Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-d]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors

 , , , , and
, , , , and
Abstract
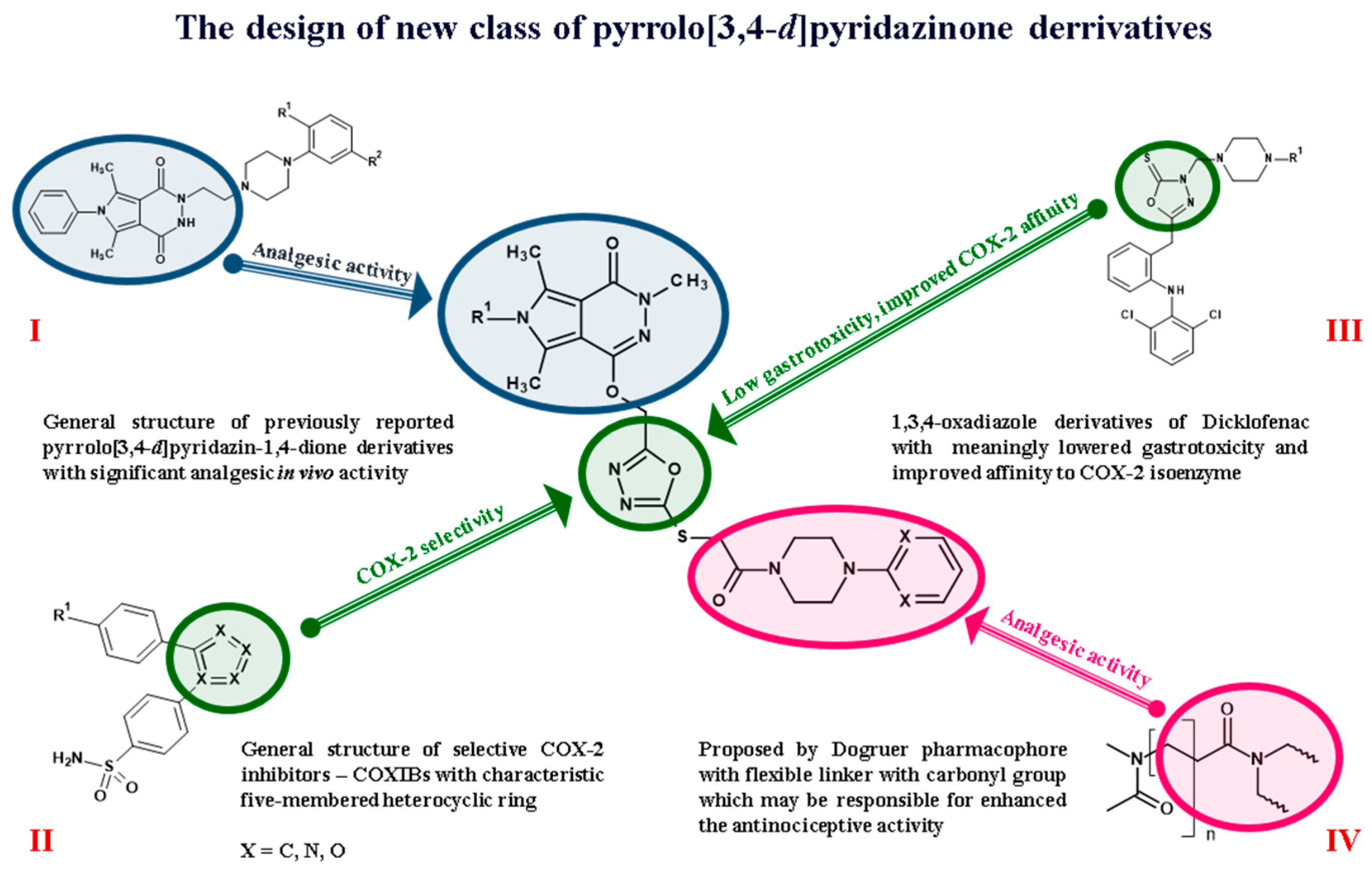
:1. Introduction
2. Results and Discussion
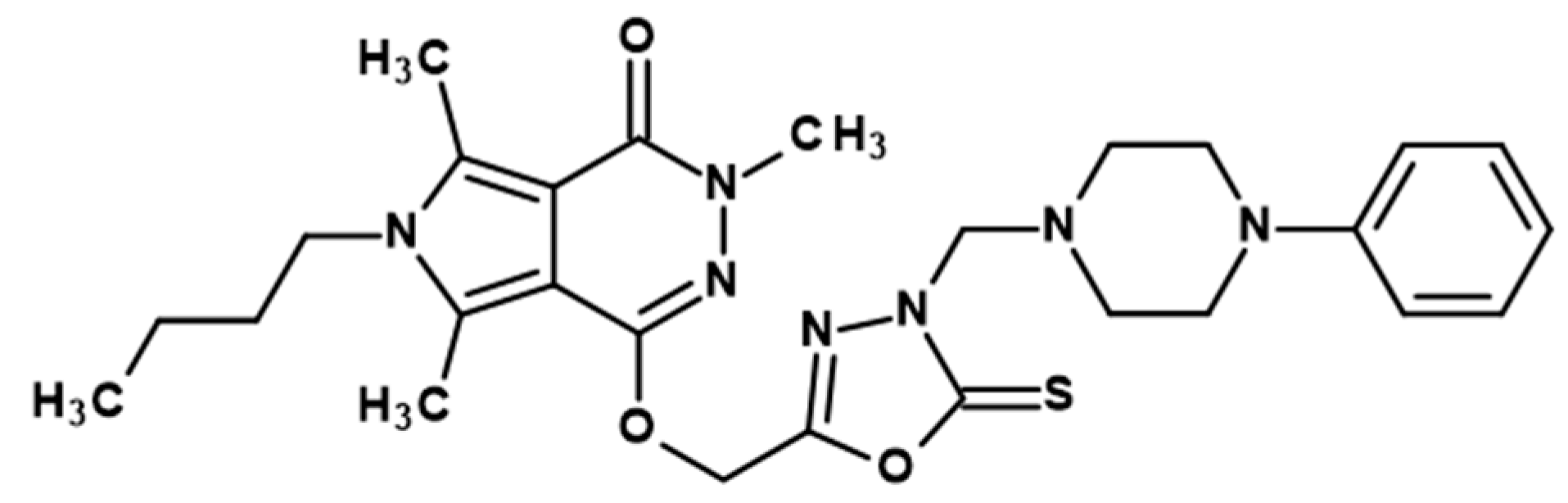
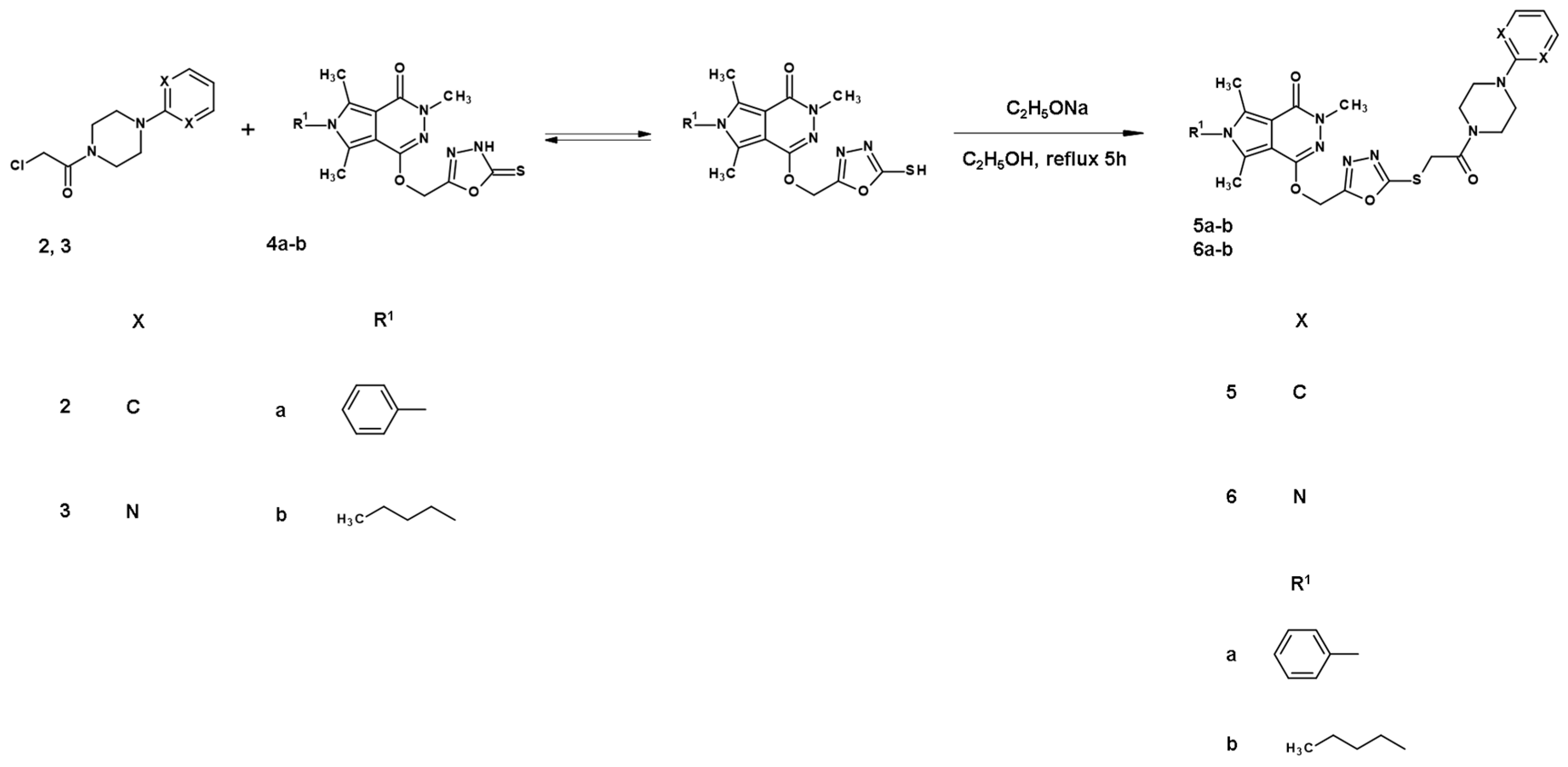
2.1. Chemistry
2.2. Cyclooxygenase (COX-1, COX-2) Inhibition Studies
2.2.1. In Vitro Cyclooxygenase Inhibition Assay
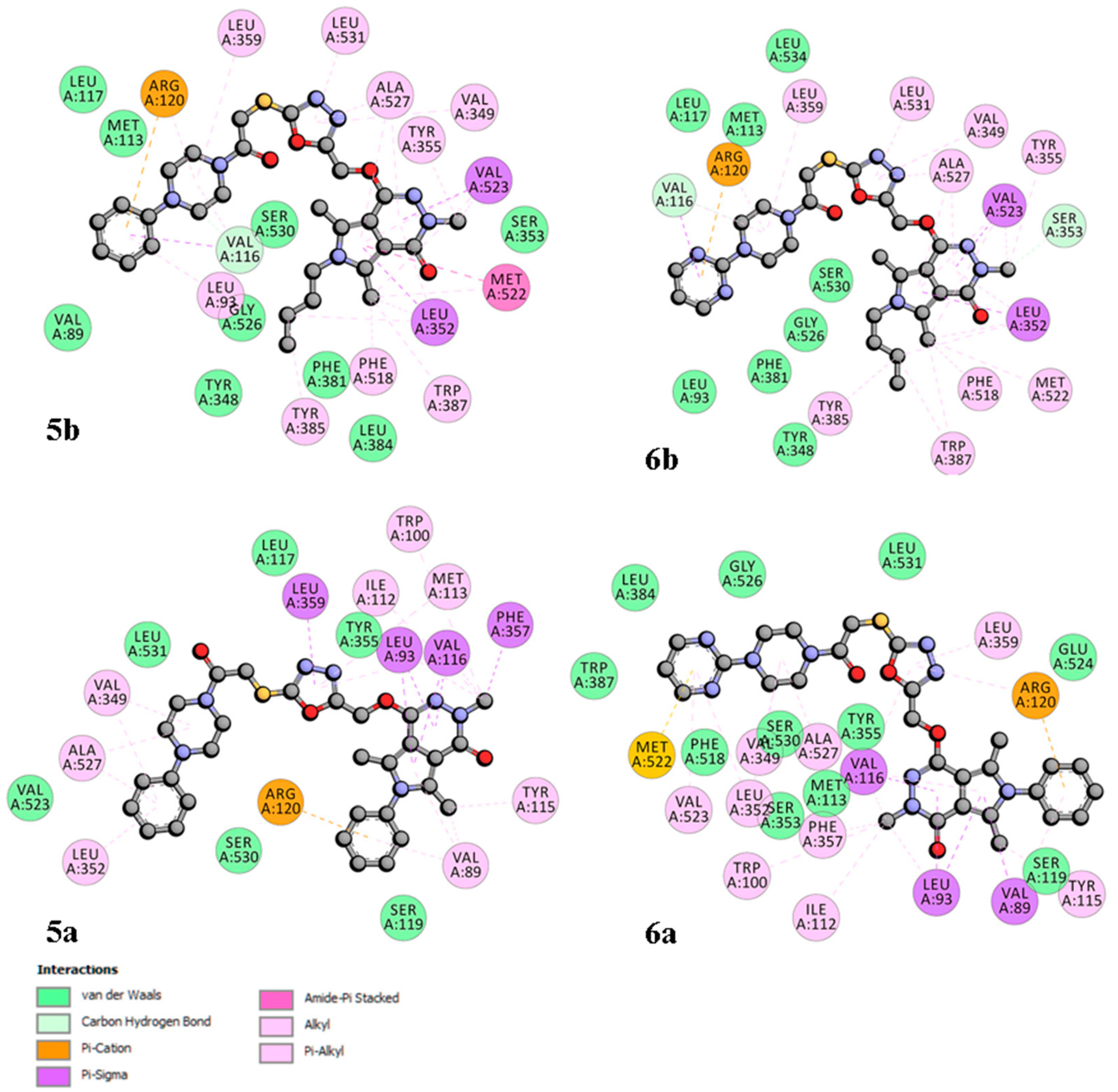
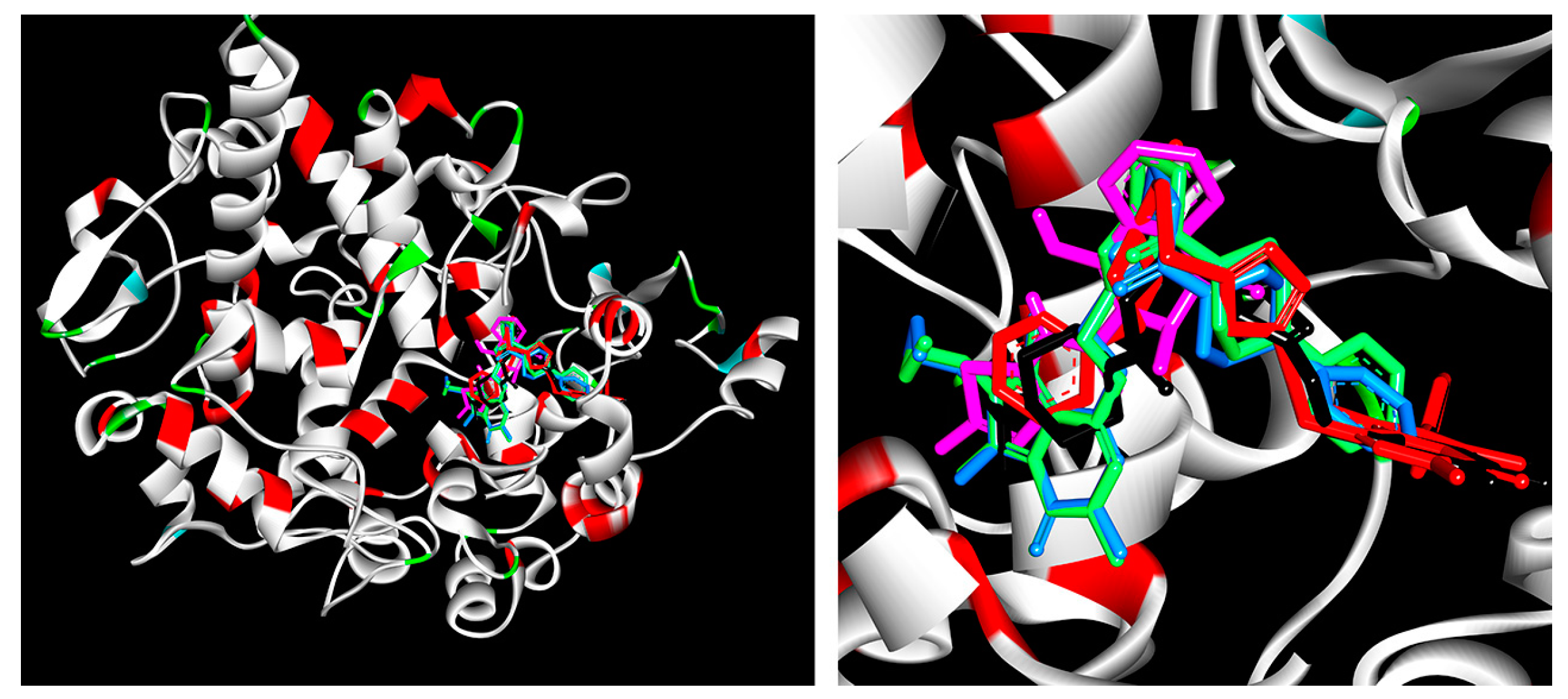
2.2.2. Cyclooxygenase Molecular Docking Study
2.3. Evaluation of Viability
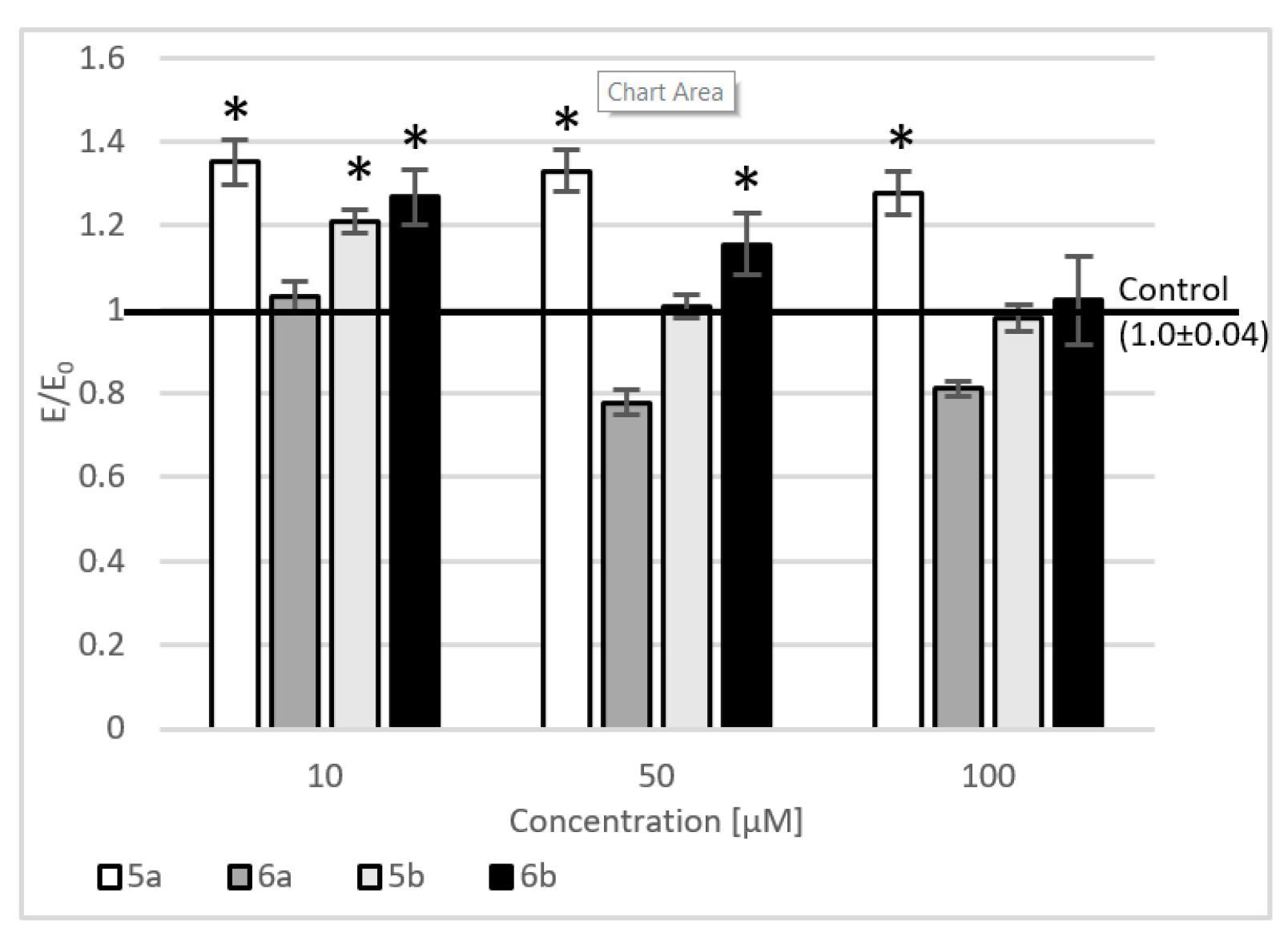
2.4. Level of Intracellular Reactive Oxygen Species, Nitric Oxide, and DNA Damage
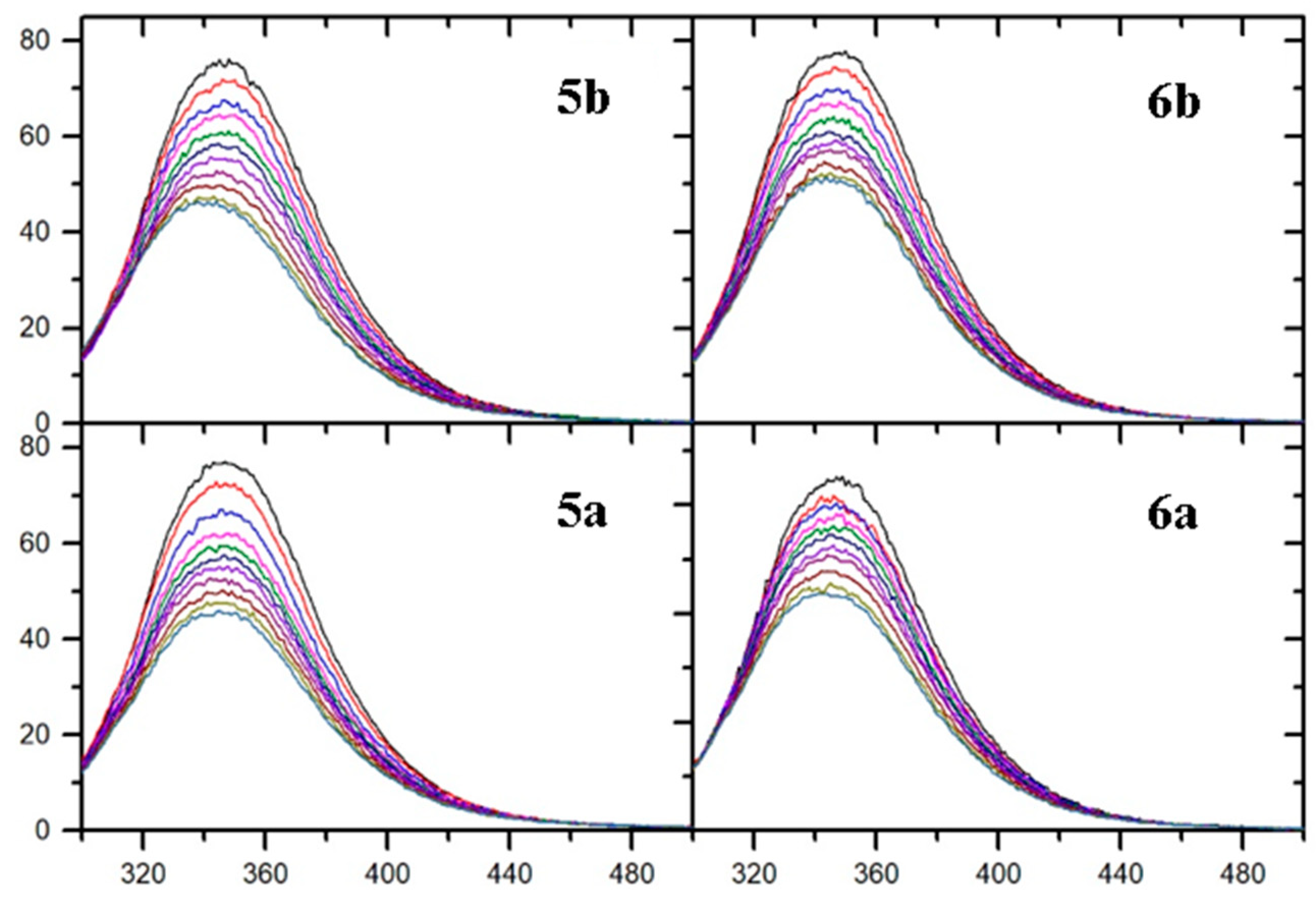
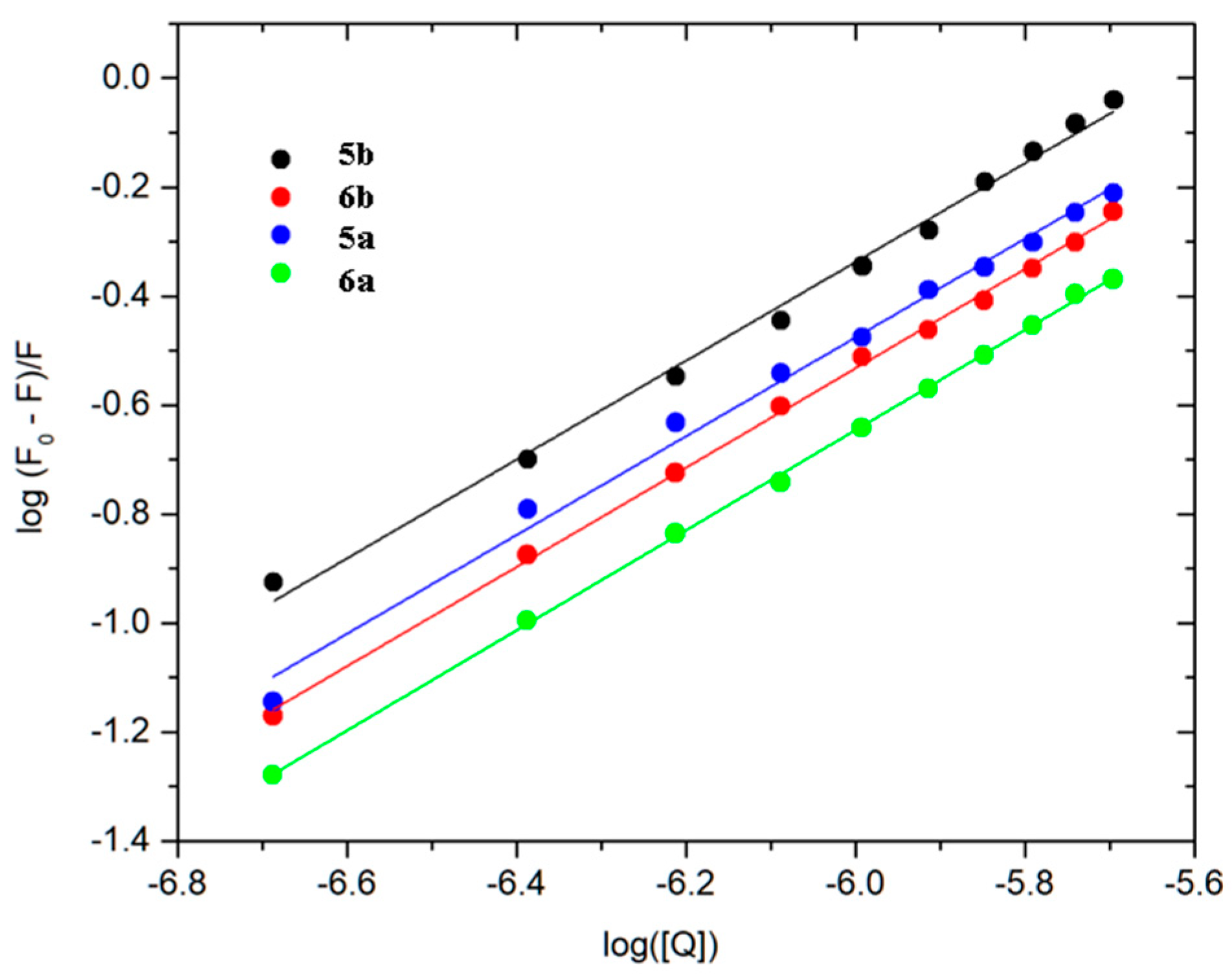
2.5. Fluorescence Quenching and Binding Constants
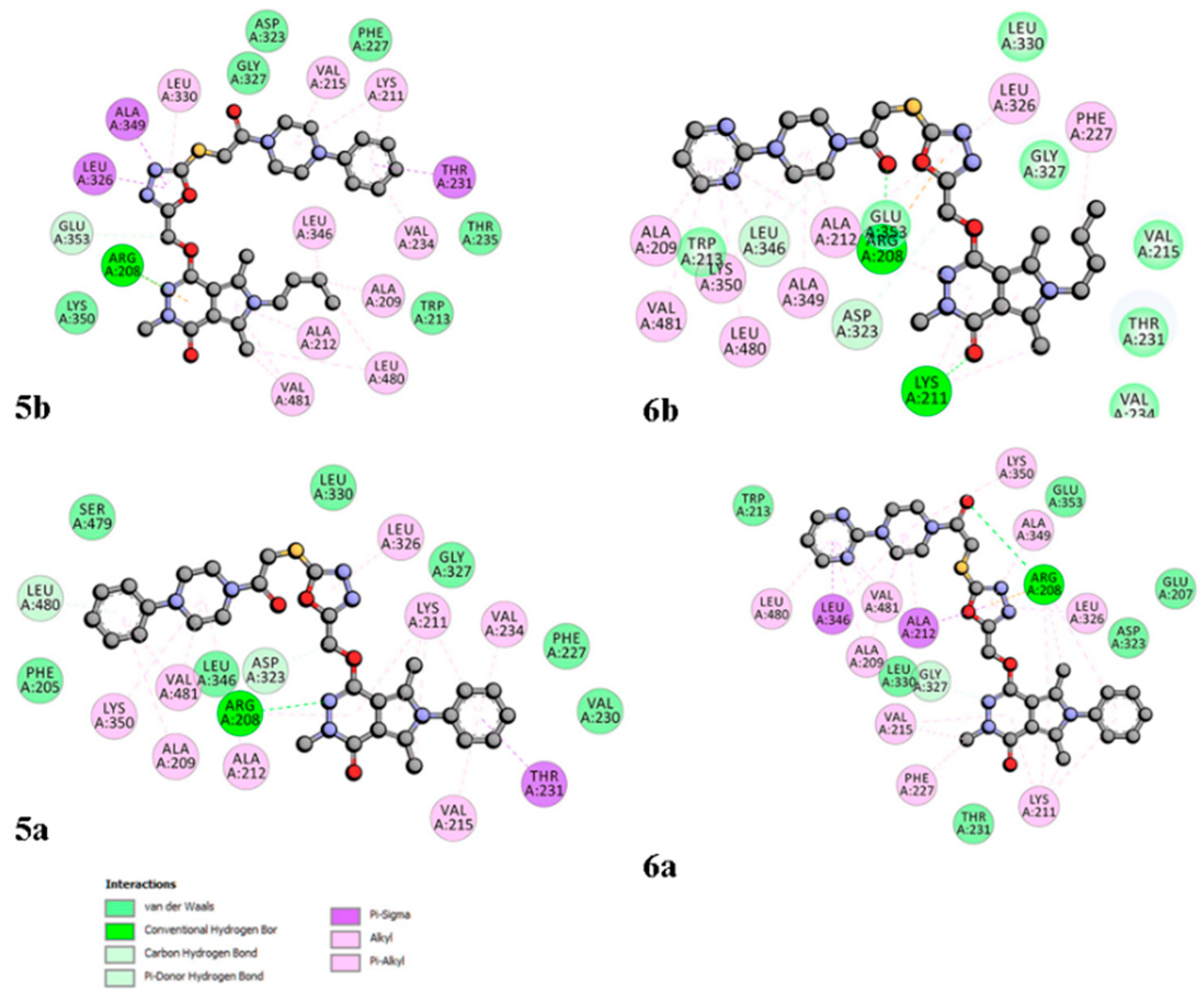
2.6. Site Markers Studies and Molecular Docking
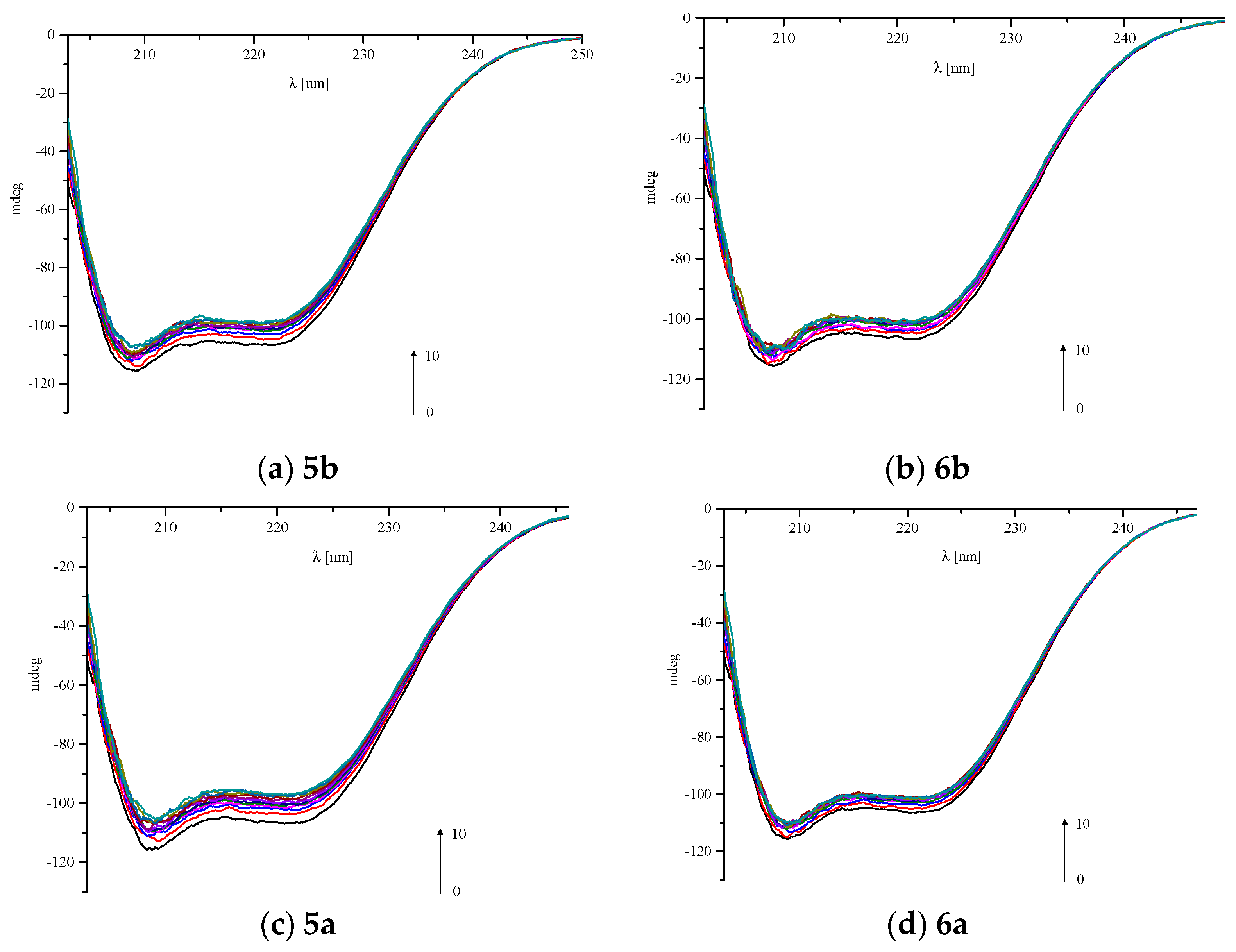
2.7. Circular Dichroism Spectra
3. Materials and Methods
3.1. Chemistry
3.1.1. Instrumentation and Chemicals
3.1.2. Chemical Synthesis
- General procedure for preparation of title derivatives of pyrrolo[3,4-d]pyridazinone 5a,b–6a,b.
3.2. Cell Line
3.3. Cell Culture Media
3.4. Tested Compounds
3.5. Experimental Design
3.6. MTT Assay
3.7. Level of Reactive Oxygen Species
3.8. Griess Assay
3.9. Fast Halo Assay
3.10. Fluorescence Spectroscopic Studies
3.11. Molecular Docking
3.12. Circular Dichroism
