
PDGF Receptor Alpha Signaling Is Key for Müller Cell Homeostasis Functions

 , ,
, ,
Abstract
:1. Introduction
2. Results
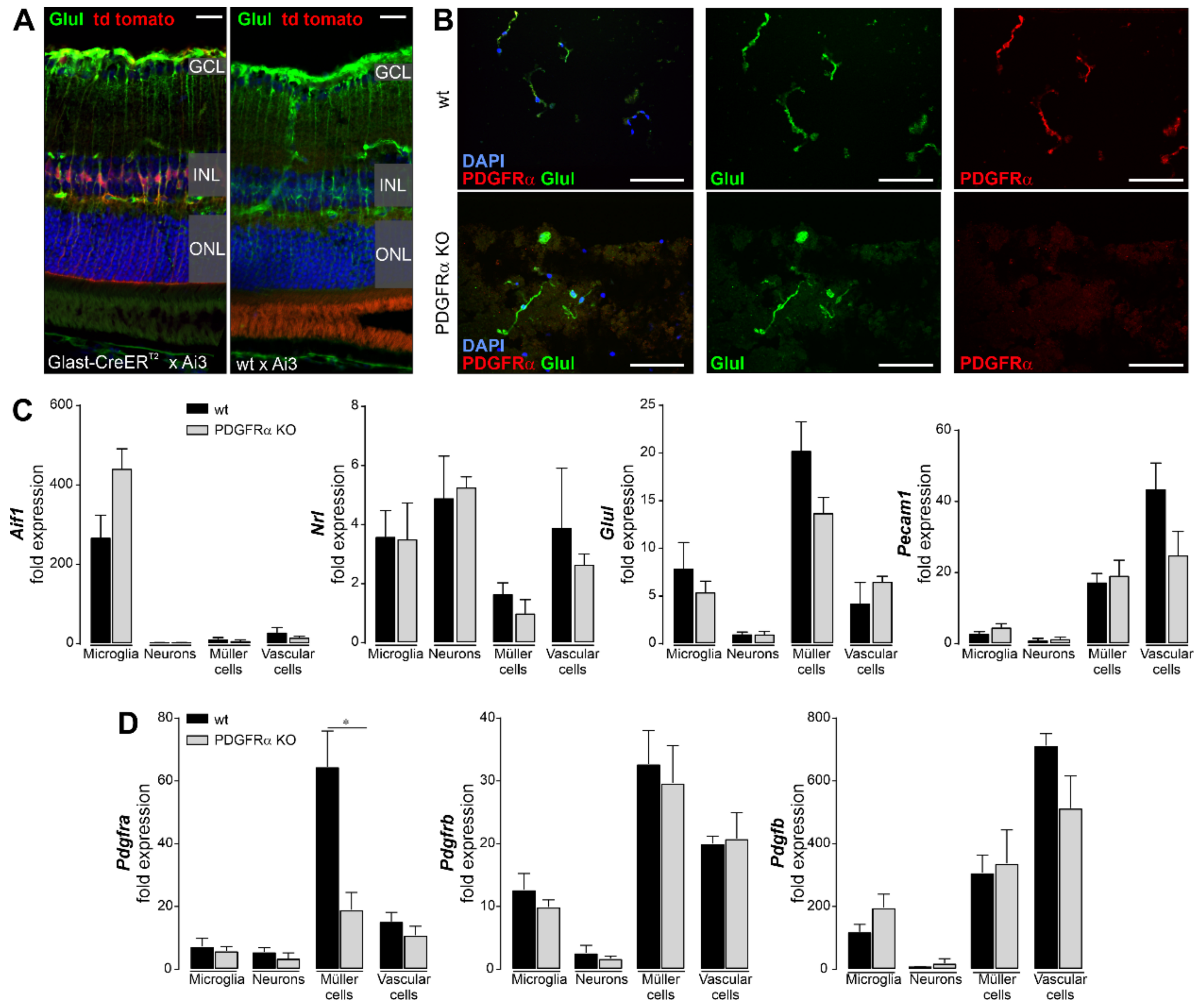
2.1. Induced Müller Cell-Specific Knockout of PDGFRα
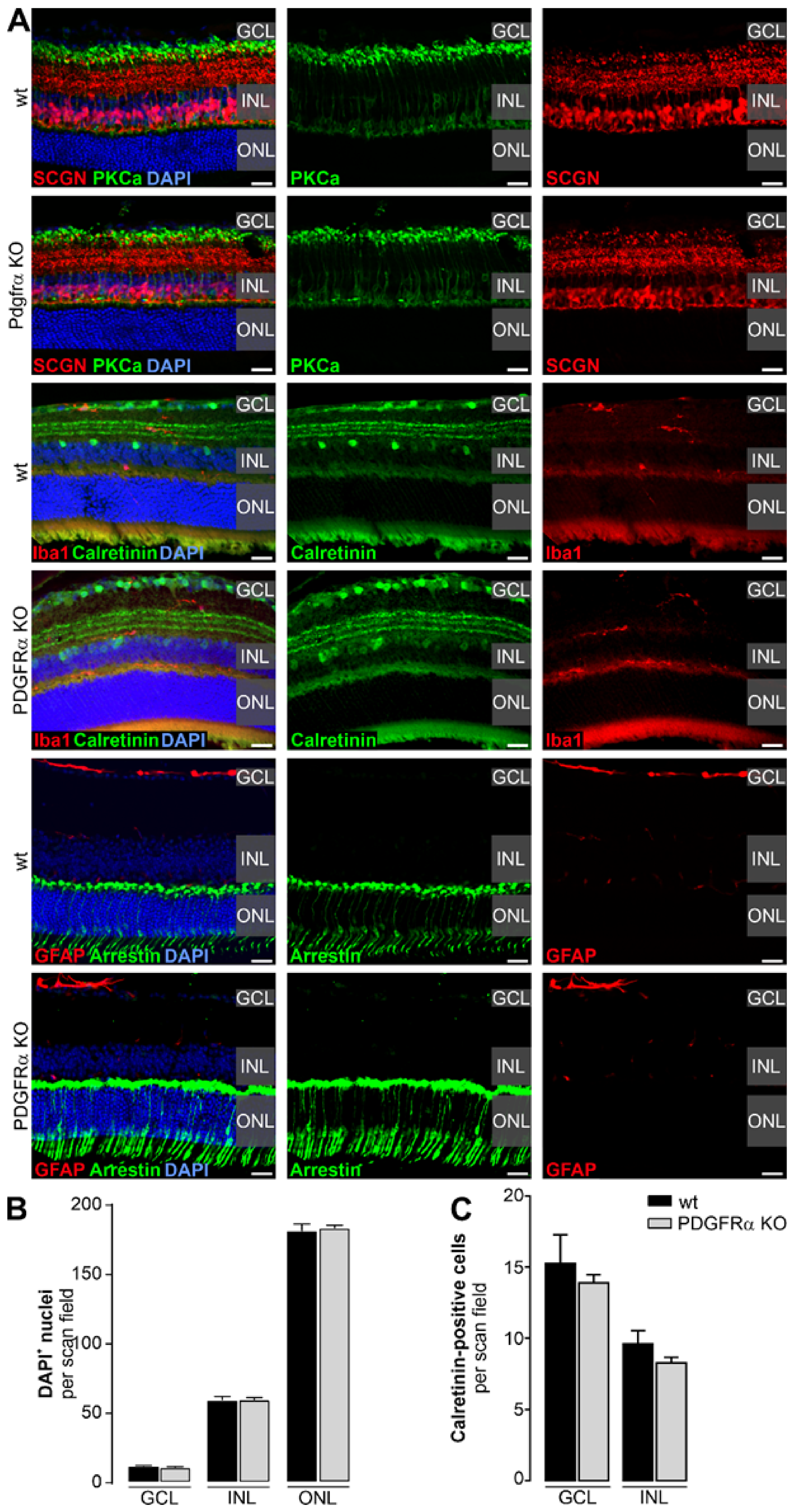
2.2. Normal Retinal Morphology in Müller Cell-Specific PDGFRα KO Mice
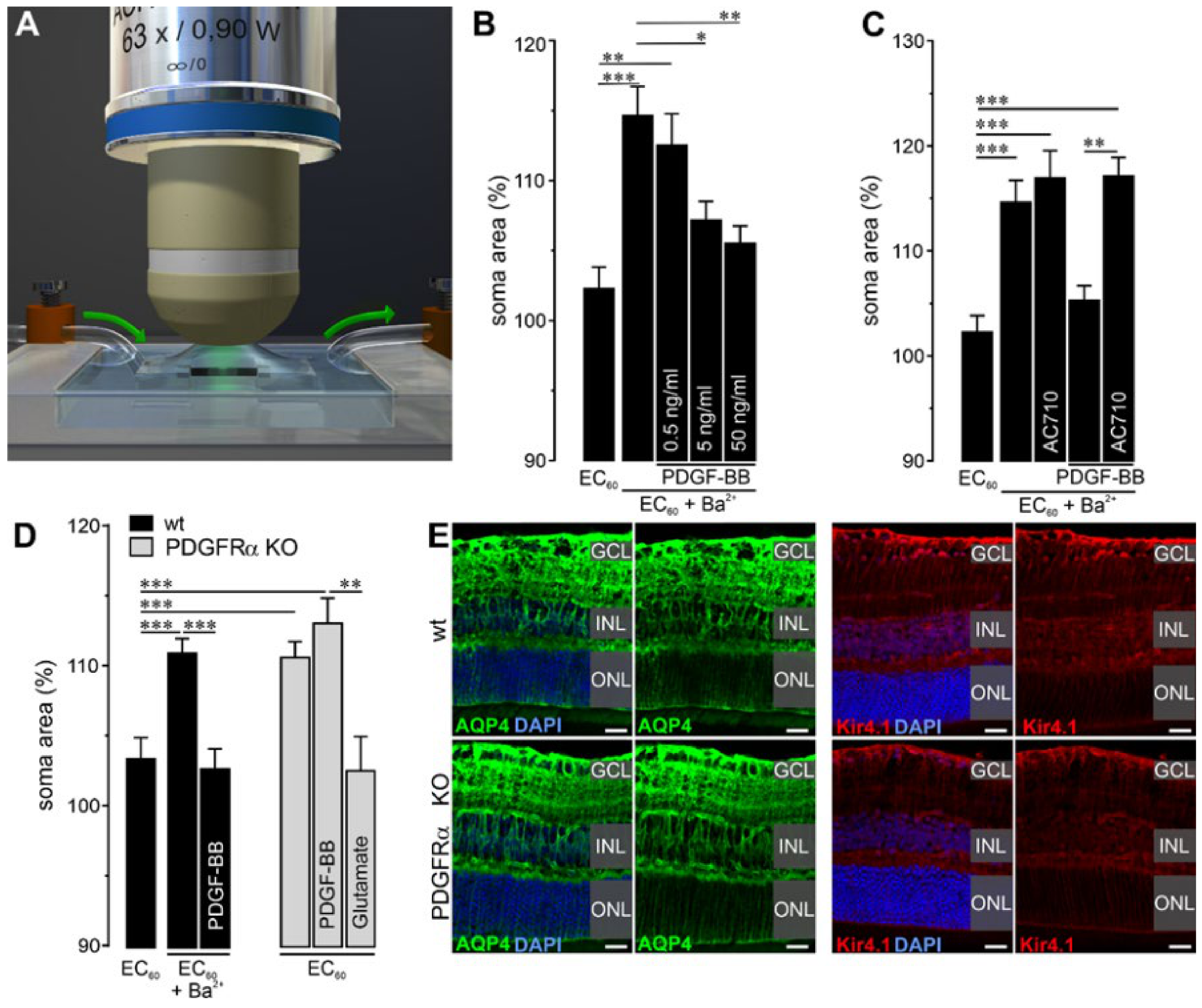
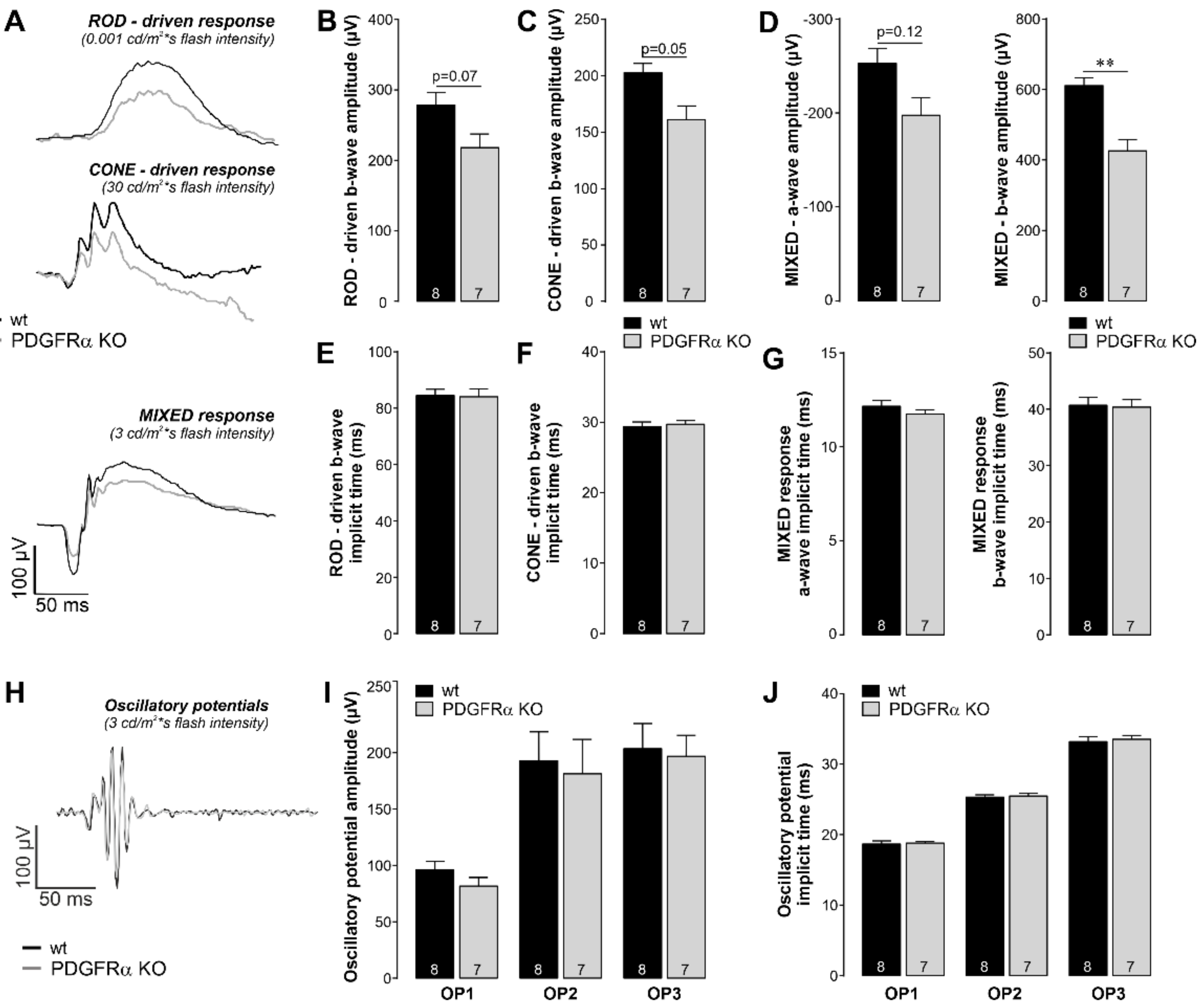
2.3. Functional Disruption in Retinae of Müller Cell-Specific PDGFRα KO Mice
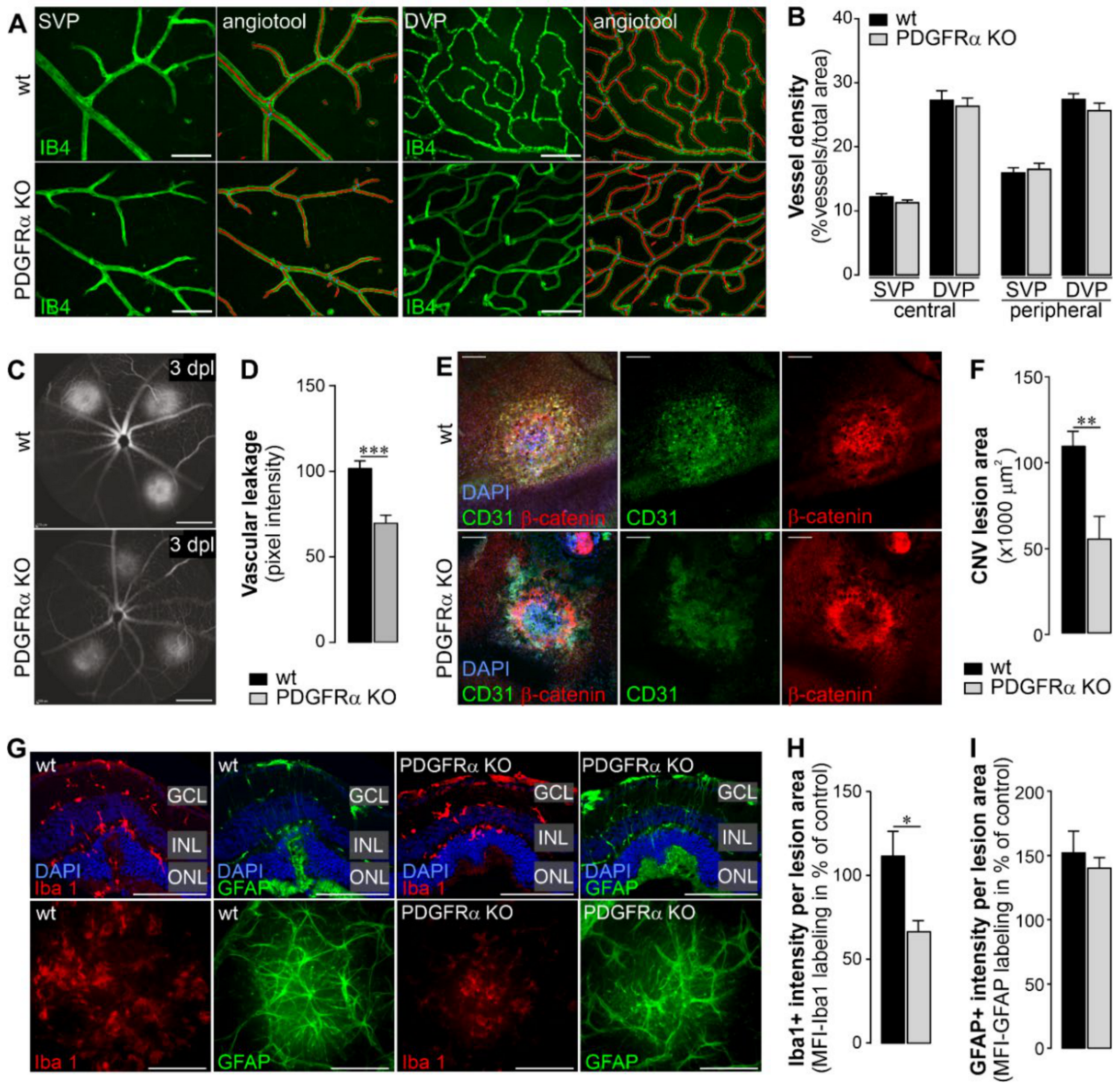
2.4. Vascular Effects of Müller Cell-Specific PDGFRα Knockout in Normal Retina and in a Model of Choroidal Neovascularization (CNV)
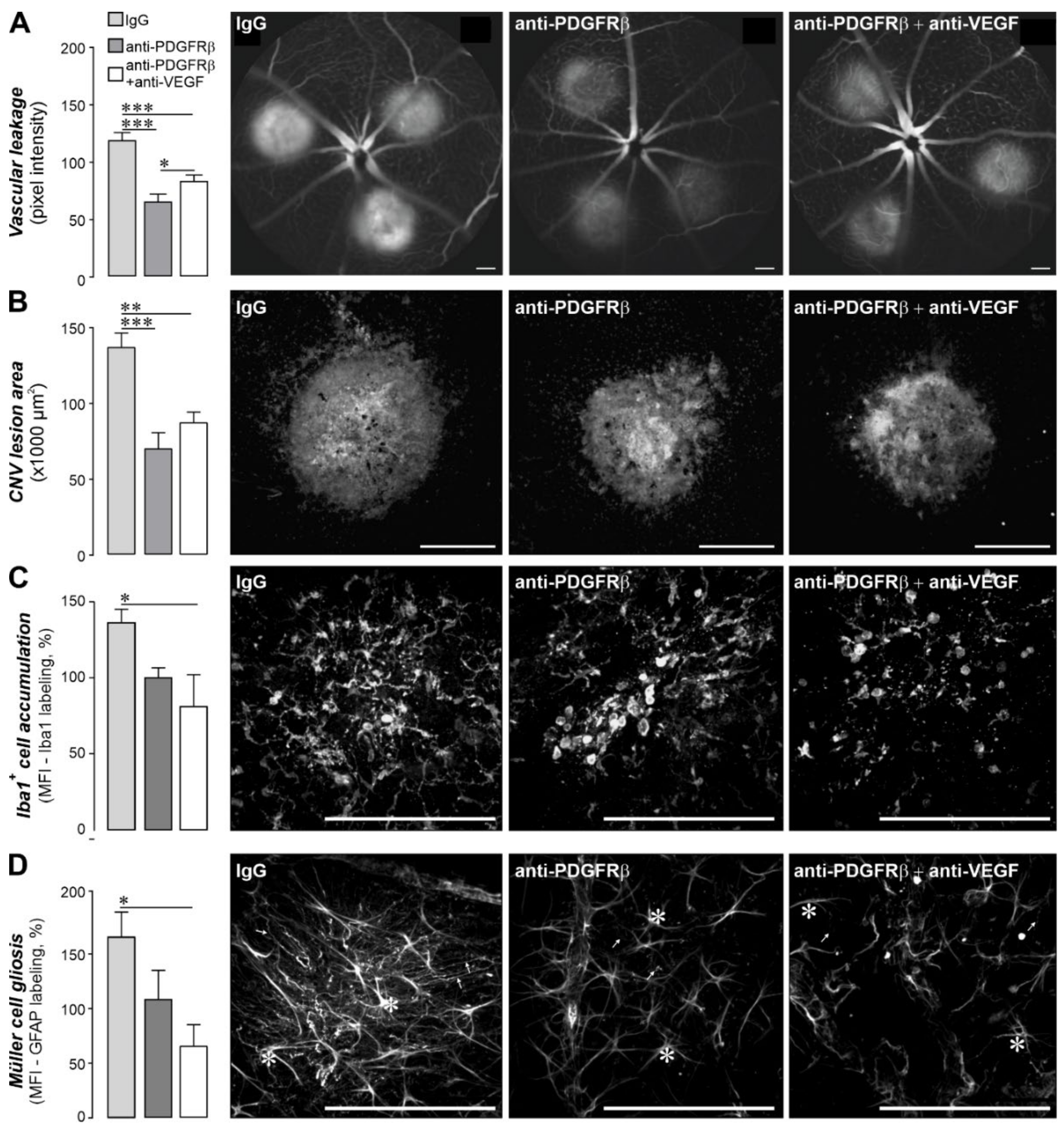
2.5. Pharmacological Inhibition of PDGF Signaling Had Similar Effects as the Müller Cell-Specific PDGFRα Knockout in the CNV Model
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Recombination Induced by Tamoxifen Administration
4.3. In Vivo Laser Model for Choroidal Neovascularization (CNV)
4.4. Intravitreal Drug Administration
4.5. Fluorescein Angiography (FA)
4.6. Immunohistochemical Analysis of Retinal Tissue and Image Analysis
4.7. Retinal Vasculature Labeling and Quantification
4.8. CNV Lesion Area Quantification
4.9. Müller Cell Reactivity and Macrophage/Microglia Accumulation Evaluation
4.10. Müller Cell Soma Swelling and Volume Regulation
4.11. Magnetic-Activated Cell Sorting (MACS) of Retinal Cell Types
4.12. Gene Expression Analyses
4.13. Electroretinogram Recording
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AQP4 | aquaporin 4 |
| CNS | central nervous system |
| CNV | choroidal neovascularization |
| ERG | electroretinogram |
| GCL | ganglion cell layer |
| IPL | inner plexiform layer |
| INL | inner nuclear layer |
| OPL | outer plexiform layer |
| ONL | outer nuclear layer |
| PDGF | platelet-derived growth factor |
| PDGFR | platelet-derived growth factor receptor |
| PDGFRα KO | Müller cell-specific platelet-derived growth factor receptor alpha knockout |
| RPE | retinal pigment epithelium |
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VEGF | vascular endothelial growth factor |
| wt | wildtype |
References
- Hammes, H.P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joussen, A.M. Molecular pathogenesis of ocular vascular disease—Anti-angiogenesis as a therapeutic concept. Dtsch. Med. Wochenschr. 2007, 132, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, S.; Chekarova, I.; Ejaz, A.; Sohail, A.; Lim, C.W. Importance of pericytes and mechanisms of pericyte loss during diabetes retinopathy. Diabetes Obes. Metab. 2008, 10, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Kim, L.A.; D’Amore, P.A. A brief history of anti-VEGF for the treatment of ocular angiogenesis. Am. J. Pathol. 2012, 181, 376–379. [Google Scholar] [CrossRef] [Green Version]
