
Development of MTH1-Binding Nucleotide Analogs Based on 7,8-Dihalogenated 7-Deaza-dG Derivatives


Abstract
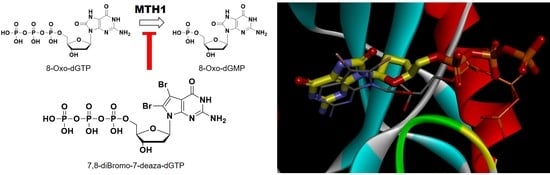
:
1. Introduction
2. Results and Discussion
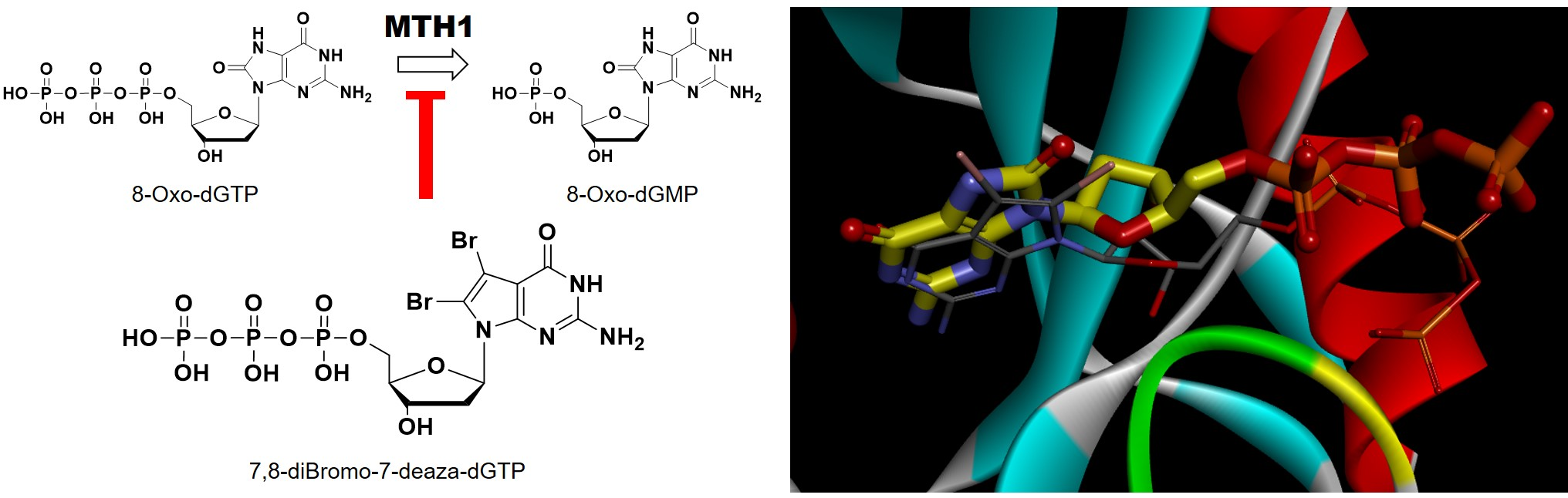
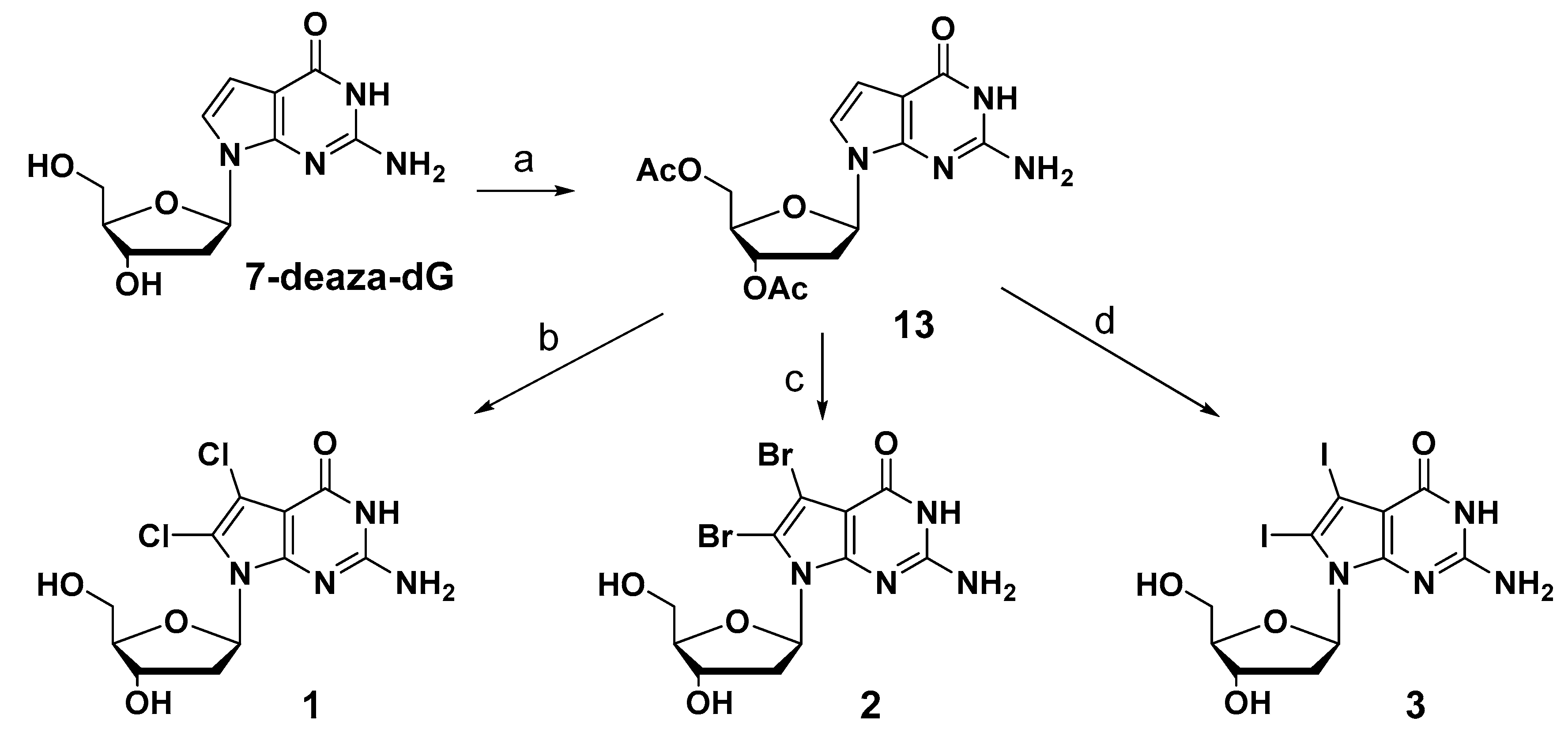
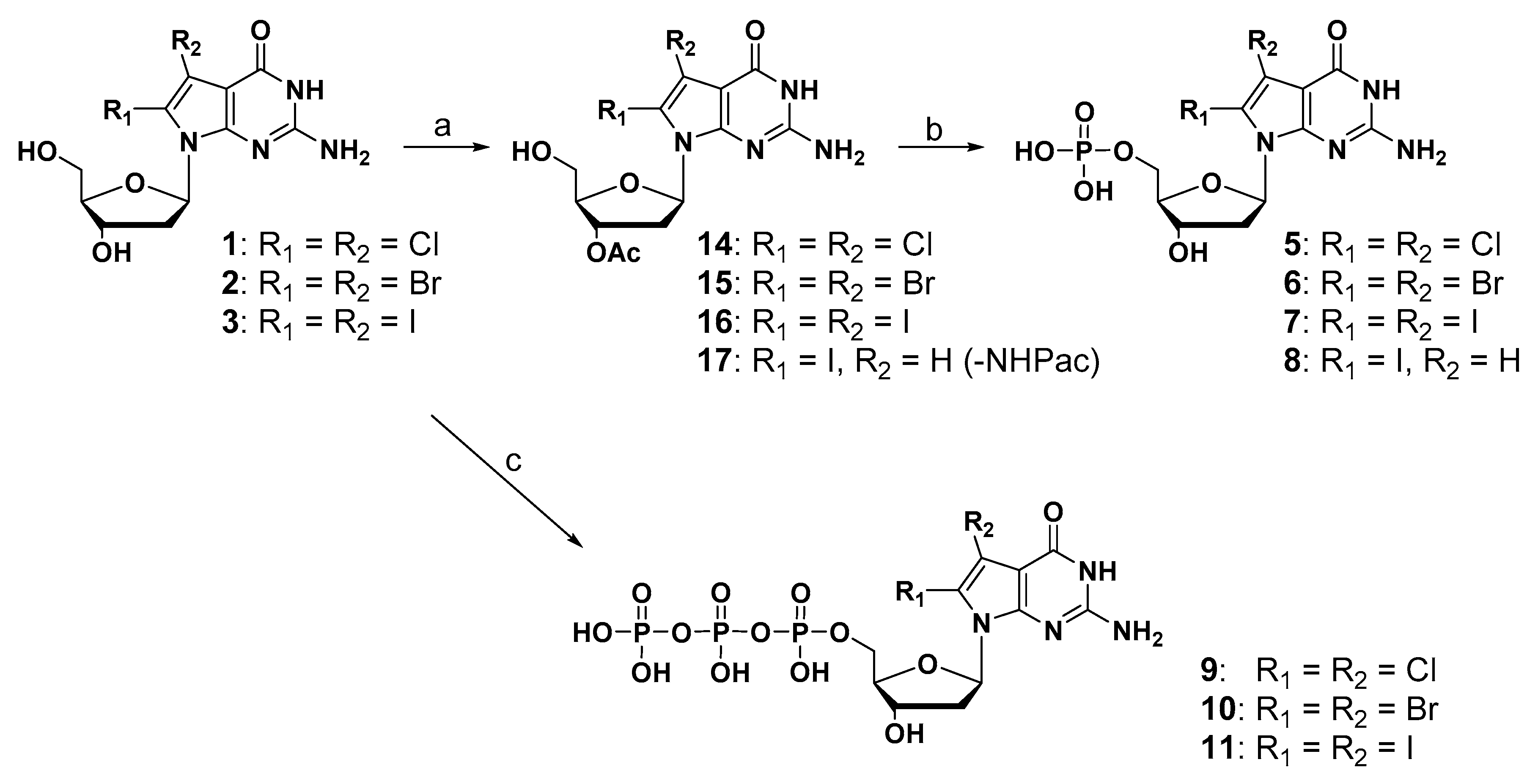
2.1. Synthesis of New 7-Deaza-dG Derivatives
2.2. Binding Properties of Dihalogenated-deaza-dG Derivatives for MTH1
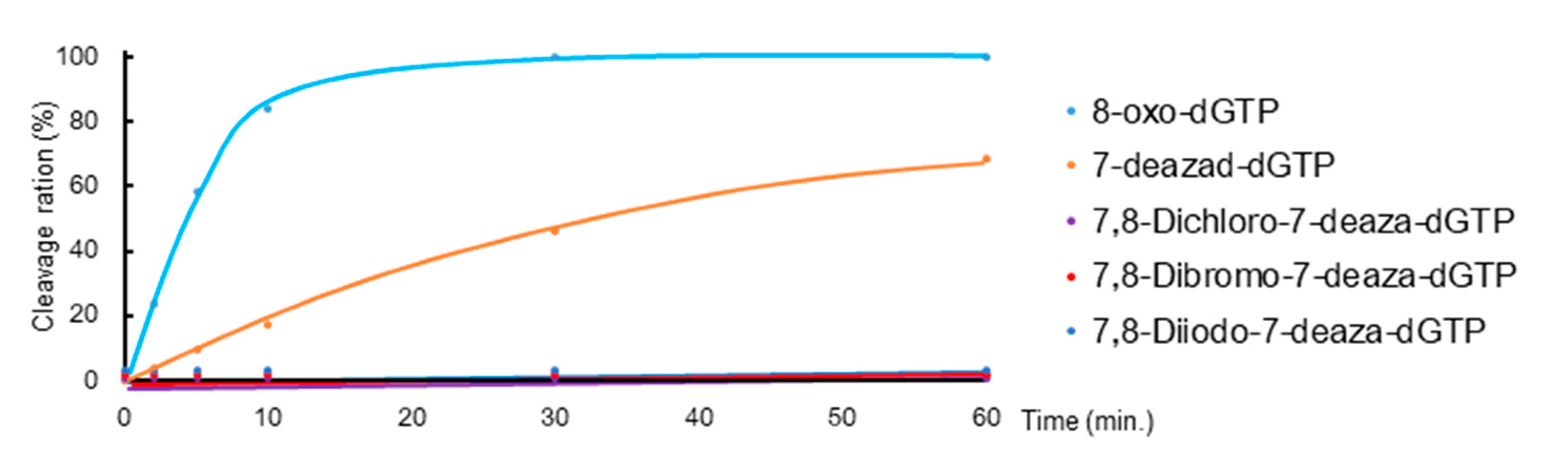
2.2.1. Hydrolytic Properties of New 7-Deaza-dGTP Derivatives by MTH1
2.2.2. Inhibitory Effects of New 7-Deaza-dG Derivatives on the Hydrolytic Activity of MTH1
2.2.3. Assessment of Inhibition Modes of New 7-Deaza-dG Derivatives
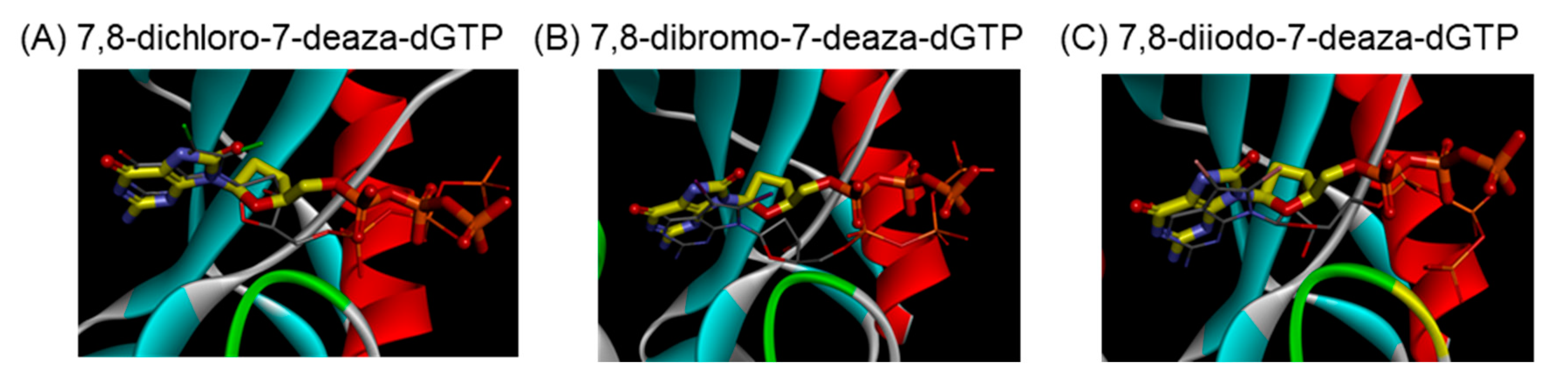
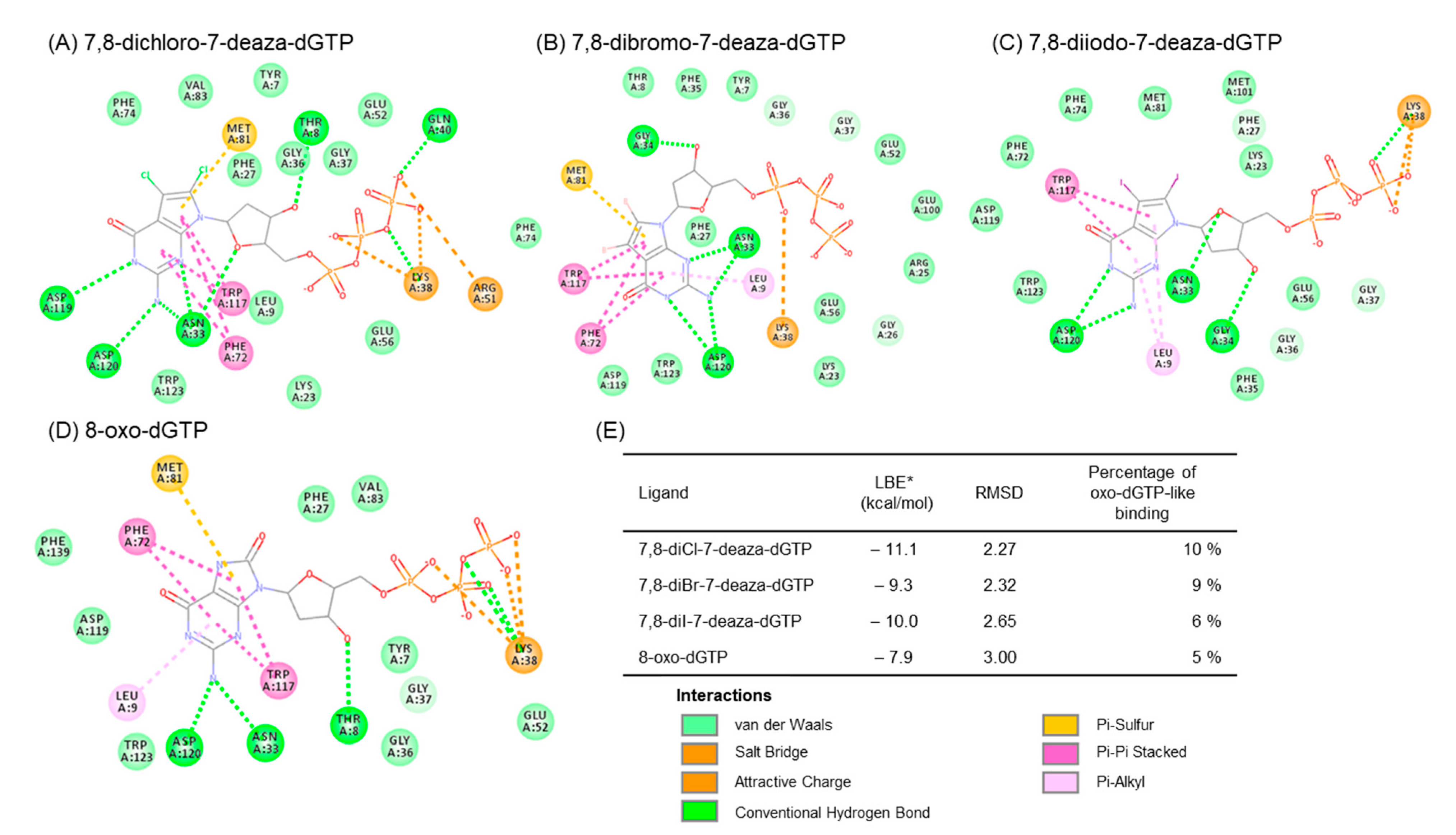
2.2.4. Prediction of Binding Properties of New 7-Deaza-dG Derivatives with MTH1
3. Materials and Methods
3.1. Synthesis of New 7-Deaza-dG Derivatives
3.1.1. General Procedure for the Synthesis of 7,8-Dihalogeneted-7-deaza-2′-deoxy-guanosine
3.1.2. General Procedure for the Synthesis of 3′-O-Acetylated Compounds
3.1.3. General Procedure for the Synthesis of 5′-Monophosphate Compounds
3.1.4. General Procedure for the Synthesis of 5′-Triphosphate Compounds