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marnett, L.J. Cyclooxygenase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J.; Hancock, A.B. PerspectiVe Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vane, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 2S–8S. [Google Scholar]
- Smith, W.L.; Urade, Y.; Jakobsson, J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [Green Version]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Bidaut-Russell, M.; Gabriel, S.E. Adverse gastrointestinal effects of NSAIDs: Consequences and costs. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 739–753. [Google Scholar] [CrossRef]
- Laine, L. Gastrointestinal effects of NSAIDs and coxibs. J. Pain Symptom Manag. 2003, 25, 32–40. [Google Scholar] [CrossRef]
- Wallace, J.L.; Devchand, P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekharan, N.V.; Dai, H.; Lamar, K.; Roos, T.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression PCOX-1 proteins). COX-3 and one of the PCOX-1 proteins (PCOX-1a) are made from the COX-1 gene but retain intron 1 in their mRNAs. PCOX-1 proteins additionally contain an in-frame deletion of exons 5-8 of the COX-1 mRNA. COX-3 and PCOX mRNAs. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar]
- Soll, A.H.; McCarthy, D. NSAID-related gastrointestinal complications. Clin. Cornerstone 1999, 1, 42–56. [Google Scholar] [CrossRef]
- Wallace, J.L. NSAID gastropathy and enteropathy: Distinct pathogenesis likely necessitates distinct prevention strategies. Br. J. Pharmacol. 2012, 165, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García Rodríguez, L.A.; Barreales Tolosa, L. Risk of Upper Gastrointestinal Complications Among Users of Traditional NSAIDs and COXIBs in the General Population. Gastroenterology 2007, 132, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K. Pathogenesis of NSAID-induced gastric damage: Importance of cyclooxygenase inhibition and gastric hypermotility. World J. Gastroenterol. 