- Jo, N.; Mailhos, C.; Ju, M.; Cheung, E.; Bradley, J.; Nishijima, K.; Robinson, G.S.; Adamis, A.P.; Shima, D.T. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am. J. Pathol. 2006, 168, 2036–2053. [Google Scholar] [CrossRef] [Green Version]
- Spaide, R.F. Rationale for combination therapy in age-related macular degeneration. Retina 2009, 29, S5–S7. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Ciulla, T.A.; Ciardella, A.P.; Devin, F.; Dugel, P.U.; Eandi, C.M.; Masonson, H.; Mones, J.; Pearlman, J.A.; Quaranta-El Maftouhi, M.; et al. Dual Antagonism of PDGF and VEGF in Neovascular Age-Related Macular Degeneration: A Phase IIb, Multicenter, Randomized Controlled Trial. Ophthalmology 2017, 124, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Dong, A.; Seidel, C.; Snell, D.; Ekawardhani, S.; Ahlskog, J.K.; Baumann, M.; Shen, J.; Iwase, T.; Tian, J.; Stevens, R.; et al. Antagonism of PDGF-BB suppresses subretinal neovascularization and enhances the effects of blocking VEGF-A. Angiogenesis 2014, 17, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF is required for visual function: Evidence for a survival role on muller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [Green Version]
- Mudhar, H.S.; Pollock, R.A.; Wang, C.; Stiles, C.D.; Richardson, W.D. PDGF and its receptors in the developing rodent retina and optic nerve. Development 1993, 118, 539–552. [Google Scholar] [PubMed]
- Cox, O.T.; Simpson, D.A.; Stitt, A.W.; Gardiner, T.A. Sources of PDGF expression in murine retina and the effect of short-term diabetes. Mol. Vis. 2003, 9, 665–672. [Google Scholar] [PubMed]
- Biswas, S.K.; Zhao, Y.; Nagalingam, A.; Gardner, T.W.; Sandirasegarane, L. PDGF- and insulin/IGF-1-specific distinct modes of class IA PI 3-kinase activation in normal rat retinas and RGC-5 retinal ganglion cells. Investig. Ophthalmol Vis. Sci. 2008, 49, 3687–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Tout, S.; Chan-Ling, T.; Hollander, H.; Stone, J. The role of Muller cells in the formation of the blood-retinal barrier. Neuroscience 1993, 55, 291–301. [Google Scholar] [CrossRef]
- Ikuno, Y.; Hibino, S.; Bando, H.; Kawasaki, Y.; Nakamura, T.; Tano, Y. Retinal glial cells stimulate microvascular pericyte proliferation via fibroblast growth factor and platelet-derived growth factor in vitro. Jpn. J. Ophthalmol. 2002, 46, 413–418. [Google Scholar] [CrossRef]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef]
