Homozygous HESX1 and COL1A1 Gene Variants in a Boy with Growth Hormone Deficiency and Early Onset Osteoporosis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
Clinical Description
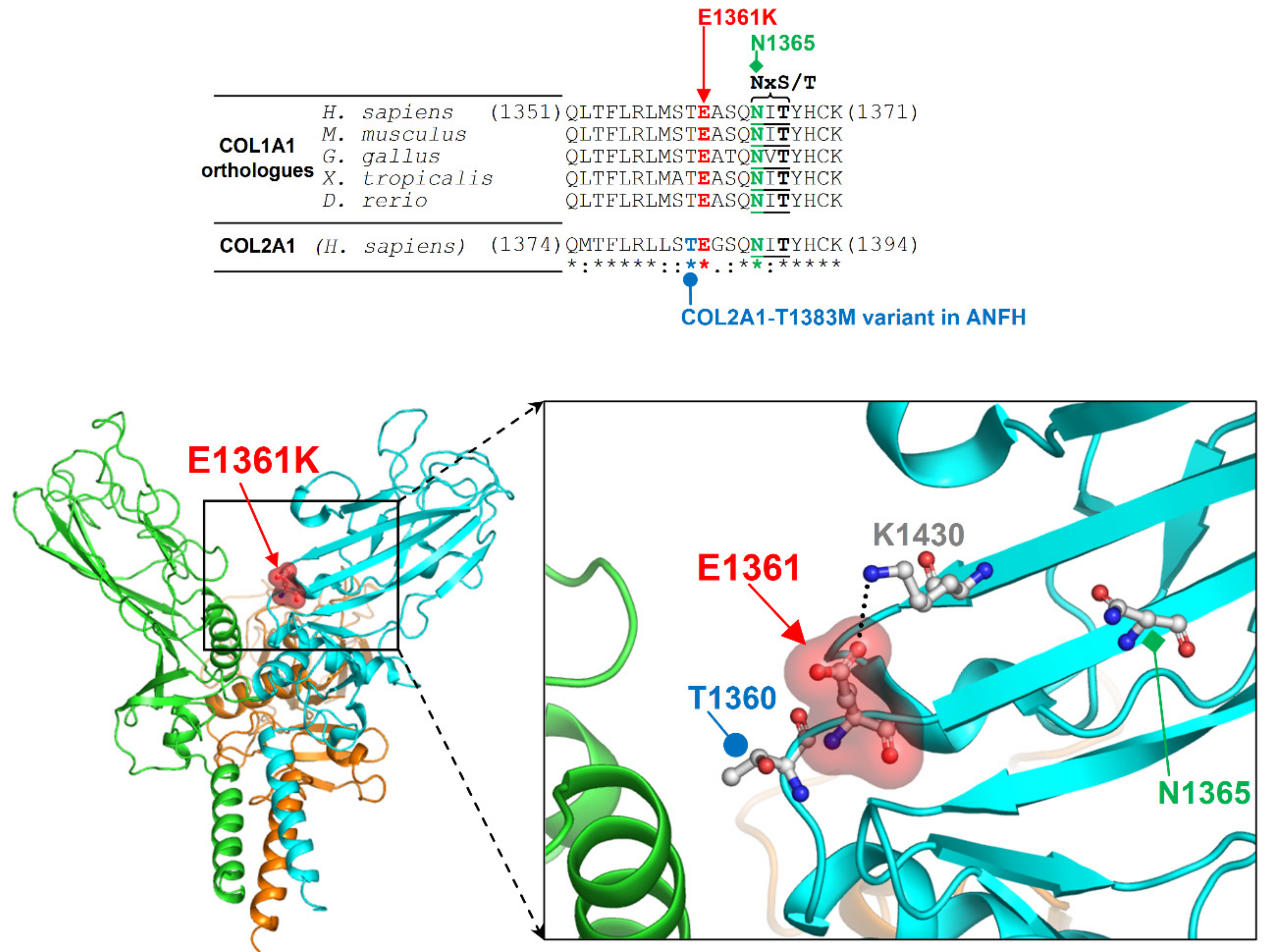
2. Results
3. Discussion
4. Materials and Methods
4.1. SNP-Array Analysis (Single Nucleotide Polymorphism–Array Analysis)
4.2. Target Exome Sequencing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alesi, V.; Capolino, R.; Genovese, S.; Capriati, T.; Loddo, S.; Calvieri, G.; Calacci, C.; Diociaiuti, A.; Diamanti, A.; Novelli, A.; et al. An additional patient with a homozygous mutation in DCPS contributes to the delination of Al-Raqad syndrome. Am. J. Med Genet. Part A 2018, 176, 2781–2786. [Google Scholar] [CrossRef] [PubMed]
- Harripaul, R.; Vasli, N.; Mikhailov, A.; Rafiq, M.A.; Mittal, K.; Windpassinger, C.; Sheikh, T.I.; Noor, A.; Mahmood, H.; Downey, S.; et al. Mapping autosomal recessive intellectual disability: Combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol. Psychiatry 2017, 23, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, G.; Aversa, T.; Zirilli, G.; De Luca, F.; Gallizzi, R.; Wasniewska, M. Congenital hypopituitarism: How to select the patients for genetic analyses. Ital. J. Pediatr. 2018, 44, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangat, M.; Radovick, S. Pituitary Hypoplasia. Endocrinol. Metab. Clin. N. Am. 2017, 46, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M. Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, R.E.; Costantini, A.; Kämpe, A.; Alm, J.J.; Mäkitie, O. New Insights into Monogenic Causes of Osteoporosis. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.Q.; Dattani, M.T.; Brickman, J.M.; McNay, D.E.G.; Warne, G.L.; Zacharin, M.; Cameron, F.; Hurst, J.; Woods, K.; Dunger, D.B.; et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum. Mol. Genet. 2001, 10, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.; Carrique, L.; Goff, S.V.-L.; Mariano, N.; Georges, R.-N.; Delolme, F.; Koivunen, P.; Myllyharju, J.; Moali, C.; Aghajari, N.; et al. Structural basis of homo- and heterotrimerization of collagen I. Nat. Commun. 2017, 8, 14671. [Google Scholar] [CrossRef] [PubMed]
- Kannu, P.; O’Rielly, D.; Hyland, J.; Kokko, L.A. Avascular necrosis of the femoral head due to a novel C propeptide mutation in COL2A1. Am. J. Med. Genet. Part A 2011, 155, 1759–1762. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.R.; Woods, K.S.; Mendonca, B.B.; Marcal, N.; Zamparini, A.L.; Stifani, S.; Brickman, J.M.; Arnhold, I.J.; Dattani, M.T. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. J. Clin. Investig. 2003, 112, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Cohen, R.N.; Cohen, L.E.; Botero, D.; Yu, C.; Sagar, A.; Jurkiewicz, M.; Radovick, S. Enhanced Repression by HESX1 as a Cause of Hypopituitarism and Septooptic Dysplasia. J. Clin. Endocrinol. Metab. 2003, 88, 4832–4839. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Hattorri, T.; Nakajima, T.; Okuhara, K.; Sato, K.; Abe, S.; Nakae, J.; Fujieda, K. Sporadic Heterozygous Frameshift Mutation ofHESX1Causing Pituitary and Optic Nerve Hypoplasia and Combined Pituitary Hormone Deficiency in a Japanese Patient. J. Clin. Endocrinol. Metab. 