2012, 18, 2147–2160. [Google Scholar] [CrossRef]
- Esposito, G.; Pagano, E.; Ii, F.; Kaji, I.I.; Scarpignato, C.; Colucci, R.; Pellegrini, C.; Fornai, M.; Tirotta, E.; Antonioli, L.; et al. Pathophysiology of NSAID-Associated Intestinal Lesions in the Rat: Luminal Bacteria and Mucosal Inflammation as Targets for Prevention. Front. Pharmacol. 2018, 9, 1340. [Google Scholar]
- Koksal, M.; Ozkan-Dagliyan, I.; Ozyazici, T.; Kadioglu, B.; Sipahi, H.; Bozkurt, A.; Bilge, S.S. Some Novel Mannich Bases of 5-(3,4-Dichlorophenyl)-1,3,4-oxadiazole-2(3H)-one and Their Anti-Inflammatory Activity. Arch. Pharm. 2017, 350. [Google Scholar] [CrossRef]
- El-Sayed, N.A.; Nour, M.S.; Salem, M.A.; Arafa, R.K. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: Design, synthesis, cytotoxicity evaluation and in silico studies. Eur. J. Med. Chem. 2019, 183, 111693. [Google Scholar] [CrossRef]
- Banerjee, A.G.; Das, N.; Shengule, S.A.; Sharma, P.A.; Srivastava, R.S.; Shrivastava, S.K. Design, synthesis, evaluation and molecular modelling studies of some novel 5,6-diphenyl-1,2,4-triazin-3(2H)-ones bearing five-member heterocyclic moieties as potential COX-2 inhibitors: A hybrid pharmacophore approach. Bioorg. Chem. 2016, 69, 102–120. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo[3,4-d]pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Wakulik, K.; Wiatrak, B.; Szczukowski, Ł.; Bodetko, D.; Szandruk-Bender, M.; Dobosz, A.; Piotr’, P.; Atek, P.; Asiorowski, K.G. Effect of Novel Pyrrolo[3,4-d]pyridazinone Derivatives on Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. Artic. 2020, 21, 2575. [Google Scholar] [CrossRef] [Green Version]