- Powner, M.B.; Gillies, M.C.; Tretiach, M.; Scott, A.; Guymer, R.H.; Hageman, G.S.; Fruttiger, M. Perifoveal muller cell depletion in a case of macular telangiectasia type 2. Ophthalmology 2010, 117, 2407–2416. [Google Scholar] [CrossRef] [Green Version]
- Powner, M.B.; Gillies, M.C.; Zhu, M.; Vevis, K.; Hunyor, A.P.; Fruttiger, M. Loss of Muller’s cells and photoreceptors in macular telangiectasia type 2. Ophthalmology 2013, 120, 2344–2352. [Google Scholar] [CrossRef]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellstrom, M.; Backstrom, G.; Fredriksson, S.; Landegren, U.; Nystrom, H.C.; et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003, 17, 1835–1840. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.S.; Okamoto, N.; Vinores, M.A.; Vinores, S.A.; Hackett, S.F.; Yamada, H.; Yamada, E.; Derevjanik, N.L.; LaRochelle, W.; Zack, D.J.; et al. Photoreceptor-specific expression of platelet-derived growth factor-B results in traction retinal detachment. Am. J. Pathol. 2000, 157, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Vinores, S.A.; Seo, M.S.; Derevjanik, N.L.; Campochiaro, P.A. Photoreceptor-specific overexpression of platelet-derived growth factor induces proliferation of endothelial cells, pericytes, and glial cells and aberrant vascular development: An ultrastructural and immunocytochemical study. Brain Res. Dev. Brain Res. 2003, 140, 169–183. [Google Scholar] [CrossRef]
- Edqvist, P.H.; Niklasson, M.; Vidal-Sanz, M.; Hallbook, F.; Forsberg-Nilsson, K. Platelet-derived growth factor over-expression in retinal progenitors results in abnormal retinal vessel formation. PLoS ONE 2012, 7, e42488. [Google Scholar] [CrossRef]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosche, A.; Hauser, A.; Lepper, M.F.; Mayo, R.; von Toerne, C.; Merl-Pham, J.; Hauck, S.M. The Proteome of Native Adult Muller Glial Cells from Murine Retina. Mol. Cell Proteom. 2016, 15, 462–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mages, K.; Grassmann, F.; Jagle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflammation 2019, 16, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurm, A.; Pannicke, T.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Glial cell-derived glutamate mediates autocrine cell volume regulation in the retina: Activation by VEGF. J. Neurochem. 2008, 104, 386–399. [Google Scholar] [CrossRef]
- Bringmann, A.; Faude, F.; Reichenbach, A. Mammalian retinal glial (Muller) cells express large-conductance Ca(2+)-activated K+ channels that are modulated by Mg2+ and pH and activated by protein kinase A. Glia 1997, 19, 311–323. [Google Scholar] [CrossRef]
