Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
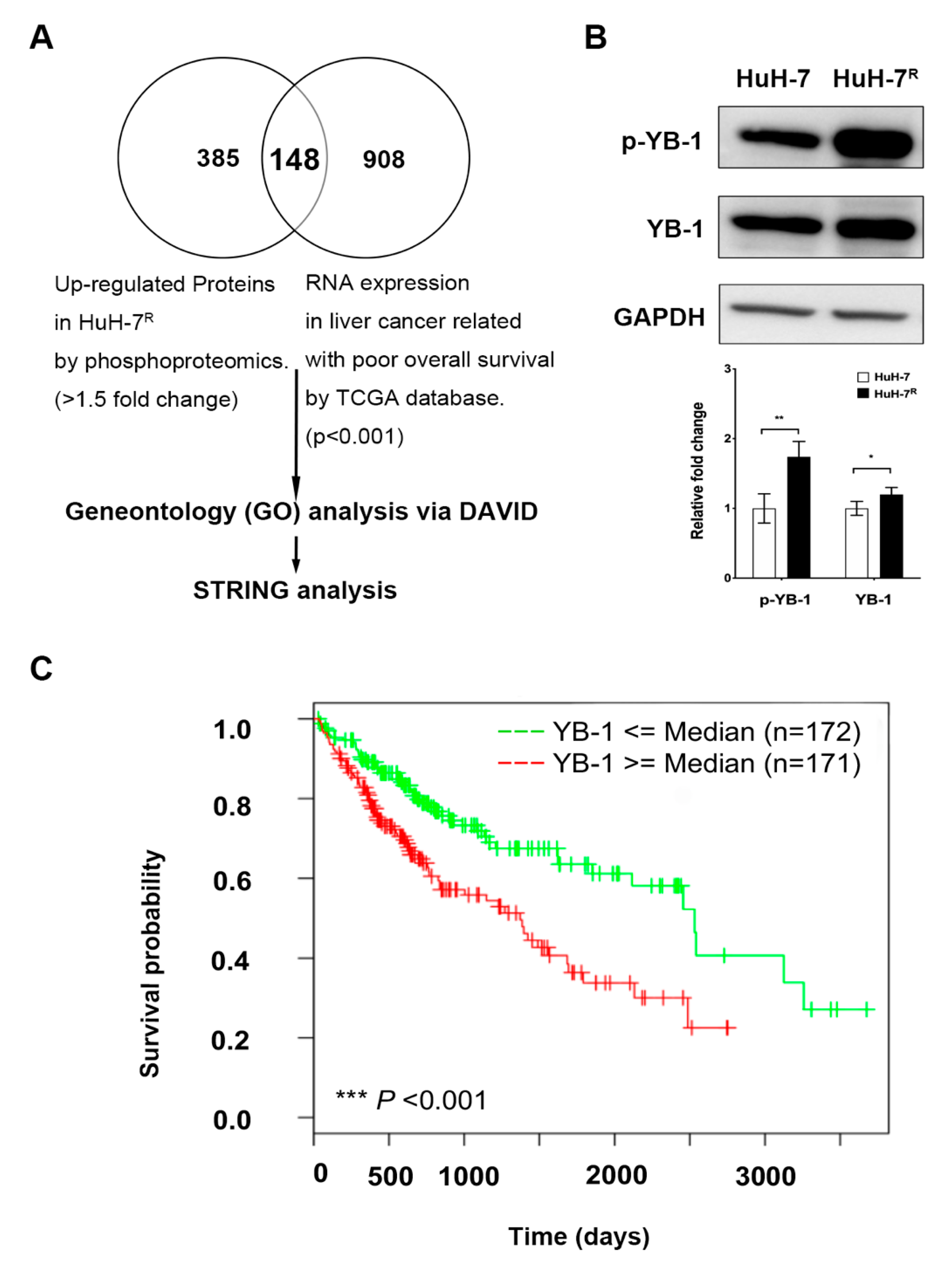
2.1. Overexpression of YB-1 in HuH-7R Cells Is Associated with Poor Prognosis of Liver Cancer
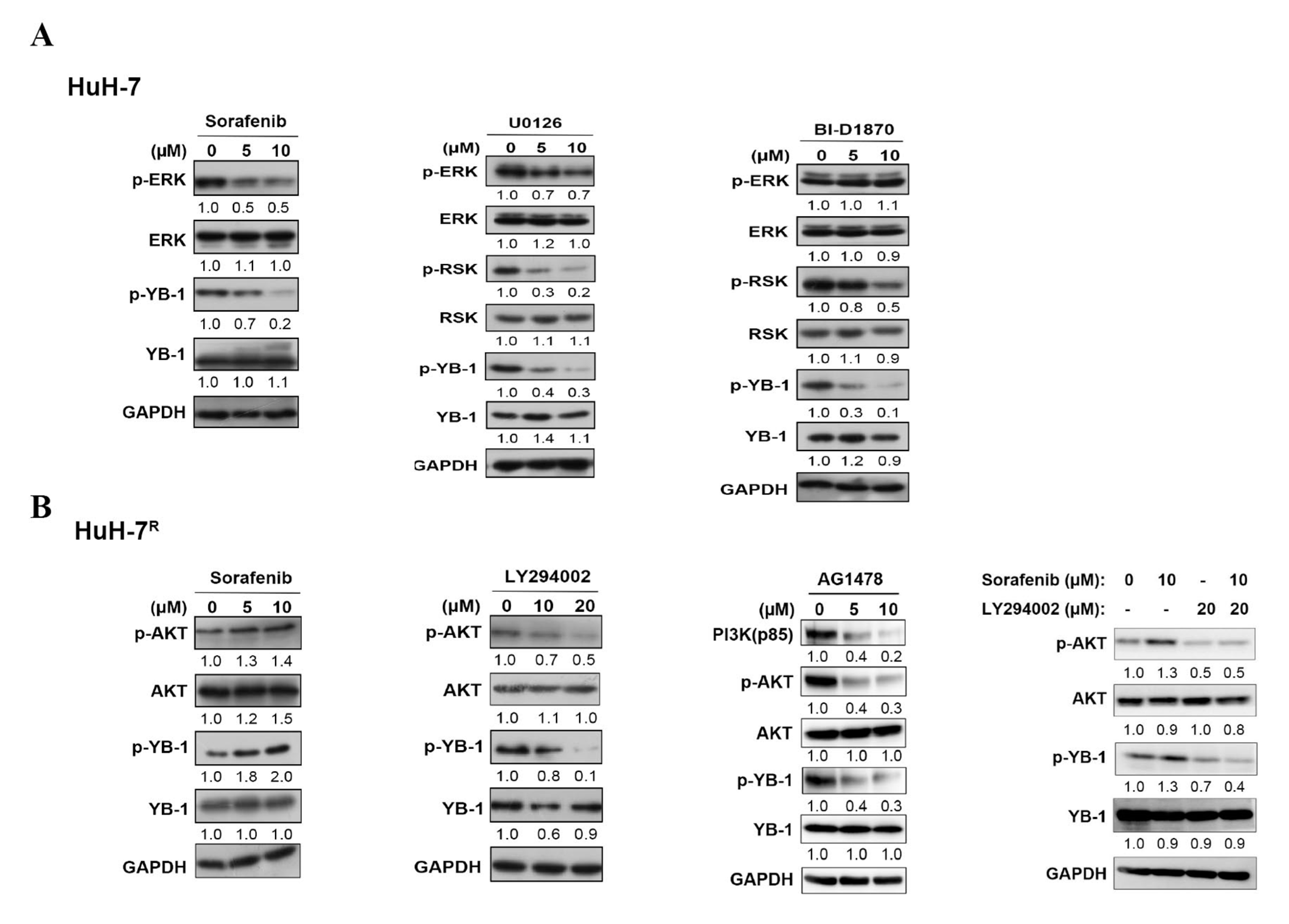
2.2. YB-1 Phosphorylation Is Regulated by Different Signaling Pathways Induced by Sorafenib in HuH-7 Cells and HuH-7R Cells
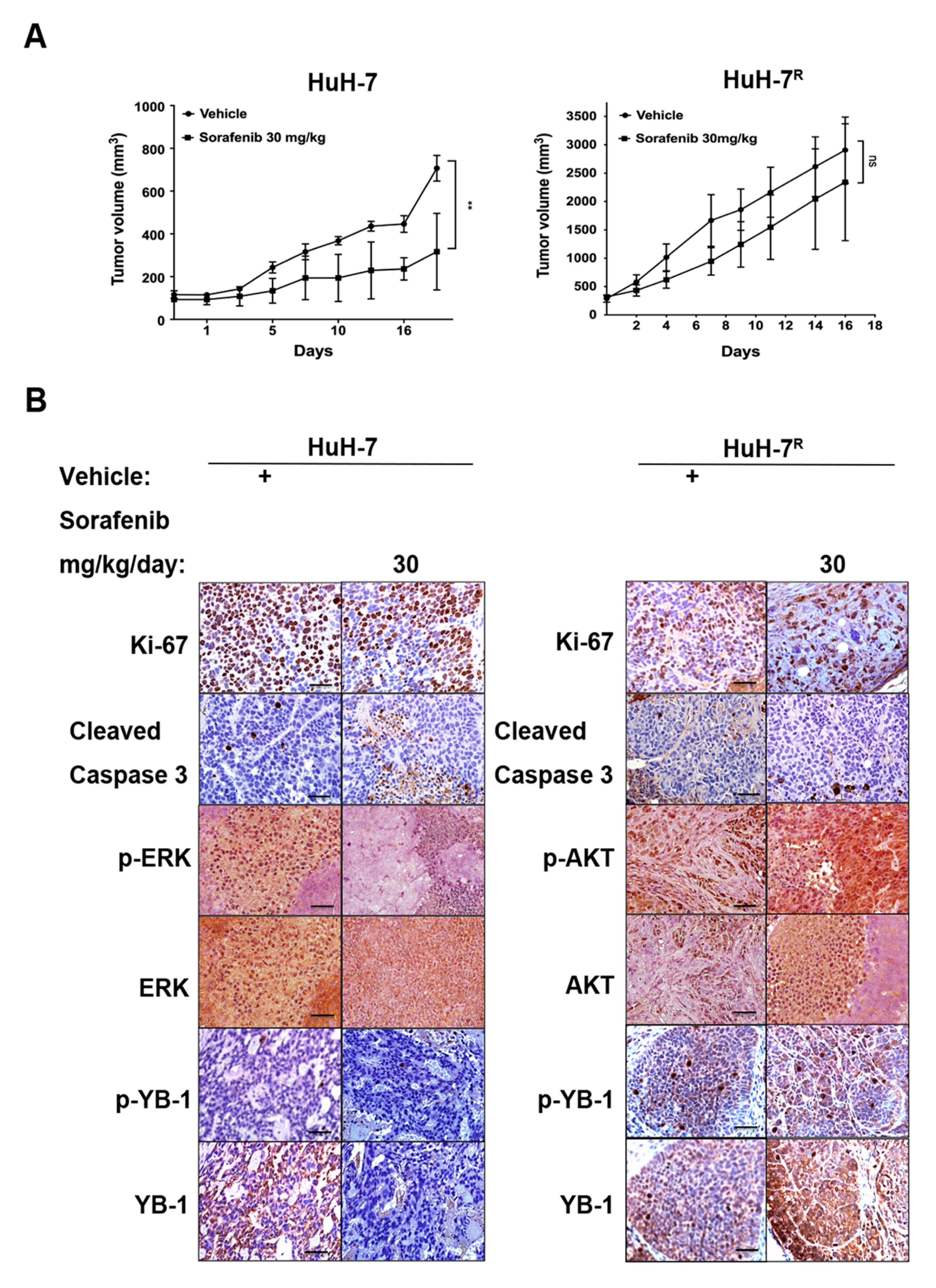
2.3. Immunohistochemical Staining of HuH-7 and HuH-7R Tumor-Bearing Mice with and Without Sorafenib Treatment
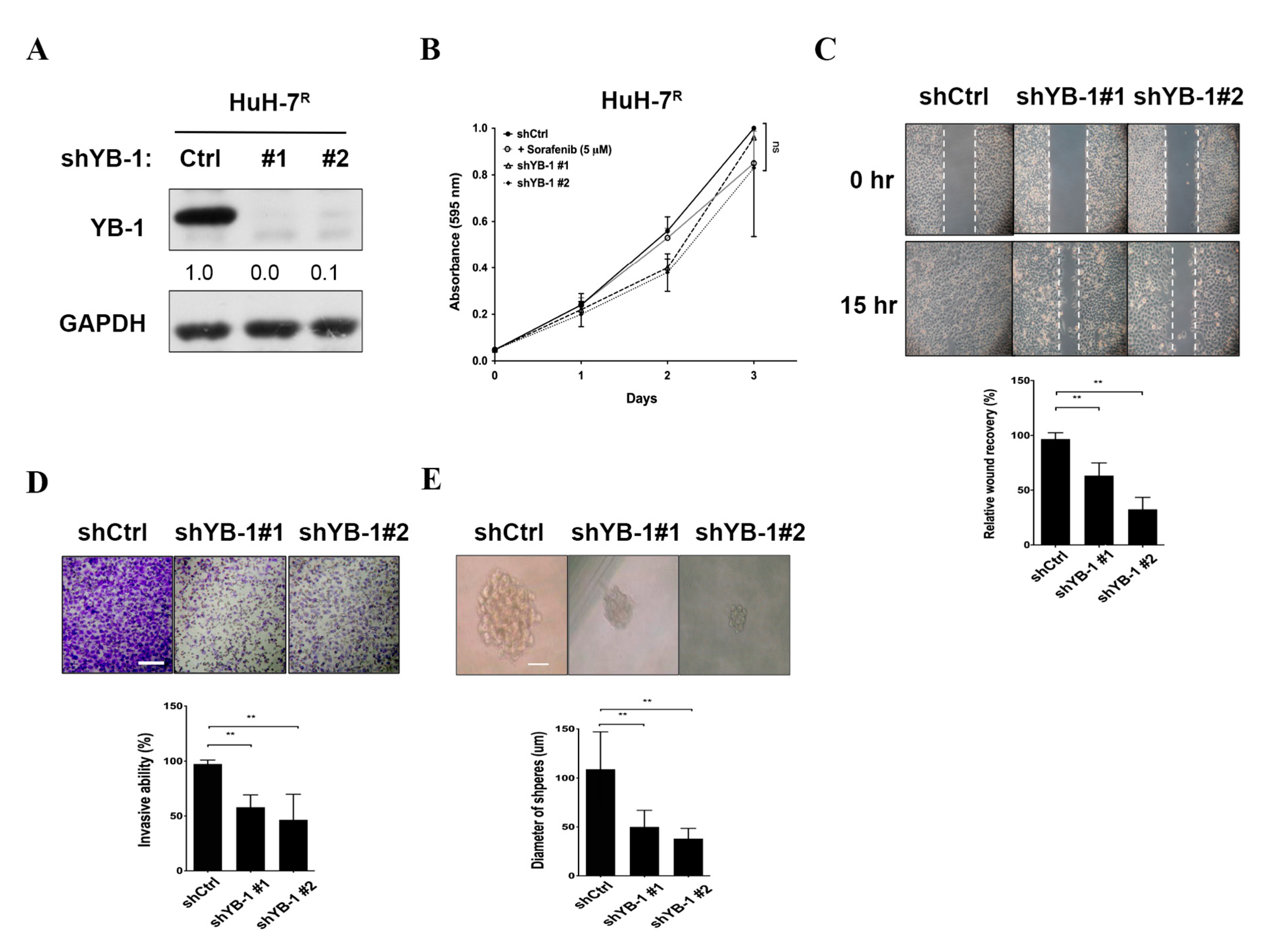
2.4. YB-1 Knockdown Does Not Affect Proliferation, But Suppresses Migration, Invasion and Sphere Formation in HuH-7R Cells
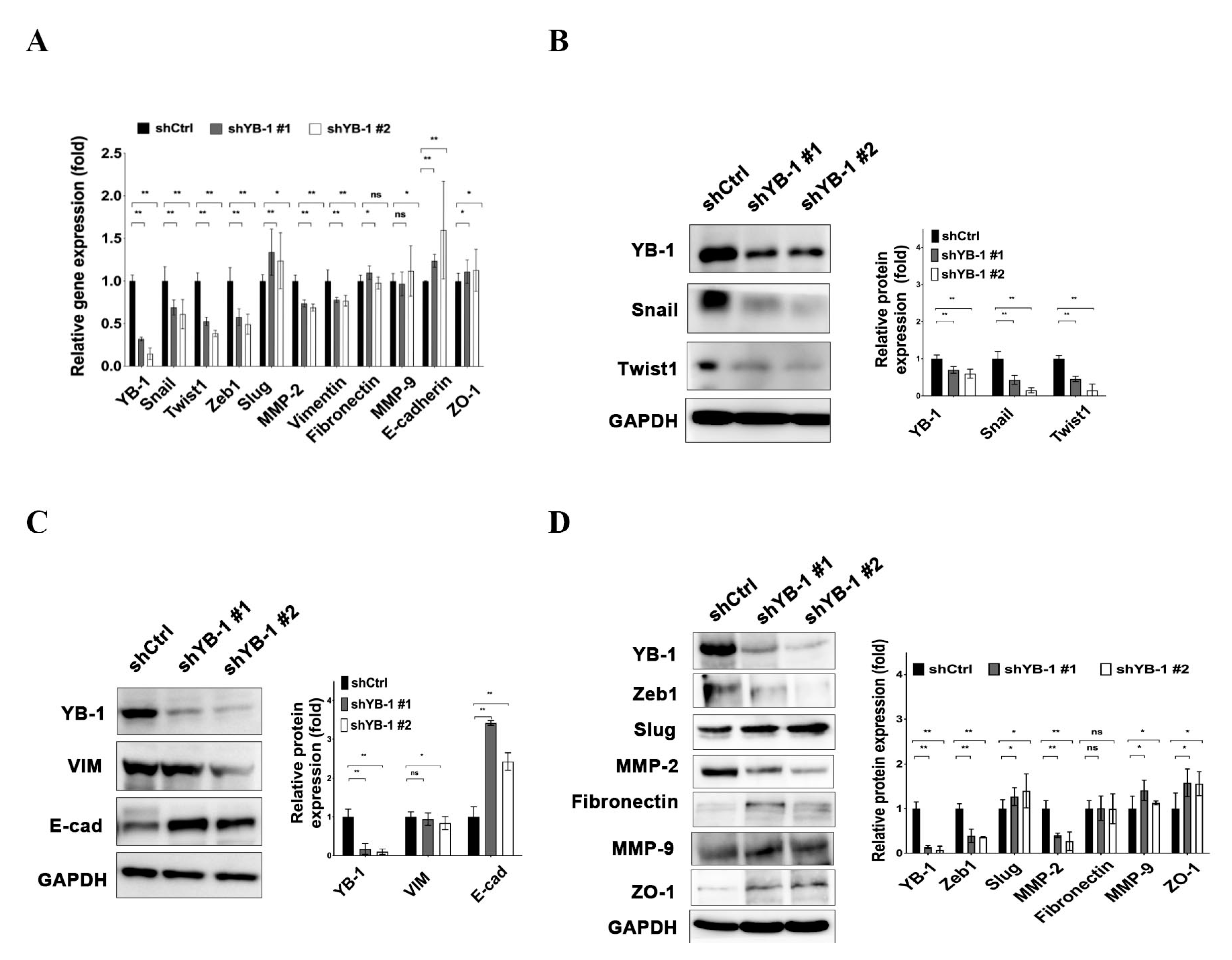
2.5. YB-1 Enhances Expression of EMT-Related Molecules in HuH-7R Cells
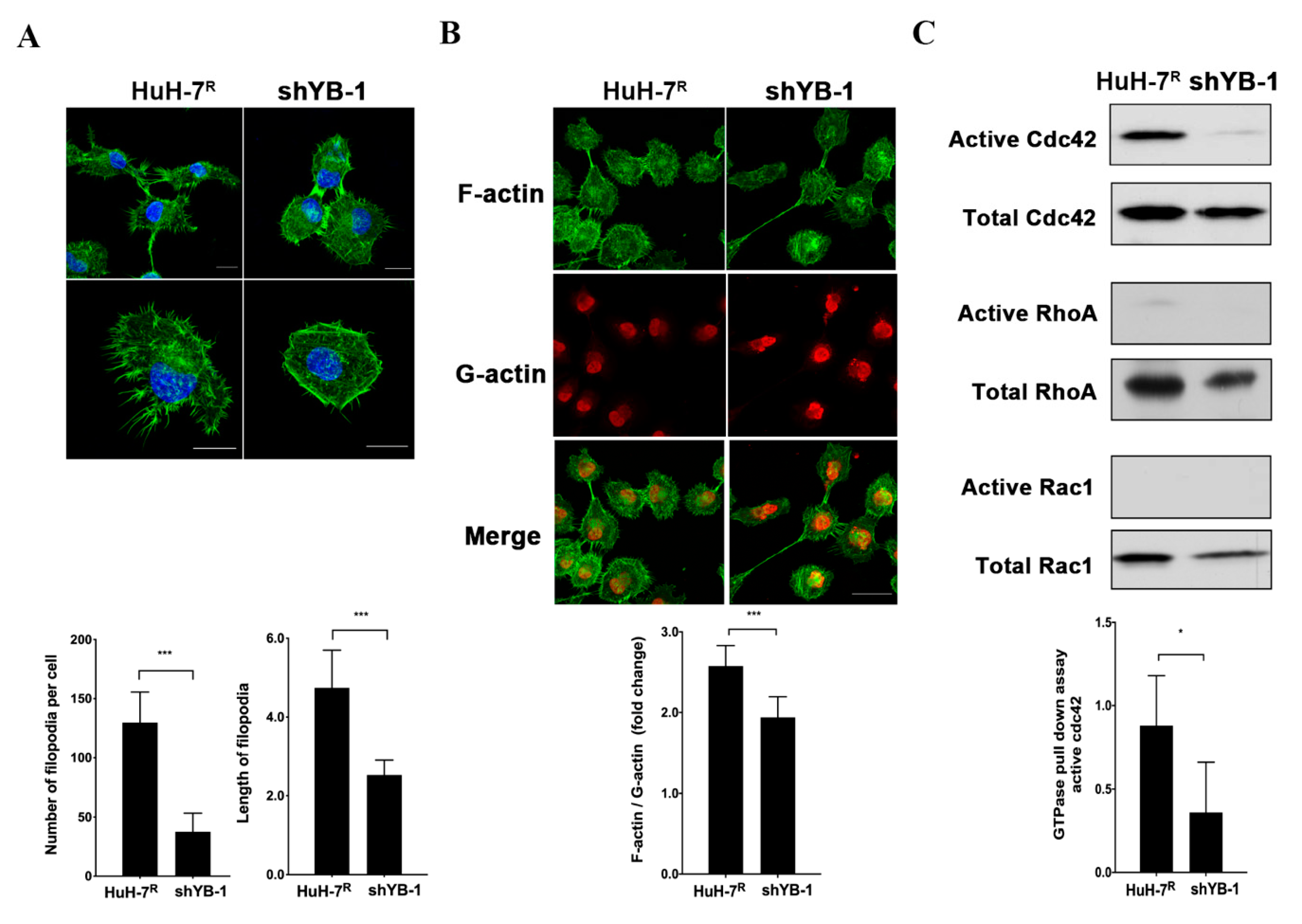
2.6. YB-1 Promotes Cdc42-Mediated F-Actin Regulation and Filopodia Formation in HuH-7R Cells
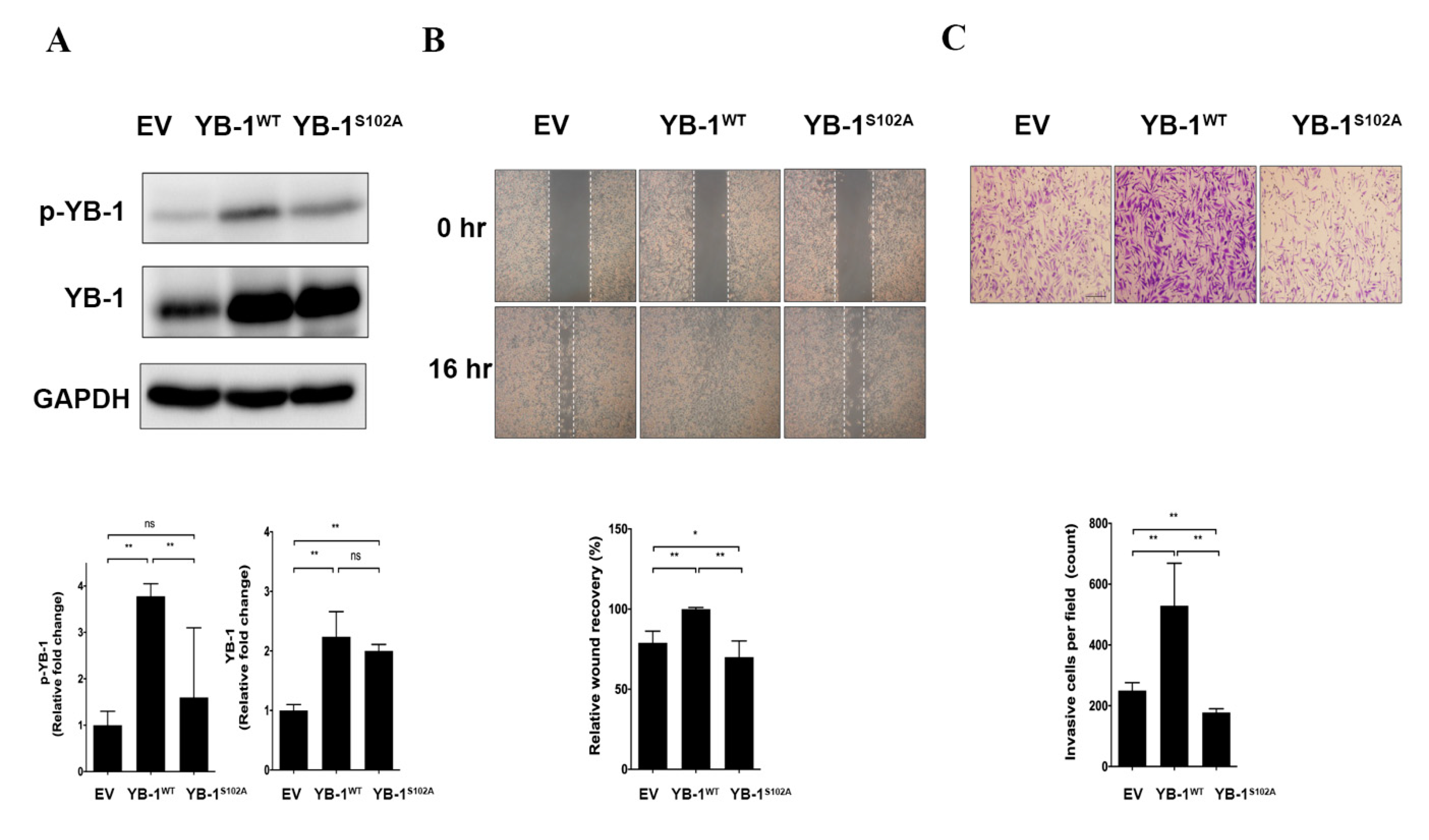
2.7. YB-1 Ser102 Phosphorylation Is Crucial for Migratory Potential and Invasiveness of HuH-7R Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture, YB-1 Knockdown, and Mutant YB-1 Expression
4.2. Bioinformatic Analysis
4.3. Immunoblotting, Immunohistochemistry (IHC) and Immunofluorescent Staining
4.4. Cell Proliferation, Viability, Wound Healing, Invasion, and Sphere Formation Assays
4.5. Quantitative Real-Time PCR (qPCR), Chromatin Immunoprecipitation (ChIP), and Pull-Down Assays
4.6. Subcutaneous Xenograft Tumor Models
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| YB-1 | Y-box binding protein-1 |
| HCC | Hepatocellular carcinoma |
| EMT | epithelial-mesenchymal transition |
| PTTG1 | pituitary tumor transforming gene 1 |
| PA2G4 | proliferation-associated protein 2G4 |
| MAPK | mitogen-activated protein kinase |
| RSK | ribosomal protein s6 kinase |
| EGFR | epidermal growth factor receptor |
| PI3K | phosphoinositide-3-kinase |
| AKT | protein kinase B |
| Snail | zinc-finger protein SNAI1 |
| Slug | zinc-finger protein SNAI2 |
| Twist1 | twist-related protein 1 |
| Zeb1 | zinc-finger E-box-binding homeobox 1 |
| MMP-2 | matrix metalloproteinase-2 |
| MMP-9 | matrix metalloproteinase-9 |
| VIM | vimentin |
References
- Verslype, C.; Van Cutsem, E.; Dicato, M.; Arber, N.; Berlin, J.D.; Cunningham, D.; De Gramont, A.; Diaz-Rubio, E.; Ducreux, M.; Gruenberger, T.; et al. The management of hepatocellular carcinoma. Current expert opinion and recommendations derived from the 10th World Congress on Gastrointestinal Cancer, Barcelona, 2008. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2009, 20 (Suppl. 7), vii1–vii6. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, T.; Chen, J.; Ge, C.; Zhao, F.; Zhu, M.; Chen, T.; Cui, Y.; Tian, H.; Yao, M.; et al. HIF-1alpha-induced RIT1 promotes liver cancer growth and metastasis and its deficiency increases sensitivity to sorafenib. Cancer Lett. 2019, 460, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.L.; Qin, S.K. Progress in systemic therapy of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Blivet-Van Eggelpoel, M.J.; Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Barbu, V.; Rey, C.; Priam, S.; Housset, C.; Rosmorduc, O.; Desbois-Mouthon, C.; et al. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J. Hepatol. 2012, 57, 108–115. [Google Scholar] [CrossRef]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Koch, A.; Evers, B.M. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010, 30, 4951–4958. [Google Scholar]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhou, X.; Shen, H.; Wang, D.; Wang, Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: Evidence from an in vitro study. BMC Med. 2009, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.K.; Ng, L.; Lam, C.S.; Wong, S.K.; Wan, T.M.; Cheng, N.S.; Yau, T.C.; Poon, R.T.; Pang, R.W. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS ONE 2013, 8, e78675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Okumura, N.; Wei, L.; Fuchs, B.C.; Fujii, T.; Sugimoto, H.; Nomoto, S.; Takeda, S.; Tanabe, K.K.; Kodera, Y. Epithelial to mesenchymal transition is associated with shorter disease-free survival in hepatocellular carcinoma. Ann. Surg. Oncol. 2014, 21, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lingala, S.; Khoobyari, S.; Nolta, J.; Zern, M.A.; Wu, J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 2011, 55, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.T.; Liao, L.Z.; Lu, C.H.; Huang, Y.H.; Lin, Y.K.; Lin, J.H.; Chow, L.P. Quantitative phosphoproteomic analysis identifies the potential therapeutic target EphA2 for overcoming sorafenib resistance in hepatocellular carcinoma cells. Exp. Mol. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Maurya, P.K.; Mishra, A.; Yadav, B.S.; Singh, S.; Kumar, P.; Chaudhary, A.; Srivastava, S.; Murugesan, S.N.; Mani, A. Role of Y Box Protein-1 in cancer: As potential biomarker and novel therapeutic target. J. Cancer 2017, 8, 1900–1907. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Stratford, A.L.; Astanehe, A.; Dunn, S.E. YB-1 is a Transcription/Translation Factor that Orchestrates the Oncogenome by Hardwiring Signal Transduction to Gene Expression. Transl. Oncogenomics 2007, 2, 49–65. [Google Scholar]
- Yasen, M.; Kajino, K.; Kano, S.; Tobita, H.; Yamamoto, J.; Uchiumi, T.; Kon, S.; Maeda, M.; Obulhasim, G.; Arii, S.; et al. The up-regulation of Y-box binding proteins (DNA binding protein A and Y-box binding protein-1) as prognostic markers of hepatocellular carcinoma. Clin. Cancer Res. 2005, 11, 7354–7361. [Google Scholar] [CrossRef] [Green Version]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.W.; Kucab, J.; Wu, J.; Lee, C.; Cheang, M.C.; Yorida, E.; Turbin, D.; Dedhar, S.; Nelson, C.; Pollak, M.; et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene 2005, 24, 4281–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.F.; Tai, W.T.; Liu, T.H.; Huang, H.P.; Lin, Y.C.; Shiau, C.W.; Li, P.K.; Chen, P.J.; Cheng, A.L. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin. Cancer Res. 2010, 16, 5189–5199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasewicz, E.; Rivas, L.; Hamdan, R.; Dokic, D.; Parimi, V.; Bernabe, B.P.; Thomas, A.; Shea, L.D.; Jeruss, J.S. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle 2014, 13, 3191–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinsch, A.; Chaker, A.; Burdelski, C.; Koop, C.; Tsourlakis, M.C.; Steurer, S.; Rink, M.; Eichenauer, T.S.; Wilczak, W.; Wittmer, C.; et al. betaIII-tubulin overexpression is linked to aggressive tumor features and genetic instability in urinary bladder cancer. Hum. Pathol. 2017, 61, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Li, X.; Ye, F.; Lu, C.; Huang, H.; Wang, F.; Cao, X.; Zhong, C. High KIF2A expression promotes proliferation, migration and predicts poor prognosis in lung adenocarcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Neilson, L.J.; Zhong, H.; Murray, P.S.; Zanivan, S.; Zaidel-Bar, R. E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci. Signal. 2014, 7, rs7. [Google Scholar] [CrossRef] [Green Version]
- Garcin, C.; Straube, A. Microtubules in cell migration. Essays Biochem. 2019, 63, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Li, H.P.; Peng, C.C.; Wu, C.C.; Chen, C.H.; Shih, M.J.; Huang, M.Y.; Lai, Y.R.; Chen, Y.L.; Chen, T.W.; Tang, P. Inactivation of the tight junction gene CLDN11 by aberrant hypermethylation modulates tubulins polymerization and promotes cell migration in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 102. [Google Scholar] [CrossRef]
- Meunier, S.; Vernos, I. Microtubule assembly during mitosis—From distinct origins to distinct functions? J. Cell Sci. 2012, 125 Pt 12, 2805–2814. [Google Scholar] [CrossRef] [Green Version]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef]
- Saito, Y.; Takasawa, A.; Takasawa, K.; Aoyama, T.; Akimoto, T.; Ota, M.; Magara, K.; Murata, M.; Hirohashi, Y.; Hasegawa, T.; et al. Aldolase A promotes epithelial-mesenchymal transition to increase malignant potentials of cervical adenocarcinoma. Cancer Sci. 2020, 111, 3071–3081. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Idichi, T.; Seki, N.; Wada, M.; Yamada, Y.; Fukuhisa, H.; Toda, H.; Kita, Y.; Kawasaki, Y.; Tanoue, K.; et al. Gene Regulation by Antitumor miR-204-5p in Pancreatic Ductal Adenocarcinoma: The Clinical Significance of Direct RACGAP1 Regulation. Cancers (Basel) 2019, 11, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohrman, A.Q.; Matus, D.Q. Divide or Conquer: Cell Cycle Regulation of Invasive Behavior. Trends Cell Biol. 2017, 27, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ci, C.; Tang, B.; Lyu, D.; Liu, W.; Qiang, D.; Ji, X.; Qiu, X.; Chen, L.; Ding, W. Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. Int. J. Mol. Med. 2019, 43, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.; Noh, B.J.; Lee, S.; Lee, H.Y.; Kim, Y.; Lim, J.; Ju, H.; Yu, H.Y.; Ryu, C.M.; Lee, P.C.; et al. Phosphorylation of TFCP2L1 by CDK1 is required for stem cell pluripotency and bladder carcinogenesis. EMBO Mol. Med. 2020, 12, e10880. [Google Scholar] [CrossRef]
- Caldon, C.E. Cdk2 regulates metastasis suppressor BRMS1. Cell Cycle 2016, 15, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Cai, J.; Meng, F.; Sui, C.; Jiang, Y. MiR-144 suppresses proliferation, invasion, and migration of breast cancer cells through inhibiting CEP55. Cancer Biol. Ther. 2018, 19, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Yu, Y.; Chen, J.; He, Y.; Chen, X.; Ren, Z.; Xue, C.; Liu, L.; Hu, Q.; Li, J.; et al. MiR-126 negatively regulates PLK-4 to impact the development of hepatocellular carcinoma via ATR/CHEK1 pathway. Cell Death Dis. 2018, 9, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Shen, Y.; Zou, Y.; Qi, Z.; Huang, G.; Xia, S.; Gao, R.; Li, F.; Huang, Z. TOP2A Promotes Cell Migration, Invasion and Epithelial–Mesenchymal Transition in Cervical Cancer via Activating the PI3K/AKT Signaling. Cancer Manag. Res. 2020, 12, 3807–3814. [Google Scholar] [CrossRef]
- Fujii, T.; Nomoto, S.; Koshikawa, K.; Yatabe, Y.; Teshigawara, O.; Mori, T.; Inoue, S.; Takeda, S.; Nakao, A. Overexpression of pituitary tumor transforming gene 1 in HCC is associated with angiogenesis and poor prognosis. Hepatology 2006, 43, 1267–1275. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, J.; Zhong, D.; Zhou, Y.; Zhang, W.; Wu, W.; Zhao, Z.; Wang, W.; Xu, W.; He, L.; et al. Overexpression of SMC4 activates TGFβ/Smad signaling and promotes aggressive phenotype in glioma cells. Oncogenesis 2017, 6, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, J.J.; Lu, Y.Z.; Sun, L.P.; Liu, J.W.; Gong, Y.H.; Xu, Q.; Dong, N.N.; Yuan, Y. Epistatic SNP interaction of ERCC6 with ERCC8 and their joint protein expression contribute to gastric cancer/atrophic gastritis risk. Oncotarget 2017, 8, 43140–43152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, H.; Zhang, S.; Buss, C.G.; Fish, L.; Tavazoie, S.; Tavazoie, S.F. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nat. Cell Biol. 2014, 513, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, M.; Jiang, Y.; Wang, R.; Liu, L.; Rode, S.; Bonahoom, M.; Kim, S.; Yang, Z.Q. Pan-cancer analysis of RNA methyltransferases identifies FTSJ3 as a potential regulator of breast cancer progression. RNA Biol. 2020, 17, 474–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieron, P.; Hader, C.; Hatina, J.; Engers, R.; Wlazlinski, A.; Muller, M.; Schulz, W.A. DKC1 overexpression associated with prostate cancer progression. Br. J. Cancer 2009, 101, 1410–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassin, E.R.; Abdul-Nabi, A.M.; Takeda, A.; Yaseen, N.R. Effects of the NUP98–DDX10 oncogene on primary human CD34+ cells: Role of a conserved helicase motif. Leukemia 2010, 24, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yu, S.; Yuan, X.; Xiong, J.; Kuang, D.; Pestell, R.G.; Wu, K. DACH1 suppresses breast cancer as a negative regulator of CD44. Sci. Rep. 2017, 7, 4361. [Google Scholar] [CrossRef]
- Wu, H.; Qin, W.; Lu, S.; Wang, X.; Zhang, J.; Sun, T.; Hu, X.; Li, Y.; Chen, Q.; Wang, Y.; et al. Long noncoding RNA ZFAS1 promoting small nucleolar RNA-mediated 2′-O-methylation via NOP58 recruitment in colorectal cancer. Mol. Cancer 2020, 19, 1–22. [Google Scholar] [CrossRef]
- Itoh, G.; Sugino, S.; Ikeda, M.; Mizuguchi, M.; Kanno, S.; Amin, M.A.; emura, K.; Yasui, A.; Hirota, T.; Tanaka, K. Nucleoporin Nup188 is required for chromosome alignment in mitosis. Cancer Sci. 2013, 104, 871–879. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype. Oxidative Med. Cell. Longev. 2018, 2018, 1027453. [Google Scholar] [CrossRef] [Green Version]
- Aydin, B.; Arga, K.Y. Co-expression Network Analysis Elucidated a Core Module in Association With Prognosis of Non-functioning Non-invasive Human Pituitary Adenoma. Front. Endocrinol. 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Wang, W.; Huang, H.; Jiang, Y.; Guo, W.; Liu, H.; Yu, J.; Hu, Y.; Wan, J.; Li, G. Prognostic Significance of PSMD1 Expression in Patients with Gastric Cancer. J. Cancer 2019, 10, 4357–4367. [Google Scholar] [CrossRef] [PubMed]
- Kusonmano, K.; Halle, M.K.; Wik, E.; Hoivik, E.A.; Krakstad, C.; Mauland, K.K.; Tangen, I.L.; Berg, A.; Werner, H.M.J.; Trovik, J.; et al. Identification of highly connected and differentially expressed gene subnetworks in metastasizing endometrial cancer. PLoS ONE 2018, 13, e0206665. [Google Scholar] [CrossRef] [PubMed]
- Nyati, S.; Gregg, B.S.; Xu, J.; Young, G.; Kimmel, L.; Nyati, M.K.; Ray, D.; Speers, C.; Rehemtulla, A. TGFBR2 mediated phosphorylation of BUB1 at Ser-318 is required for transforming growth factor-beta signaling. Neoplasia 2020, 22, 163–178. [Google Scholar] [CrossRef]
- Harvey, S.E.; Xu, Y.; Lin, X.; Gao, X.D.; Qiu, Y.; Ahn, J.; Xiao, X.; Cheng, C. Coregulation of alternative splicing by hnRNPM and ESRP1 during EMT. RNA 2018, 24, 1326–1338. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; He, M.; Huang, S.; Lin, R.; Zhan, M.; Yang, D.; Shen, H.; Xu, S.; Cheng, W.; Yu, J.; et al. Circular RNA circERBB2 promotes gallbladder cancer progression by regulating PA2G4-dependent rDNA transcription. Mol. Cancer 2019, 18, 166. [Google Scholar] [CrossRef] [Green Version]