- Malinka, W.; Redzicka, A.; Jastrzbska Wiȩsek, M.; Filipek, B.; Dybała, M.; Karczmarzyk, Z.; Urbańczyk-Lipkowska, Z.; Kalicki, P. Derivatives of pyrrolo[3,4-d]pyridazinone, a new class of analgesic agents. Eur. J. Med. Chem. 2011, 46, 4992–4999. [Google Scholar] [CrossRef]
- Mogilski, S.; Kubacka, M.; Redzicka, A.; Kazek, G.; Dudek, M.; Malinka, W.; Filipek, B. Antinociceptive, anti-inflammatory and smooth muscle relaxant activities of the pyrrolo[3,4-d]pyridazinone derivatives: Possible mechanisms of action. Pharmacol. Biochem. Behav. 2015, 133, 99–110. [Google Scholar] [CrossRef]
- Kohara, Y.; Imamiya, E.; Kubo, K.; Wada, T.; Inada, Y.; Naka, T. A new class of angiotensin II receptor antagonists with a novel acidic bioisostere. Bioorganic Med. Chem. Lett. 1995, 5, 1903–1908. [Google Scholar] [CrossRef]
- Kohara, Y.; Kubo, K.; Imamiya, E.; Wada, T.; Inada, Y.; Naka, T. Synthesis and Angiotensin II Receptor Antagonistic Activities of Benzimidazole Derivatives Bearing Acidic Heterocycles as Novel Tetrazole Bioisosteres 1. J. Med. Chem. 1996, 39, 5228–5235. [Google Scholar] [CrossRef]
- Tagad, H.D.; Hamada, Y.; Nguyen, J.T.; Hamada, T.; Abdel-Rahman, H.; Yamani, A.; Nagamine, A.; Ikari, H.; Igawa, N.; Hidaka, K.; et al. Design of pentapeptidic BACE1 inhibitors with carboxylic acid bioisosteres at P1′ and P4 positions. Bioorganic Med. Chem. 2010, 18, 3175–3186. [Google Scholar] [CrossRef]