2003, 88, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Q.; Benedetti, A.F.F.; Ma, Q.; Gregory, L.; Li, J.Z.; Dattani, M.; Sadeghi-Nejad, A.; Arnhold, I.J.; De Mendonça, B.B.; Camper, S.A.; et al. HESX1mutations in patients with congenital hypopituitarism: Variable phenotypes with the same genotype. Clin. Endocrinol. 2016, 85, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattani, M.T.; Martinez-Barbera, J.-P.; Thomas, P.Q.; Brickman, J.M.; Gupta, R.; Mårtensson, I.-L.; Toresson, H.; Fox, M.; Wales, J.K.H.; Hindmarsh, P.C.; et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat. Genet. 1998, 19, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Corneli, G.; Vivenza, D.; Prodam, F.; Di Dio, G.; Vottero, A.; Rapa, A.; Bellone, S.; Bernasconi, S.; Bona, G. Heterozygous mutation of HESX1 causing hypopituitarism and multiple anatomical malformations without features of septo-optic dysplasia. J. Endocrinol. Investig. 2008, 31, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Newbern, K.; Natrajan, N.; Kim, H.-G.; Chorich, L.P.; Halvorson, L.M.; Cameron, R.S.; Layman, L.C. Identification of HESX1 mutations in Kallmann syndrome. Fertil. Steril. 2013, 99, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- De Rienzo, F.; Mellone, S.; Bellone, S.; Babu, D.; Fusco, I.; Prodam, F.; Petri, A.; Muniswamy, R.; De Luca, F.; Salerno, M.; et al. Italian Study Group on Genetics of CPHD. Frequency of genetic defects in combined pituitary hormone deficiency: A systematic review and analysis of a multicentre Italian cohort. Clin. Endocrinol. 2015, 83, 849–860. [Google Scholar] [CrossRef]
- Barnes, A.M.; Ashok, A.; Makareeva, E.N.; Brusel, M.; Cabral, W.A.; Weis, M.; Moali, C.; Bettler, E.; Eyre, D.R.; Cassella, J.P.; et al. COL1A1 C-propeptide mutations cause ER mislocalization of procollagen and impair C-terminal procollagen processing. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2210–2223. [Google Scholar] [CrossRef]
- Symoens, S.; Hulmes, D.J.S.; Bourhis, J.-M.; Coucke, P.J.; De Paepe, A.; Malfait, F. Type I Procollagen C-Propeptide Defects: Study of Genotype-Phenotype Correlation and Predictive Role of Crystal Structure. Hum. Mutat. 2014, 35, 1330–1341. [Google Scholar] [CrossRef]
- Nicholls, A.C.; Osse, G.; Schloon, H.G.; Lenard, H.G.; Deak, S.; Myers, J.C.; Prockop, D.J.; Weigel, W.R.; Fryer, P.; Pope, F.M. The clinical features of homozygous alpha 2(I) collagen deficient osteogenesis imperfecta. J. Med. Genet. 1984, 21, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Pihlajaniemi, T.; Dickson, L.A.; Pope, F.M.; Korhonen, V.R.; Nicholls, A.; Prockop, D.J.; Myers, J.C. Osteogenesis imperfecta: Cloning of a pro-alpha 2(I) collagen gene with a frameshift mutation. J. Biol. Chem. 1984, 259, 12941–12944. [Google Scholar] [CrossRef]
- Spotila, L.D.; Sereda, L.; Prockop, D.J. Partial isodisomy for maternal chromosome 7 and short stature in an individual with a mutation at the COL1A2 locus. Am. J. Hum. Genet. 1992, 51, 1396–1405. [Google Scholar] [PubMed]
- Marini, J.; Lewis, M.; Wang, Q.; Chen, K.; Orrison, B. Serine for glycine substitutions in type I collagen in two cases of type IV osteogenesis imperfecta (OI). Additional evidence for a regional model of OI pathophysiology. J. Biol. Chem. 1993, 268, 2667–2673. [Google Scholar] [CrossRef]
