hirankarn, N.; Palaga, T.; Pisitkun, P.; Pisitkun, T.; Leelahavanichkul, A. Fc Gamma Receptor IIB Deficient Mice: A Lupus Model with Increased Endotoxin Tolerance-Related Sepsis Susceptibility. Shock 2017, 47, 743–752. [Google Scholar] [CrossRef]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [Green Version]
- Akira, S. Toll receptor families: Structure and function. Semin. Immunol. 2004, 16, 1–2. [Google Scholar] [CrossRef]
- Koenderman, L. Inside-Out Control of Fc-Receptors. Front. Immunol. 2019, 10, 544. [Google Scholar] [CrossRef]
- Eppensteiner, J.; Kwun, J.; Scheuermann, U.; Barbas, A.; Limkakeng, A.T.; Kuchibhatla, M.; Elster, E.A.; Kirk, A.D.; Lee, J. Damage- and pathogen-associated molecular patterns play differential roles in late mortality after critical illness. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, Z.; Yu, A.M.; Wang, W.; Wei, Z.; Akhter, E.; Maurer, K.; Costa Reis, P.; Song, L.; Petri, M.; et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PLoS ONE 2014, 9, e93846. [Google Scholar] [CrossRef]
- Issara-Amphorn, J.; Chancharoenthana, W.; Visitchanakun, P.; Leelahavanichkul, A. Syk Inhibitor Attenuates Polymicrobial Sepsis in FcgRIIb-Deficient Lupus Mouse Model, the Impact of Lupus Characteristics in Sepsis. J. Innate Immun. 2020, 1–19. [Google Scholar] [CrossRef]
- Issara-Amphorn, J.; Somboonna, N.; Pisitkun, P.; Hirankarn, N.; Leelahavanichkul, A. Syk inhibitor attenuates inflammation in lupus mice from FcgRIIb deficiency but not in pristane induction: The influence of lupus pathogenesis on the therapeutic effect. Lupus 2020, 29, 1248–1262. [Google Scholar] [CrossRef]
- Engstad, C.S.; Engstad, R.E.; Olsen, J.O.; Osterud, B. The effect of soluble beta-1,3-glucan and lipopolysaccharide on cytokine production and coagulation activation in whole blood. Int. Immunopharmacol. 2002, 2, 1585–1597. [Google Scholar] [CrossRef]
- Ferwerda, G.; Meyer-Wentrup, F.; Kullberg, B.J.; Netea, M.G.; Adema, G.J. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell. Microbiol. 2008, 10, 2058–2066. [Google Scholar] [CrossRef]
- Kikkert, R.; Bulder, I.; de Groot, E.R.; Aarden, L.A.; Finkelman, M.A. Potentiation of Toll-like receptor-induced cytokine production by (1→3)-beta-D-glucans: Implications for the monocyte activation test. J. Endotoxin Res. 2007, 13, 140–149. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Giroux, M.; Descoteaux, A. Cyclooxygenase-2 expression in macrophages: Modulation by protein kinase C-alpha. J. Immunol. 2000, 165, 3985–3991. [Google Scholar] [CrossRef] [Green Version]
- Meek, I.L.; Van de Laar, M.A.F.J.; Vonkeman, H.E. Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks. Pharmaceuticals 2010, 3, 2146–2162. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, C.; Nath, R.K.; Perkinson, J.; Somasundaram, S.G.; Bjarnason, I. NSAID-induced gut inflammation and vasoconstriction: Causes and potential reversal with beta-CGRP—A hypothesis. Biosci. Hypotheses 2009, 2, 290–294. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [Green Version]
- Charbonney, E.; Tsang, J.Y.; Li, Y.; Klein, D.; Duque, P.; Romaschin, A.; Marshall, J.C. Endotoxemia Following Multiple Trauma: Risk Factors and Prognostic Implications. Crit. Care Med. 2016, 44, 335–341. [Google Scholar] [CrossRef]