- Palkar, M.B.; Singhai, A.S.; Ronad, P.M.; Vishwanathswamy, A.H.M.; Boreddy, T.S.; Veerapur, V.P.; Shaikh, M.S.; Rane, R.A.; Karpoormath, R. Synthesis, pharmacological screening and in silico studies of new class of Diclofenac analogues as a promising anti-inflammatory agents. Bioorganic Med. Chem. 2014, 22, 2855–2866. [Google Scholar] [CrossRef]
- Manjunatha, K.; Poojary, B.; Lobo, P.L.; Fernandes, J.; Kumari, N.S. Synthesis and biological evaluation of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5225–5233. [Google Scholar] [CrossRef] [PubMed]
- Dogruer, D.S.; Sahin, M.F.; Ünlü, S.; Ito, S. Studies on some 3(2H)-pyridazinone derivatives with antinociceptive activity. Arch. Pharm. 2000, 333, 79–86. [Google Scholar] [CrossRef]
- Dogruer, D.S.; Kupeli, E.; Yesilada, E.; Sahin, M.F. Synthesis of New 2-[1(2H)-Phthalazinon-2-yl]acetamide and 3-[1(2H)-Phthalazinon-2-yl]propanamide Derivatives as Antinociceptive and Anti-inflammatory Agents. Arch. Pharm. 2004, 337, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Dekhane, D.V.; Pawar, S.S.; Gupta, S.; Shingare, M.S.; Patil, C.R.; Thore, S.N. Synthesis and anti-inflammatory activity of some new 4,5-dihydro-1,5- diaryl-1H-pyrazole-3-substituted-heteroazole derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 6527–6532. [Google Scholar] [CrossRef]
- Bansal, S.; Bala, M.; Suthar, S.K.; Choudhary, S.; Bhattacharya, S.; Bhardwaj, V.; Singla, S.; Joseph, A. Design and synthesis of novel 2-phenyl-5-(1,3-diphenyl-1H-pyrazol-4-yl)-1, 3,4-oxadiazoles as selective COX-2 inhibitors with potent anti-inflammatory