- Kofuji, P.; Biedermann, B.; Siddharthan, V.; Raap, M.; Iandiev, I.; Milenkovic, I.; Thomzig, A.; Veh, R.W.; Bringmann, A.; Reichenbach, A. Kir potassium channel subunit expression in retinal glial cells: Implications for spatial potassium buffering. Glia 2002, 39, 292–303. [Google Scholar] [CrossRef]
- Nagelhus, E.A.; Horio, Y.; Inanobe, A.; Fujita, A.; Haug, F.M.; Nielsen, S.; Kurachi, Y.; Ottersen, O.P. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 1999, 26, 47–54. [Google Scholar] [CrossRef]
- Weymouth, A.E.; Vingrys, A.J. Rodent electroretinography: Methods for extraction and interpretation of rod and cone responses. Prog. Retin. Eye Res. 2008, 27, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Gambardella, L.; Kurcz, C.; Vermeren, S. A computational tool for quantitative analysis of vascular networks. PLoS ONE 2011, 6, e27385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balser, C.; Wolf, A.; Herb, M.; Langmann, T. Co-inhibition of PGF and VEGF blocks their expression in mononuclear phagocytes and limits neovascularization and leakage in the murine retina. J. Neuroinflammation 2019, 16, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, M.A.; Hanout, M.; Sarwar, S.; Hassan, M.; Agarwal, A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Platelet-Derived Growth Factor Inhibitors: A Potential Therapeutic Approach for Ocular Neovascularization. Dev. Ophthalmol. 2016, 55, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Takahama, S.; Adetunji, M.O.; Zhao, T.; Chen, S.; Li, W.; Tomarev, S.I. Retinal Astrocytes and GABAergic Wide-Field Amacrine Cells Express PDGFRalpha: Connection to Retinal Ganglion Cell Neuroprotection by PDGF-AA. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4703–4711. [Google Scholar] [CrossRef] [Green Version]
- Kanamoto, T.; Rimayanti, U.; Okumuchi, H.; Kiuchi, Y. Platelet-Derived Growth Factor Receptor Alpha Is Associated with Oxidative Stress-Induced Retinal Cell Death. Curr. Eye Res. 2011, 36, 336–340. [Google Scholar] [CrossRef]
- Velez, G.; Weingarden, A.R.; Tucker, B.A.; Lei, H.; Kazlauskas, A.; Young, M.J. Retinal Pigment Epithelium and Müller Progenitor Cell Interaction Increase Müller Progenitor Cell Expression of PDGFR. Stem Cells Int. 2012, 2012, 106486. [Google Scholar] [CrossRef] [Green Version]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef] [Green Version]
- Leveen, P.; Pekny, M.; Gebre-Medhin, S.; Swolin, B.; Larsson, E.; Betsholtz, C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994, 8, 1875–1887. [Google Scholar] [CrossRef] [Green Version]