3.2. Hydrolysis of 7,8-Dihalogenated-7-deaza-dGTP, 8-CF3-7-deaza-dGTP, and 8-oxo-dGTP by MTH1
3.3. Inhibitory Effects of New 7-Deaza-dG Derivatives on the Hydrolytic Activity of MTH1
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MTH1 | human mutt homolog 1, also called NUDT1 |
| 7-deaza-dG | 7-deaza-2′-deoxyguanosine |
| 8-oxo-dGTP | 8-oxo-2′-deoxyguanosine-5′-triphosphate |
| 8-oxo-dGMP | 8-oxo-2′-deoxyguanosine-5′-monophosphate |
| ROS | reactive oxygen species |
| IC50 | half maximal inhibitory concentration |
| Ki | inhibition constant |
| NCS | N-chlorosuccinimide |
| NBC | N-bromosuccinimide |
| NIS | N-iodosuccinimide |
| POCl3 | phosphoryl chloride |
| TBSCl | tert-butylchlorodimethylsilane |
| DMAP | 4-dimethylaminopyridine |
| Et3N-3HF | triethylamine trihydrofluoride |
| TEA | triethylamine |
| DCC | N,N’-dicyclohexylcarbodiimide |
| DMF | N,N-dimethylformamide |
| HPLC | high performance liquid chromatography |
| Tris-HCl | tris(hydroxymethyl)aminomethane hydrochloride |
| BSA | bovine serum albumin |
| DTT | dithiothreitol |
| KM | michaelis constant |
| kcat | turnover number |
| Asp | aspartic acid |
| Trp | tryptophan |
| Lys | lysine |
References
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, B.A.; Crapo, J.D. Biology of disease: Free radicals and tissue injury. Lab. Investig. 1982, 47, 412–426. [Google Scholar] [PubMed]