- De Paepe, A.; Nuytinck, L.; Raes, M.; Fryns, J.-P. Homozygosity by descent for a COL1A2 mutation in two sibs with severe osteogenesis imperfecta and mild clinical expression in the heterozygotes. Qual. Life Res. 1997, 99, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Byers, P.H.; Pyott, S.M. Recessively Inherited Forms of Osteogenesis Imperfecta. Annu. Rev. Genet. 2012, 46, 475–497. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Tournis, S.; Kämpe, A.; Ain, N.U.; Taylan, F.; Doulgeraki, A.; Mäkitie, O. Autosomal Recessive Osteogenesis Imperfecta Caused by a Novel Homozygous COL1A2 Mutation. Calcif. Tissue Int. 2018, 103, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udomchaiprasertkul, W.; Kuptanon, C.; Porntaveetus, T.; Shotelersuk, V. A family with homozygous and heterozygous p.Gly337Ser mutations in COL1A2. Eur. J. Med. Genet. 2020, 63, 103896. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Authors | Status Variant (18G > C, Q6H) | Family Unaffected Heterozygous Carriers | Pituitary Gland | GH | TSH | ACTH | LH/FSH | Optic Nerve | Corpus Callosum Septum Pellucidum | Other Clinical Features |
|---|---|---|---|---|---|---|---|---|---|---|
| Thomas 2001 | Heterozygous | Father | Anterior pituitary hypoplasia and ectopic posterios pituitary | Deficient | Deficient by age of 7 years | Normal | Deficient | Normal | Normal | Small penis |
| Corneli 2007 (also described by Vivenza 2011 and De Rienzo 2015) | Heterozygous | Mother | Anterior pituitary hypoplasia and ectopic posterior pituitary | Deficient | Deficient | Deficient | Prepubertal at diagnosis | Normal | Normal | Narrow biparietal diameter, mandibular hypoplasia, bilateral cryptorchidism, micropenis |
| Newbern 2013 | Heterozygous | Unknown | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported | Not reported | Idiopathic hypogonadotropic hypogonadism, anosmia |
| Our patient | Homozygous | Father, mother, two sisters | Normal | Deficient | Normal | Normal | Normal | Normal | Corpus callosum hypoplasia | Low-grade glioma in the right cerebral peduncle, spastic paraparesis, osteoporosis, scoliosis, pubertal retardation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alesi, V.; Dentici, M.L.; Genovese, S.; Loddo, S.; Bellacchio, E.; Orlando, V.; Di Tommaso, S.; Catino, G.; Calacci, C.; Calvieri, G.; et al. Homozygous HESX1 and COL1A1 Gene Variants in a Boy with Growth Hormone Deficiency and Early Onset Osteoporosis. Int. J. Mol. Sci. 2021, 22, 750. https://doi.org/10.3390/ijms22020750
Alesi V, Dentici ML, Genovese S, Loddo S, Bellacchio E, Orlando V, Di Tommaso S, Catino G, Calacci C, Calvieri G, et al. Homozygous HESX1 and COL1A1 Gene Variants in a Boy with Growth Hormone Deficiency and Early Onset Osteoporosis. International Journal of Molecular Sciences. 2021; 22(2):750. https://doi.org/10.3390/ijms22020750
Chicago/Turabian StyleAlesi, Viola, Maria Lisa Dentici, Silvia Genovese, Sara Loddo, Emanuele Bellacchio, Valeria Orlando, Silvia Di Tommaso, Giorgia Catino, Chiara Calacci, Giusy Calvieri, and et al. 2021. "Homozygous HESX1 and COL1A1 Gene Variants in a Boy with Growth Hormone Deficiency and Early Onset Osteoporosis" International Journal of Molecular Sciences 22, no. 2: 750. https://doi.org/10.3390/ijms22020750