- Enge, M.; Bjarnegard, M.; Gerhardt, H.; Gustafsson, E.; Kalen, M.; Asker, N.; Hammes, H.P.; Shani, M.; Fassler, R.; Betsholtz, C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002, 21, 4307–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadeja, S.; Mort, R.L.; Keighren, M.; Hart, A.W.; Joynson, R.; Wells, S.; Potter, P.K.; Jackson, I.J. A CNS-specific hypomorphic Pdgfr-beta mutant model of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3569–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruttiger, M.; Calver, A.R.; Kruger, W.H.; Mudhar, H.S.; Michalovich, D.; Takakura, N.; Nishikawa, S.; Richardson, W.D. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron 1996, 17, 1117–1131. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Smith, L.E.H. Retinal Vasculature in Development and Diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122. [Google Scholar] [CrossRef]
- Zhang, X.; Serb, J.M.; Greenlee, M.H. Mouse retinal development: A dark horse model for systems biology research. Bioinform. Biol. Insights 2011, 5, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Amini, R.; Rocha-Martins, M.; Norden, C. Neuronal Migration and Lamination in the Vertebrate Retina. Front. Neurosci. 2017, 11, 742. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Muller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Weuste, M.; Wurm, A.; Iandiev, I.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. HB-EGF: Increase in the ischemic rat retina and inhibition of osmotic glial cell swelling. Biochem. Biophys. Res. Commun. 2006, 347, 310–318. [Google Scholar] [CrossRef]
- Pannicke, T.; Frommherz, I.; Biedermann, B.; Wagner, L.; Sauer, K.; Ulbricht, E.; Hartig, W.; Krugel, U.; Ueberham, U.; Arendt, T.; et al. Differential effects of P2Y1 deletion on glial activation and survival of photoreceptors and amacrine cells in the ischemic mouse retina. Cell Death Dis. 2014, 5, e1353. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Harada, C.; Watanabe, M.; Inoue, Y.; Sakagawa, T.; Nakayama, N.; Sasaki, S.; Okuyama, S.; Watase, K.; Wada, K.; et al. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc. Natl. Acad. Sci. USA 1998, 95, 4663–4666. [Google Scholar] [CrossRef] [Green Version]
- Pannicke, T.; Wurm, A.; Iandiev, I.; Hollborn, M.; Linnertz, R.; Binder, D.K.; Kohen, L.; Wiedemann, P.; Steinhauser, C.; Reichenbach, A.; et al. Deletion of aquaporin-4 renders retinal glial cells more susceptible to osmotic stress. J. Neurosci. Res. 2010, 88, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Patil, R.V.; Verkman, A.S. Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Investig. Ophthalmol. Vis. Sci. 2002, 43, 573–579. [Google Scholar]