- Topal, M.D.; Baker, M.S. DNA precursor pool: A significant target for N-methyl-N-nitrosourea in C3H/10T1/2 clone 8 cells. Proc. Natl Acad. Sci. USA 1982, 79, 2211–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenken, S.; Jovanovic, S.V. One-electron redox potentials of purines and pyrimidines. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Burrows, C.J.; Muller, J.G. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Reaction of HO· with guanine derivatives in aqueous solution: Formation of two different redox-active OH-adduct radicals and their unimolecular transformation reactions. Properties of G(-H). Chem. Eur. J. 2000, 6, 475–484. [Google Scholar] [CrossRef]
- Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Influence of the oxidatively damaged adduct 8-oxodeoxyguanosine on the conformation, energetics, and thermodynamic stability of a DNA duplex. Biochemistry 1995, 34, 16148–16160. [Google Scholar] [CrossRef]
- Gannett, P.M.; Sura, T.P.; Virginia, W. Base pairing of 8-oxoguanosine and 8-oxo-2’-deoxyguanosine with 2’-deoxyadenosine, 2’-deoxycytosine, 2’-deoxyguanosine, and thymidine. Chem. Res. Toxicol. 1993, 6, 690–700. [Google Scholar] [CrossRef]
- McAuley-Hecht, K.E.; Leonard, G.A.; Gibson, N.J.; Thomson, J.B.; Watson, W.P.; Hunter, W.N.; Brown, T. Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry 1994, 33, 10266–10270. [Google Scholar] [CrossRef]
- Culp, S.J.; Cho, B.P.; Kadlubar, F.F.; Evans, F.E. Structural and conformational analyses of 8-hydroxy-2’-deoxyguanosine. Chem. Res. Toxicol. 1989, 2, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Goddard, W.A.; Noyes, K.T.; Sowers, L.C.; Hwang, S.; Chung, D.S. First principles calculations of the tautomers and pKa values of 8-oxoguanine: implications for mutagenicity and repair. Chem. Res. Toxicol. 2002, 15, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Takeshita, M.; Grollaman, A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Hsu, G.W.; Ober, M.; Carell, T.; Beese, L.S. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature 2004, 431, 217–221. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001, 477, 59–70. [Google Scholar] [CrossRef]
- Sakai, Y.; Furuichi, M.; Takahashi, M.; Mishima, M.; Iwai, S.; Shirakawa, M.; Nakabeppu, Y. A molecular basis for the selective recognition of 2-hydroxy-dATPand 8-oxo-dGTP by human MTH1. J. Biol. Chem. 2002, 277, 8579–8587. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, H.; Yakushiji, H.; Dugue, L.; Tanimoto, M.; Pochet, S.; Nakabeppu, Y.; Harashima, H. Probing the substrate recognition mechanism of the human MTH1 protein by nucleotide analogs. J. Mol. Biol. 2004, 336, 843–850. [Google Scholar] [CrossRef]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Ström, C.E.; Svensson, L.M.; Schultz, N.; Lundbäck, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar] [CrossRef]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Chen, F. Targeting human MutT homolog 1 (MTH1) for cancer eradication: Current progress and perspectives. Acta Pharm. Sin. B 2020, 10, 2259–2271. [Google Scholar] [CrossRef]
- Berglund, U.W.; Sanjiv, K.; Gad, H.; Kalderén, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Moukengue, B.; Brown, H.K.; Charrier, C.; Battaglia, S.; Baud’huin, M.; Quillard, T.; Pham, T.M.; Pateras, I.S.; Gorgoulis, V.G.; Helleday, T.; et al. TH1579, MTH1 inhibitor, delays tumour growth and inhibits metastases development in osteosarcoma mode. EBioMedicine 2020, 53, 102704. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Sanjiv, K.; Gad, H.; Pham, T.; Gokturk, C.; Rasti, A.; Zhao, Z.; He, K.; Feng, M.; Zang, Y.; et al. Karonudib is a promising anticancer therapy in hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrocchi, A.; Leo, E.; Reyna, N.J.; Hamilton, M.M.; Shi, X.; Parker, C.A.; Mseeh, F.; Bardenhagen, J.P.; Leonard, P.; Cross, J.B.; et al. Identification of potent and selective MTH1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1503–1507. [Google Scholar] [CrossRef]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [CrossRef]
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Taniguchi, Y.; Sasaki, S. Synthesis of 8-halogenated-7-deaza-2’-deoxyguanosine as an 8-oxo-2’-deoxyguanosine analogue and evaluation of its base pairing properties. Tetrahedron 2014, 70, 2040–2047. [Google Scholar] [CrossRef]
- Yin, Y.; Sasaki, S.; Taniguchi, Y. Inhibitory effect of 8-halogenated 7-deaza-2′-deoxyguanosine triphosphates on human 8-oxo-2′-deoxyguanosine triphosphatase, hMTH1, activities. ChemBioChem 2016, 17, 566–569. [Google Scholar] [CrossRef]
- Yin, Y.; Sasaki, S.; Taniguchi, Y. Effects of 8-halo-7-deaza-2’-deoxyguanosine triphosphate on DNA synthesis by DNA polymerase and cell proliferation. Bioorg. Med. Chem. 2016, 24, 3856–3861. [Google Scholar] [CrossRef]
- Thavoncharoensub, N.; Maruyama, K.; Heh, C.H.; Leong, K.H.; Shi, H.; Shigematsu, Y.; Sasaki, S.; Taniguchi, Y. Synthesis of γ-N-modified 8-oxo-2′-deoxyguanosine triphosphate and its characterization. Nucleosides Nucleotides Nucleic Acids 2019, 38, 578–589. [Google Scholar] [CrossRef]