activity. Eur. J. Med. Chem. 2014, 80, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Pandey, D.; Mandalapu, D.; Bala, V.; Sharma, V.; Shukla, M.; Yadav, S.K.; Singh, N.; Jaiswal, S.; Maikhuri, J.P.; et al. Design, synthesis and biological profiling of aryl piperazine based scaffolds for the management of androgen sensitive prostatic disorders. Medchemcomm 2016, 7, 2111–2121. [Google Scholar] [CrossRef]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 inhibition activities and molecular docking study of isothiazolopyridine derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Z.; Huang, X.Z.; Xu, J.H.; Zneng, Z.Z.; Wang, Z.B. The Methods of Fluorescence Analysis, 2ed ed.; Science Press: Beijing, China, 1990. [Google Scholar]
- Wani, T.A.; Bakheit, A.H.; Zargar, S.; Bhat, M.A.; Al-Majed, A.A. Molecular docking and experimental investigation of new indole derivative cyclooxygenase inhibitor to probe its binding mechanism with bovine serum albumin. Bioorg. Chem. 2019, 89, 103010. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. (Ed.) Principles of Fluorescence Spectroscopy, 3rd ed.; Springer US: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Mohammadnia, F.; Fatemi, M.H.; Taghizadeh, S.M. Study on the interaction of anti-inflammatory drugs with human serum albumin using molecular docking, quantitative structure–activity relationship, and fluorescence spectroscopy. Luminescence 2020, 35, 266–273. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta Gen. Subj. 