- Tachecí, I.; Bradna, P.; Douda, T.; Baštecká, D.; Kopáčová, M.; Rejchrt, S.; Bureš, J. NSAID-Induced Enteropathy in Rheumatoid Arthritis Patients with Chronic Occult Gastrointestinal Bleeding: A Prospective Capsule Endoscopy Study. Gastroenterol. Res. Pract. 2013, 2013, 268382. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.P.; Gunasekara, D.B.; Speer, J.; Reed, M.I.; Peña, A.N.; Midkiff, B.R.; Magness, S.T.; Bultman, S.J.; Allbritton, N.L.; Redinbo, M.R. Nonsteroidal Anti-Inflammatory Drug-Induced Leaky Gut Modeled Using Polarized Monolayers of Primary Human Intestinal Epithelial Cells. ACS Infect. Dis. 2018, 4, 46–52. [Google Scholar] [CrossRef]
- Gault, M.H.; Barrett, B.J. Analgesic nephropathy. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1998, 32, 351–360. [Google Scholar] [CrossRef]
- Kleinknecht, D. Interstitial nephritis, the nephrotic syndrome, and chronic renal failure secondary to nonsteroidal anti-inflammatory drugs. Semin. Nephrol. 1995, 15, 228–235. [Google Scholar]
- Amornphimoltham, P.; Yuen, P.S.T.; Star, R.A.; Leelahavanichkul, A. Gut Leakage of Fungal-Derived Inflammatory Mediators: Part of a Gut-Liver-Kidney Axis in Bacterial Sepsis. Dig. Dis. Sci. 2019, 64, 2416–2428. [Google Scholar] [CrossRef]
- Deng, G.M.; Tsokos, G.C. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J. Immunol. 2008, 181, 4019–4026. [Google Scholar] [CrossRef]
- Podolska, M.J.; Biermann, M.H.; Maueröder, C.; Hahn, J.; Herrmann, M. Inflammatory etiopathogenesis of systemic lupus erythematosus: An update. J. Inflamm. Res. 2015, 8, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Thim-Uam, A.; Surawut, S.; Issara-Amphorn, J.; Jaroonwitchawan, T.; Hiengrach, P.; Chatthanathon, P.; Wilantho, A.; Somboonna, N.; Palaga, T.; Pisitkun, P.; et al. Leaky-gut enhanced lupus progression in the Fc gamma receptor-IIb deficient and pristane-induced mouse models of lupus. Sci. Rep. 2020, 10, 777. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S. The Pharmacology of Indomethacin. Headache 2016, 56, 436–446. [Google Scholar] [CrossRef]
- Nalamachu, S.; Wortmann, R. Role of indomethacin in acute pain and inflammation management: A review of the literature. Postgrad. Med. 2014, 126, 92–97. [Google Scholar] [CrossRef]
- Summ, O.; Andreou, A.P.; Akerman, S.; Holland, P.R.; Hoffmann, J.; Goadsby, P.J. Differential actions of indomethacin: Clinical relevance in headache. Pain 2020. [Google Scholar] [CrossRef]
- Drugs for gout. Med. Lett. Drugs Ther. 2019, 61, 33–37.
- Drugs for osteoarthritis. Med. Lett. Drugs Ther. 2020, 62, 57–62.
- Lambrechts, M.J.; Cook, J.L. Nonsteroidal Anti-Inflammatory Drugs and Their Neuroprotective Role after an Acute Spinal Cord Injury: A Systematic Review of Animal Models. Glob. Spine J. 2020, 2192568220901689. [Google Scholar] [CrossRef] [Green Version]
- Rekatsina, M.; Paladini, A.; Cifone, M.G.; Lombardi, F.; Pergolizzi, J.V.; Varrassi, G. Influence of Microbiota on NSAID Enteropathy: A Systematic Review of Current Knowledge and the Role of Probiotics. Adv. Ther. 2020, 37, 1933–1945. [Google Scholar] [CrossRef] [Green Version]
- Nishio, N.; Ito, S.; Suzuki, H.; Isobe, K. Antibodies to wounded tissue enhance cutaneous wound healing. Immunology 2009, 128, 369–380. [Google Scholar] [CrossRef]
- Stewart, R.; Hammond, S.A.; Oberst, M.; Wilkinson, R.W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. ImmunoTher. Cancer 2014, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Flierl, M.A.; Day, D.E.; Nadeau, B.A.; Zetoune, F.S.; Sarma, J.V.; Werner, C.M.; Wanner, G.A.; Simmen, H.P.; Huber-Lang, M.S.; et al. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS Pathog. 2009, 5, e1000464. [Google Scholar] [CrossRef] [Green Version]