- Taktakishvili, M.; Nair, V. A new method for the phosphorylation of nucleosides. Tetrahedron Lett. 2000, 41, 7173–7176. [Google Scholar] [CrossRef]
- Caton-Williams, J.; Lin, L.; Smith, M.; Huang, Z. Convenient synthesis of nucleoside 5′-triphosphates for RNA transcription. Chem. Commun. 2011, 47, 8142–8144. [Google Scholar] [CrossRef] [PubMed]
- Caton-Williams, J.; Hoxhaj, R.; Fiaz, B.; Huang, Z. Use of a novel 5′-regioselective phosphitylating reagent for one-pot synthesis of nucleoside 5′-triphosphates from unprotected nucleosides. Curr. Protoc. Nucleic Acid Chem. 2013, 52, 1.30.1–1.30.21. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1= | R2= | R3= | Compound | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | Cl | Cl | H | 1 | 22.5 |
| 2 | Br | Br | H | 2 | 10.7 |
| 3 | I | I | H | 3 | 17.0 |
| 4 | I | H | H | 42 | 44.8 |
| 5 | Cl | Cl | PO3H2 | 5 | 0.13 |
| 6 | Br | Br | PO3H2 | 6 | 0.26 |
| 7 | I | I | PO3H2 | 7 | 0.13 |
| 8 | I | H | PO3H2 | 8 | 0.84 |
| 9 | Cl | Cl | P3O9H4 | 9 | 0.16 |
| 10 | Br | Br | P3O9H4 | 10 | 0.11 |
| 11 | I | I | P3O9H4 | 11 | 0.13 |
| 12 | I | H | P3O9H4 | 122 | 0.42 |
| 13 | - | - | - | TH2872 | 0.01 |