- Yoon, C.-H.; Kim, M.-J.; Lee, H.; Kim, R.-K.; Lim, E.-J.; Yoo, K.-C.; Lee, G.-H.; Cui, Y.-H.; Oh, Y.S.; Gye, M.C.; et al. PTTG1Oncogene Promotes Tumor Malignancy via Epithelial to Mesenchymal Transition and Expansion of Cancer Stem Cell Population. J. Biol. Chem. 2012, 287, 19516–19527. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-M.; Huang, H.-W.; Kwang-Huei, L.; Lee, K.-F.; Tsai, C.-Y.; Lu, P.-H.; Chi, H.-C.; Lin, Y.-H.; Kuo, L.-M.; Lin, K.-H. MicroRNA-26b inhibits tumor metastasis by targeting the KPNA2/c-jun pathway in human gastric cancer. Oncotarget 2016, 7, 39511–39526. [Google Scholar] [CrossRef] [Green Version]
- Habibi, G.; Leung, S.; Law, J.H.; Gelmon, K.; Masoudi, H.; Turbin, D.; Pollak, M.; Nielsen, T.O.; Huntsman, D.G.; Dunn, S.E. Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res. 2008, 10, R86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Woodford, N.; Xia, X.; Hamburger, A.W. Repression of E2F1-mediated transcription by the ErbB3 binding protein Ebp1 involves histone deacetylases. Nucleic Acids Res. 2003, 31, 2168–2177. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.X.T.; Lee, Y.; Urbani, L.; Utz, P.J.; Hamburger, A.W.; Sunwoo, J.B.; Mitchell, B.S. Regulation of ribosomal RNA synthesis in T cells: Requirement for GTP and Ebp1. Blood 2015, 125, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.Q.; Hoang, D.H.; Vo, T.T.N.; Huynh, V.; Ghoda, L.; Marcucci, G.; Nguyen, L.X.T. The role of ErbB3 binding protein 1 in cancer: Friend or foe? J. Cell. Physiol. 2018, 233, 9110–9120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Liu, S.; Xu, Z.; Huang, S.; Tan, X.; Sun, C.; Lu, L. Pituitary tumor-transforming gene 1 (PTTG1) is overexpressed in oral squamous cell carcinoma (OSCC) and promotes migration, invasion and epithelial–mesenchymal transition (EMT) in SCC15 cells. Tumor Biol. 2014, 35, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.-M.; Huang, H.-X.; Chang, P.-H.; Tseng, K.-C.; Miyajima, A.; Chern, E. Y-box binding protein-1 promotes hepatocellular carcinoma-initiating cell progression and tumorigenesis via Wnt/β-catenin pathway. Oncotarget 2017, 8, 2604–2616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Q.; Liu, J.; Cao, H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 9377–9384. [Google Scholar] [CrossRef]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Fei, Z.; Zhang, G. Synergistic anticancer activity of 20(S)-Ginsenoside Rg3 and Sorafenib in hepatocellular carcinoma by modulating PTEN/Akt signaling pathway. Biomed. Pharmacother. 2018, 97, 1282–1288. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef]