- Jaroonwitchawan, T.; Visitchanakun, P.; Dang, P.C.; Ritprajak, P.; Palaga, T.; Leelahavanichkul, A. Dysregulation of Lipid Metabolism in Macrophages Is Responsible for Severe Endotoxin Tolerance in FcgRIIB-Deficient Lupus Mice. Front. Immunol. 2020, 11, 959. [Google Scholar] [CrossRef]
- Allantaz-Frager, F.; Turrel-Davin, F.; Venet, F.; Monnin, C.; De Saint Jean, A.; Barbalat, V.; Cerrato, E.; Pachot, A.; Lepape, A.; Monneret, G. Identification of biomarkers of response to IFNg during endotoxin tolerance: Application to septic shock. PLoS ONE 2013, 8, e68218. [Google Scholar] [CrossRef] [Green Version]
- Vergadi, E.; Vaporidi, K.; Tsatsanis, C. Regulation of Endotoxin Tolerance and Compensatory Anti-inflammatory Response Syndrome by Non-coding RNAs. Front. Immunol. 2018, 9, 2705. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fang, S.; Li, X.; Feng, J.; Du, J.; Guo, L.; Su, Y.; Zhou, J.; Ding, G.; Bai, Y.; et al. Aspirin inhibits LPS-induced macrophage activation via the NF-κB pathway. Sci. Rep. 2017, 7, 11549. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, C.M.; Marcarino, P.; Sheldrake, T.A.; Lazzarato, L.; Cena, C.; Fruttero, R.; Gasco, A.; Fox, S.; Megson, I.L.; Rossi, A.G. A novel hybrid aspirin-NO-releasing compound inhibits TNFalpha release from LPS-activated human monocytes and macrophages. J. Inflamm. 2008, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Chen, F.; Zhang, A.; Zhu, B.; Sun, J.; Xie, Q.; Chen, Z. Aspirin inhibits lipopolysaccharide-induced COX-2 expression and PGE2 production in porcine alveolar macrophages by modulating protein kinase C and protein tyrosine phosphatase activity. BMB Rep. 2014, 47, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Fullerton, J.N.; O’Brien, A.J.; Gilroy, D.W. Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. 2014, 35, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Beehrle, D.M.; Evans, D. A review of NSAID complications: Gastrointestinal and more. Lippincott’s Prim. Care Pract. 1999, 3, 305–315. [Google Scholar]
- Ejaz, P.; Bhojani, K.; Joshi, V.R. NSAIDs and kidney. J. Assoc. Physicians India 2004, 52, 632–640. [Google Scholar]
- Schlondorff, D. Renal complications of nonsteroidal anti-inflammatory drugs. Kidney Int. 1993, 44, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Clavé, S.; Rousset-Rouvière, C.; Daniel, L.; Tsimaratos, M. The Invisible Threat of Non-steroidal Anti-inflammatory Drugs for Kidneys. Front. Pediatr. 2019, 7, 520. [Google Scholar] [CrossRef] [Green Version]
- Vadivel, N.; Trikudanathan, S.; Singh, A.K. Analgesic nephropathy. Kidney Int. 2007, 72, 517–520. [Google Scholar] [CrossRef] [Green Version]
- Mérida, E.; Praga, M. NSAIDs and Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2019, 14, 1280–1282. [Google Scholar] [CrossRef]
- Sukkummee, W.; Jittisak, P.; Wonganan, P.; Wittayalertpanya, S.; Chariyavilaskul, P.; Leelahavanichkul, A. The prominent impairment of liver/intestinal cytochrome P450 and intestinal drug transporters in sepsis-induced acute kidney injury over acute and chronic renal ischemia, a mouse model comparison. Ren. Fail. 2019, 41, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Leelahavanichkul, A.; Somparn, P.; Panich, T.; Chancharoenthana, W.; Wongphom, J.; Pisitkun, T.; Hirankarn, N.; Eiam-Ong, S. Serum miRNA-122 in acute liver injury induced by kidney injury and sepsis in CD-1 mouse models. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 1341–1352. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Panpetch, W.; Kullapanich, C.; Dang, C.P.; Visitchanakun, P.; Saisorn, W.; Wongphoom, J.; Wannigama, D.L.; Thim-Uam, A.; Patarakul, K.; Somboonna, N.; et al. Candida Administration Worsens Uremia-Induced Gut Leakage in Bilateral Nephrectomy Mice, an Impact of Gut Fungi and Organismal Molecules in Uremia. mSystems 2021, 6. [Google Scholar] [CrossRef]