| Entry | Inhibitor | kcat (s−1) | KM (μM) | Ki (nM) |
|---|---|---|---|---|
| 1 | without | 10.6 | 17.6 | - |
| 2 | 7,8-dichloro-7-deaza-dGMP (5) | 9.7 | 117.8 | 37.0 |
| 3 | 7,8- dichloro-7-deaza-dGTP (9) | 9.3 | 142.9 | 28.1 |
| 4 | 7,8-dibromo-7-deaza-dGMP (6) | 9.1 | 151.5 | 27.9 |
| 5 | 7,8-dibromo-7-deaza-dGTP (10) | 9.4 | 126.6 | 24.2 |
| 6 | 7,8-diiodo-7-deaza-dGMP (7) | 10.7 | 157.2 | 26.8 |
| 7 | 7,8-diiodo-7-deaza-dGTP (11) | 11.9 | 133.4 | 30.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, H.; Ishikawa, R.; Heh, C.H.; Sasaki, S.; Taniguchi, Y. Development of MTH1-Binding Nucleotide Analogs Based on 7,8-Dihalogenated 7-Deaza-dG Derivatives. Int. J. Mol. Sci. 2021, 22, 1274. https://doi.org/10.3390/ijms22031274
Shi H, Ishikawa R, Heh CH, Sasaki S, Taniguchi Y. Development of MTH1-Binding Nucleotide Analogs Based on 7,8-Dihalogenated 7-Deaza-dG Derivatives. International Journal of Molecular Sciences. 2021; 22(3):1274. https://doi.org/10.3390/ijms22031274
Chicago/Turabian StyleShi, Hui, Ren Ishikawa, Choon Han Heh, Shigeki Sasaki, and Yosuke Taniguchi. 2021. "Development of MTH1-Binding Nucleotide Analogs Based on 7,8-Dihalogenated 7-Deaza-dG Derivatives" International Journal of Molecular Sciences 22, no. 3: 1274. https://doi.org/10.3390/ijms22031274