- You, Y.; Zhu, L.; Zhang, T.; Shen, T.; Fontes, A.; Yiannikas, C.; Parratt, J.; Barton, J.; Schulz, A.; Gupta, V.; et al. Evidence of Muller Glial Dysfunction in Patients with Aquaporin-4 Immunoglobulin G-Positive Neuromyelitis Optica Spectrum Disorder. Ophthalmology 2019, 126, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowell, R.E.; Barabas, P.; Augustine, J.; Chevallier, O.; McCarron, P.; Chen, M.; McGeown, J.G.; Curtis, T.M. Müller glial dysfunction during diabetic retinopathy in rats is reduced by the acrolein-scavenging drug, 2-hydrazino-4,6-dimethylpyrimidine. Diabetologia 2018, 61, 2654–2667. [Google Scholar] [CrossRef] [Green Version]
- Wagner, L.; Pannicke, T.; Rupprecht, V.; Frommherz, I.; Volz, C.; Illes, P.; Hirrlinger, J.; Jagle, H.; Egger, V.; Haydon, P.G.; et al. Suppression of SNARE-dependent exocytosis in retinal glial cells and its effect on ischemia-induced neurodegeneration. Glia 2017, 65, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Cabrera-Ghayouri, S.; Christie, L.A.; Held, K.S.; Viswanath, V. Translational Preclinical Pharmacologic Disease Models for Ophthalmic Drug Development. Pharm. Res. 2019, 36, 58. [Google Scholar] [CrossRef] [Green Version]
- Balaggan, K.S.; Binley, K.; Esapa, M.; MacLaren, R.E.; Iqball, S.; Duran, Y.; Pearson, R.A.; Kan, O.; Barker, S.E.; Smith, A.J.; et al. EIAV vector-mediated delivery of endostatin or angiostatin inhibits angiogenesis and vascular hyperpermeability in experimental CNV. Gene Ther. 2006, 13, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Gong, Y.; Liegl, R.; Wang, Z.; Liu, C.H.; Meng, S.S.; Burnim, S.B.; Saba, N.J.; Fredrick, T.W.; Morss, P.C.; et al. FGF21 Administration Suppresses Retinal and Choroidal Neovascularization in Mice. Cell Rep. 2017, 18, 1606–1613. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Walsh, A.C.; Muldrew, A.; Updike, P.G.; Barbour, T.; Sadda, S.R. Quantitative fluorescein angiographic analysis of choroidal neovascular membranes: Validation and correlation with visual function. Investig. Ophthalmol. Vis. Sci. 2007, 48, 349–354. [Google Scholar] [CrossRef]
- Berger, J.W.; Yoken, J. Computer-assisted quantitation of choroidal neovascularization for clinical trials. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2286–2295. [Google Scholar]