- Shin, S.J.; Noh, C.K.; Lim, S.G.; Lee, K.M.; Lee, K.J. Non-steroidal anti-inflammatory drug-induced enteropathy. Intest. Res. 2017, 15, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Deitch, E.A. The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch. Surg. 1990, 125, 403–404. [Google Scholar] [CrossRef]
- Sugimura, N.; Otani, K.; Watanabe, T.; Nakatsu, G.; Shimada, S.; Fujimoto, K.; Nadatani, Y.; Hosomi, S.; Tanaka, F.; Kamata, N.; et al. High-fat diet-mediated dysbiosis exacerbates NSAID-induced small intestinal damage through the induction of interleukin-17A. Sci. Rep. 2019, 9, 16796. [Google Scholar] [CrossRef]
- Utzeri, E.; Usai, P. Role of non-steroidal anti-inflammatory drugs on intestinal permeability and nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 3954–3963. [Google Scholar] [CrossRef]
- Dennehy, K.M.; Ferwerda, G.; Faro-Trindade, I.; Pyz, E.; Willment, J.A.; Taylor, P.R.; Kerrigan, A.; Tsoni, S.V.; Gordon, S.; Meyer-Wentrup, F.; et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur. J. Immunol. 2008, 38, 500–506. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Worasilchai, N.; Wannalerdsakun, S.; Jutivorakool, K.; Somparn, P.; Issara-Amphorn, J.; Tachaboon, S.; Srisawat, N.; Finkelman, M.; Chindamporn, A. Gastrointestinal Leakage Detected by Serum (1→3)-β-D-Glucan in Mouse Models and a Pilot Study in Patients with Sepsis. Shock 2016, 46, 506–518. [Google Scholar] [CrossRef]
- Panpetch, W.; Hiengrach, P.; Nilgate, S.; Tumwasorn, S.; Somboonna, N.; Wilantho, A.; Chatthanathon, P.; Prueksapanich, P.; Leelahavanichkul, A. Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 2020, 11, 465–480. [Google Scholar] [CrossRef]
- Panpetch, W.; Somboonna, N.; Bulan, D.E.; Issara-Amphorn, J.; Finkelman, M.; Worasilchai, N.; Chindamporn, A.; Palaga, T.; Tumwasorn, S.; Leelahavanichkul, A. Oral administration of live- or heat-killed Candida albicans worsened cecal ligation and puncture sepsis in a murine model possibly due to an increased serum (1→3)-β-d-glucan. PLoS ONE 2017, 12, e0181439. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors: Old friends and new family members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.P.; Leelahavanichkul, A. Over-expression of miR-223 induces M2 macrophage through glycolysis alteration and attenuates LPS-induced sepsis mouse model, the cell-based therapy in sepsis. PLoS ONE 2020, 15, e0236038. [Google Scholar] [CrossRef]
- Cho, J.Y. Immunomodulatory effect of nonsteroidal anti-inflammatory drugs (NSAIDs) at the clinically available doses. Arch. Pharmacal Res. 2007, 30, 64–74. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suarez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef]
- Kent, T.H.; Cardelli, R.M.; Stamler, F.W. Small intestinal ulcers and intestinal flora in rats given indomethacin. Am. J. Pathol. 1969, 54, 237–249. [Google Scholar]