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Bakheit, A.H.; Mohamed, M.S.; Eldehna, W.M.; Abdel-Aziz, H.A.; Attia, M.I. Synthesis and biophysical insights into the binding of a potent anti-proliferative non-symmetric bis-isatin derivative with bovine serum albumin: Spectroscopic and molecular docking approaches. Appl. Sci. 2017, 7, 617. [Google Scholar] [CrossRef]
- Suryawanshi, V.D.; Walekar, L.S.; Gore, A.H.; Anbhule, P.V.; Kolekar, G.B. Spectroscopic analysis on the binding interaction of biologically active pyrimidine derivative with bovine serum albumin. J. Pharm. Anal. 2016, 6, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Wani, T.A.; Bakheit, A.H.; Al-Majed, A.R.A.; Bhat, M.A.; Zargar, S. Study of the interactions of bovine serum albumin with the new anti-inflammatory agent 4-(1,3-dsioxo-1,3-dihydro-2H-isoindol-2-yl)-N-[(4-ethoxy-phenyl) methylidene]benzohydrazide using a multi-spectroscopic approach and molecular docking. Molecules 2017, 22, 1258. [Google Scholar] [CrossRef] [Green Version]
- Krzyżak, E.; Szkatuła, D.; Wiatrak, B.; Gębarowski, T.; Marciniak, A. Synthesis, Cyclooxygenases Inhibition Activities and Interactions with BSA of N-substituted 1H-pyrrolo[3,4-c]pyridine-1,3(2H)-diones Derivatives. Molecules 2020, 25, 2934. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The Characterization of Two Specific Drug Binding Sites on Human Serum Albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Klotz, I.M.; Urquhart, J.M. The Binding of Organic Ions by Proteins. Effect of Temperature. J. Am. Chem. Soc. 1949, 71, 847–851. [Google Scholar] [CrossRef]