- Yeh, C.-C.; Mong-Hsun, T.; Shao, Y.-Y.; Ho, W.-C.; Tsai, M.-H.; Feng, W.-C.; Chow, L.-P. Integrated Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) Quantitative Proteomic Analysis Identifies Galectin-1 as a Potential Biomarker for Predicting Sorafenib Resistance in Liver Cancer. Mol. Cell. Proteom. 2015, 14, 1527–1545. [Google Scholar] [CrossRef] [Green Version]
- Sinnberg, T.; Sauer, B.; Holm, P.; Spangler, B.; Kuphal, S.; Bosserhoff, A.; Schittek, B. MAPK and PI3K/AKT mediated YB-1 activation promotes melanoma cell proliferation which is counteracted by an autoregulatory loop. Exp. Dermatol. 2012, 21, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.; Meyer, K.; Spang, R.; Schittek, B.; Bosserhoff, A.K. Melanoma inhibitory activity promotes melanoma development through activation of YBX1. Pigment. Cell Melanoma Res. 2013, 26, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Friemel, J.; Rechsteiner, M.; Frick, L.; Boehm, F.; Struckmann, K.; Egger, M.; Moch, H.; Heikenwalder, M.; Weber, A. Intratumor Heterogeneity in Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 21, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pribluda, A.; De La Cruz, C.C.; Jackson, E.L. Intratumoral Heterogeneity: From Diversity Comes Resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimova, V.; Ovchinnikov, L.P.; Sorensen, P.H. Y-box Binding Protein 1: Providing a New Angle on Translational Regulation. Cell Cycle 2006, 5, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.-J.; Chen, H.-Y.; Ye, Z.; Zhu, S.; Zeng, Z.; He, C.; Liu, M.-L.; Huang, K.; Zhong, J.-X.; Xu, F.-Y.; et al. Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene 2018, 37, 5811–5828. [Google Scholar] [CrossRef]
- Yang, M.-H.; Chen, C.-L.; Chau, G.-Y.; Chiou, S.-H.; Su, C.-W.; Chou, T.-Y.; Peng, W.-L.; Wu, J.-C. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology 2009, 50, 1464–1474. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.-M.; Chuah, L.-H.; Leong, C.-O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Pang, T.; Li, M.; Zhang, Y.; Yong, W.; Kang, H.; Yao, Y.; Hu, X. Y Box-Binding Protein 1 Promotes Epithelial-Mesenchymal Transition, Invasion, and Metastasis of Cervical Cancer via Enhancing the Expressions of Snail. Int. J. Gynecol. Cancer 2017, 27, 1753–1760. [Google Scholar] [CrossRef]
- Deng, J.-J.; Zhang, W.; Xu, X.-M.; Zhang, F.; Tao, W.-P.; Ye, J.-J.; Ge, W. Twist mediates an aggressive phenotype in human colorectal cancer cells. Int. J. Oncol. 2016, 48, 1117–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Izumi, H.; Onitsuka, T.