- Odabasoglu, F.; Cakir, A.; Suleyman, H.; Aslan, A.; Bayir, Y.; Halici, M.; Kazaz, C. Gastroprotective and antioxidant effects of usnic acid on indomethacin-induced gastric ulcer in rats. J. Ethnopharmacol. 2006, 103, 59–65. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Somparn, P.; Bootprapan, T.; Tu, H.; Tangtanatakul, P.; Nuengjumnong, R.; Worasilchai, N.; Tiranathanagul, K.; Eiam-ong, S.; Levine, M.; et al. High-dose ascorbate with low-dose amphotericin B attenuates severity of disease in a model of the reappearance of candidemia during sepsis in the mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R223–R234. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Tan, I.; Chuzhin, Y.; Reddy, B.; Budhai, L.; Holzer, A.; Gu, Y.; Davidson, A. CTLA4Ig inhibits T cell-dependent B-cell maturation in murine systemic lupus erythematosus. J. Clin. Investig. 2000, 106, 91–101. [Google Scholar] [CrossRef]
- Visitchanakun, P.; Saisorn, W.; Wongphoom, J.; Chatthanathon, P.; Somboonna, N.; Svasti, S.; Fucharoen, S.; Leelahavanichkul, A. Gut leakage enhances sepsis susceptibility in iron-overloaded β-thalassemia mice through macrophage hyperinflammatory responses. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G966–G979. [Google Scholar] [CrossRef]
- Leelahavanichkul, A.; Yan, Q.; Hu, X.; Eisner, C.; Huang, Y.; Chen, R.; Mizel, D.; Zhou, H.; Wright, E.C.; Kopp, J.B.; et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010, 78, 1136–1153. [Google Scholar] [CrossRef] [Green Version]
- Leelahavanichkul, A.; Huang, Y.; Hu, X.; Zhou, H.; Tsuji, T.; Chen, R.; Kopp, J.B.; Schnermann, J.; Yuen, P.S.; Star, R.A. Chronic kidney disease worsens sepsis and sepsis-induced acute kidney injury by releasing High Mobility Group Box Protein-1. Kidney Int. 2011, 80, 1198–1211. [Google Scholar] [CrossRef] [Green Version]
- Erben, U.; Loddenkemper, C.; Doerfel, K.; Spieckermann, S.; Haller, D.; Heimesaat, M.M.; Zeitz, M.; Siegmund, B.; Kühl, A.A. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol. 2014, 7, 4557–4576. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward | Reverse |
|---|---|---|
| Fc gamma receptor I (FcgRI) | 5′-CACAAATGCCCTTAGACCAC-3′ | 5′-ACCCTAGAGTTCCAGGGATG-3′ |
| Fc gamma receptor IIb (FcgRIIb) | 5′-TTCTCAAGCATCCCGAAGCC-3′ | 5′-TTCCCAATGCCAAGGGAGAC-3′ |
| Fc gamma receptor III (FcgRIII) | 5′-AGGGCCTCCATCTGGACTG-3′ | 5′-GTGGTTCTGGTAATCATGCTCTG-3′ |
| Fc gamma receptor IV (FcgRIV) | 5′-AACGGCAAAGGCAAGAAGTA-3′ | 5′-CCGCACAGAGAAATACAGCA-3′ |
| β2 microglobulin (β2M) | 5′CCACTGAAAAAGATGAGTATGCCT-3′ | 5′-CCAATCCAAATGCGGCATCTTCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhunyakarnjanarat, T.; Udompornpitak, K.; Saisorn, W.; Chantraprapawat, B.; Visitchanakun, P.; Dang, C.P.; Issara-Amphorn, J.; Leelahavanichkul, A. Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-cient lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut. Int. J. Mol. Sci. 2021, 22, 1377. https://doi.org/10.3390/ijms22031377
Bhunyakarnjanarat T, Udompornpitak K, Saisorn W, Chantraprapawat B, Visitchanakun P, Dang CP, Issara-Amphorn J, Leelahavanichkul A. Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-cient lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut. International Journal of Molecular Sciences. 2021; 22(3):1377. https://doi.org/10.3390/ijms22031377
Chicago/Turabian StyleBhunyakarnjanarat, Thansita, Kanyarat Udompornpitak, Wilasinee Saisorn, Bhumdhanin Chantraprapawat, Peerapat Visitchanakun, Cong Phi Dang, Jiraphorn Issara-Amphorn, and Asada Leelahavanichkul. 2021. "Prominent Indomethacin-Induced Enteropathy in Fcgriib Defi-cient lupus Mice: An Impact of Macrophage Responses and Immune Deposition in Gut" International Journal of Molecular Sciences 22, no. 3: 1377. https://doi.org/10.3390/ijms22031377