- Zhou, L.; Sun, X.; Huang, Z.; Zhou, T.; Zhu, X.; Liu, Y.; Wang, J.; Cheng, B.; Li, M.; He, C.; et al. Imatinib Ameliorated Retinal Neovascularization by Suppressing PDGFR-alpha and PDGFR-beta. Cell Physiol. Biochem. 2018, 48, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, P.; Kokona, D.; Ebneter, A.; Zinkernagel, M.S. Effect of Inhibition of Colony-Stimulating Factor 1 Receptor on Choroidal Neovascularization in Mice. Am. J. Pathol. 2020, 190, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Parlier, R.; Shen, J.K.; Lutty, G.A.; Vinores, S.A. VEGF receptor blockade markedly reduces retinal microglia/macrophage infiltration into laser-induced CNV. PLoS ONE 2013, 8, e71808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tah, V.; Orlans, H.O.; Hyer, J.; Casswell, E.; Din, N.; Sri Shanmuganathan, V.; Ramskold, L.; Pasu, S. Anti-VEGF Therapy and the Retina: An Update. J. Ophthalmol. 2015, 2015, 627674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K.; Group, S.-U.S. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Bhisitkul, R.B.; Desai, S.J.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Fellow Eye Comparisons for 7-Year Outcomes in Ranibizumab-Treated AMD Subjects from ANCHOR, MARINA, and HORIZON (SEVEN-UP Study). Ophthalmology 2016, 123, 1269–1277. [Google Scholar] [CrossRef]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, J.L.; Fawzi, A.A.; Khondkaryan, A.; Lam, L.; Chong, L.P.; Eliott, D.; Walsh, A.C.; Hwang, J.; Sadda, S.R. Bevacizumab and ranibizumab tachyphylaxis in the treatment of choroidal neovascularisation. Br. J. Ophthalmol. 2012, 96, 14–20. [Google Scholar] [CrossRef]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.L.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef]
- Askou, A.L. Development of gene therapy for treatment of age-related macular degeneration. Acta Ophthalmol. 2014, 92, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.A.; Nettesheim, E.R.; Connor, T.B.; Lipinski, D.M. Development of an inducible anti-VEGF rAAV gene therapy strategy for the treatment of wet AMD. Sci. Rep. 2018, 8, 11763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, T.A.C.; Georgiou, M.; Bainbridge, J.W.B.; Michaelides, M. Gene therapy for neovascular age-related macular degeneration: Rationale, clinical trials and future directions. Br. J. Ophthalmol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Slezak, M.; Goritz, C.; Niemiec, A.; Frisen, J.; Chambon, P.; Metzger, D.; Pfrieger, F.W. Transgenic mice for conditional gene manipulation in astroglial cells. Glia 2007, 55, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Luckoff, A.; Scholz, R.; Sennlaub, F.; Xu, H.; Langmann, T. Comprehensive analysis of mouse retinal mononuclear phagocytes. Nat. Protoc. 2017, 12, 1136–1150. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Slezak, M.; Grosche, A.; Niemiec, A.; Tanimoto, N.; Pannicke, T.; Munch, T.A.; Crocker, B.; Isope, P.; Hartig, W.; Beck, S.C.; et al. Relevance of exocytotic glutamate release from retinal glia. Neuron 2012, 74, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Uckermann, O.; Iandiev, I.; Francke, M.; Franze, K.; Grosche, J.; Wolf, S.; Kohen, L.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Selective staining by vital dyes of Muller glial cells in retinal wholemounts. Glia 2004, 45, 59–66. [Google Scholar] [CrossRef]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef] [Green Version]
- Pauly, D.; Agarwal, D.; Dana, N.; Schafer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell Rep. 2019, 29, 2835–2848. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Feuer, W.J. Lessons from Recent Phase III Trial Failures: Don’t Design Phase III Trials Based on Retrospective Subgroup Analyses from Phase II Trials. Ophthalmology 2018, 125, 1488–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Primer Sequences: Forward | Primer Sequences: Reverse | TaqMan® Probe from Roche | Accession Number |
|---|---|---|---|---|
| idh3b | 5′ gctgcggcatctcaatct 3′ | 5′ ccatgtctcgagtccgtacc 3′ | # 67 | NM_130884.4 |
| aif1 | 5′ atctgccgtccaaacttga 3′ | 5′ ctaggtgggtcttgggaacc 3′ | # 67 | NM_001361501.1 |
| glul | 5′gcccaagtgtgtggaagag 3′ | 5′aaggggtctcgaaacatgg 3′ | # 58 | NM_008131.4 |
| nrl | 5′ tgcctttctggttctgacagt 3′ | 5′ gaaagccattctgggactga 3′ | # 53 | NM_008736.3 |
| pecam1 | 5′ gctggtgctctatgcaagc 3′ | 5′ atggatgctgttgatggtga 3′ | # 64 | NM_008816.3 |
| pdgfra | 5′ cagacattgaccctgttcca 3′ | 5′ tctcttccgaagtctgtgagc 3′ | # 69 | NM_001083316.2 |
| pdgfrb | 5′ tcaagctgcaggtcaatgtc 3′ | 5′ ccattggcagggtgactc 3′ | # 67 | NM_001146268.1 |
| pdgfb | 5′ cggcctgtgactagaagtcc 3′ | 5′ gagcttgaggcgtcttgg 3′ | # 32 | NM_011057.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Lezama, N.; Wolf, A.; Koch, S.; Pfaller, A.M.; Biber, J.; Guillonneau, X.; Langmann, T.; Grosche, A. PDGF Receptor Alpha Signaling Is Key for Müller Cell Homeostasis Functions. Int. J. Mol. Sci. 2021, 22, 1174. https://doi.org/10.3390/ijms22031174
Díaz-Lezama N, Wolf A, Koch S, Pfaller AM, Biber J, Guillonneau X, Langmann T, Grosche A. PDGF Receptor Alpha Signaling Is Key for Müller Cell Homeostasis Functions. International Journal of Molecular Sciences. 2021; 22(3):1174. https://doi.org/10.3390/ijms22031174
Chicago/Turabian StyleDíaz-Lezama, Nundehui, Anne Wolf, Susanne Koch, Anna M. Pfaller, Josef Biber, Xavier Guillonneau, Thomas Langmann, and Antje Grosche. 2021. "PDGF Receptor Alpha Signaling Is Key for Müller Cell Homeostasis Functions" International Journal of Molecular Sciences 22, no. 3: 1174. https://doi.org/10.3390/ijms22031174