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Yokomizo, A.; Naito, S.; Kohno, K. Twist Promotes Tumor Cell Growth through YB-1 Expression. Cancer Res. 2008, 68, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational Activation of Snail1 and Other Developmentally Regulated Transcription Factors by YB-1 Promotes an Epithelial-Mesenchymal Transition. Cancer Cell 2009, 15, 402–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Luo, W. Matrix metalloproteinase 2 contributes to aggressive phenotype, epithelial-mesenchymal transition and poor outcome in nasopharyngeal carcinoma. OncoTargets Ther. 2019, 12, 5701–5711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, P.R.; Harendza, S.; Pollock, A.S.; Lovett, D.H. Glomerular Mesangial Cell-specific Transactivation of Matrix Metalloproteinase 2 Transcription Is Mediated by YB-1. J. Biol. Chem. 1997, 272, 22905–22912. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.-B.; Zhu, Q.-C.; Chen, H.-Q.; Peng, J.-Y.; Chao, H.-L.; Du, H.-X.; Wang, Z.-G.; Jin, Z.M. Knockdown of Y-box-binding protein-1 inhibits the malignant progression of HT-29 colorectal adenocarcinoma cells by reversing epithelial-mesenchymal transition. Mol. Med. Rep. 2014, 10, 2720–2728. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The Tight Junction Protein ZO-1 Establishes a Link between the Transmembrane Protein Occludin and the Actin Cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Ou, D.L.; Shen, Y.C.; Yu, S.L.; Chen, K.F.; Yeh, P.Y.; Fan, H.H.; Feng, W.C.; Wang, C.T.; Lin, L.I.; Hsu, C.; et al. Induction of DNA damage-inducible gene GADD45beta con-tributes to sorafenib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2010, 70, 9309–9318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.-L.; Yang, J.-C.; Lee, H.; Sheu, F.; Hsu, C.-H.; Lin, S.-L.; Chow, L.-P. The C-Terminal Disulfide Bonds ofHelicobacter pyloriGroES Are Critical for IL-8 Secretion via the TLR4-Dependent Pathway in Gastric Epithelial Cells. J. Immunol. 2015, 194, 3997–4007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-L.; Ni, J.; Feng, W.-T.; Yao, R.; Yue, S.; Zhu, Y.-N.; Tang, H.-Y.; Lv, L.-Y.; Feng, J.-F.; Zhu, W. High YBX1 expression indicates poor prognosis and promotes cell migration and invasion in nasopharyngeal carcinoma. Exp. Cell Res. 2017, 361, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhou, Y.; Zhai, B.; Liao, J.; Xu, W.; Zhang, R.; Li, J.; Zhang, Y.; Chen, L.; Qian, H.; et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011, 11, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, L.-Z.; Chen, C.-T.; Li, N.-C.; Lin, L.-C.; Huang, B.-S.; Chang, Y.-H.; Chow, L.-P. Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 224. https://doi.org/10.3390/ijms22010224
Liao L-Z, Chen C-T, Li N-C, Lin L-C, Huang B-S, Chang Y-H, Chow L-P. Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2021; 22(1):224. https://doi.org/10.3390/ijms22010224
Chicago/Turabian StyleLiao, Li-Zhu, Chih-Ta Chen, Nien-Chen Li, Liang-Chun Lin, Bo-Shih Huang, Ya-Hui Chang, and Lu-Ping Chow. 2021. "Y-Box Binding Protein-1 Promotes Epithelial-Mesenchymal Transition in Sorafenib-Resistant Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 22, no. 1: 224. https://doi.org/10.3390/ijms22010224