- Kelly, S.; Price, N. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2005, 1, 349–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Lu, Z.X.; Cui, T.; Shi, Q.L. Applications of Circular Dichroism (CD) and Optical Rotatory Dispersion (ORD) in Molecular Biology, 1st ed.; Science Press: Beijing, China, 1987. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian~16 {R}evision {A}.03 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 inhibition activities and molecular docking study of newly designed and synthesized pyrrolo[3,4-c]pyrrole Mannich bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, H.; Liu, J.; Lau, C.W.; Liu, P.; Chen, Z.Y.; Lee, H.K.; Tipoe, G.L.; Ho, H.M.; Yao, X.; et al. Cyclooxygenase-2-dependent oxidative stress mediates palmitate-induced impairment of endothelium-dependent relaxations in mouse arteries. Biochem. Pharmacol. 2015, 91, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Burdon, C.; Mann, C.; Cindrova-Davies, T.; Ferguson-Smith, A.C.; Burton, G.J. Oxidative Stress and the Induction of Cyclooxygenase Enzymes and Apoptosis in the Murine Placenta. Placenta 2007, 28, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
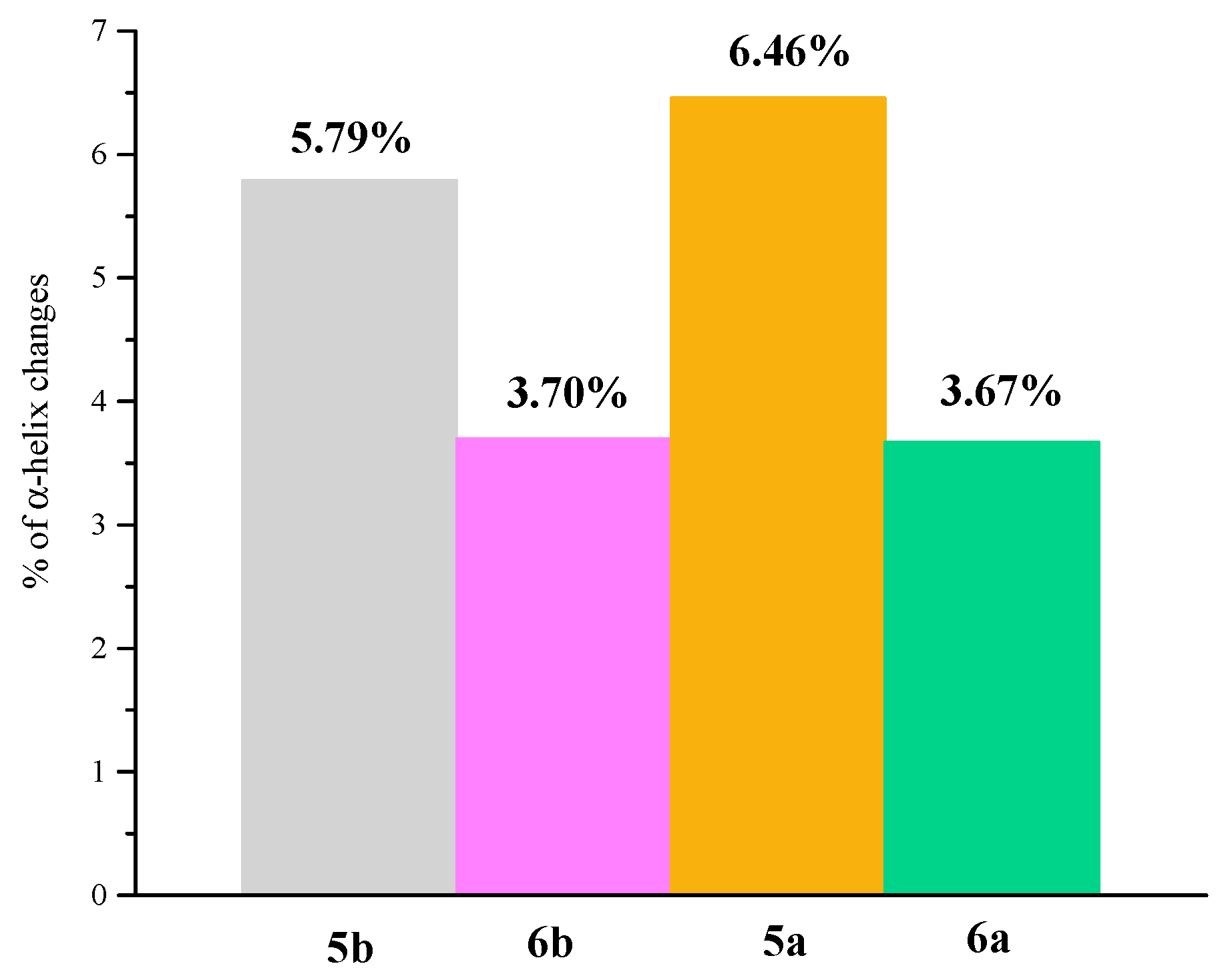{kind=link}
{kind=link}
{kind=link}
| Compound | Cyclooxygenase Inhibition Assay IC50 [µM] | |
|---|---|---|
| COX-1 | COX-2 | |
| 5a | ND 1 | 658.7 (15.0) |
| 5b | ND 1 | 257.4 (11.3) |
| 6a | ND 1 | 160.2 (6.8) |
| 6b | ND 1 | 371.0 (10.1) |
| Meloxicam | 83.7 (1.8) | 59.2 (2.4) |
| ∆G° [kJmol−1] | ∆E1 [kJmol−1] | ∆E2 [kJmol−1] | ∆E3 [kJmol−1] | Ki [μM] | |
|---|---|---|---|---|---|
| 5a | −20.44 | −30.18 | −30.43 | 0.25 | 285.75 |
| 5b | −26.75 | −39.20 | −38.87 | −0.33 | 20.39 |
| 6a | −30.54 | −40.52 | −40.88 | −0.37 | 4.44 |
| 6b | −21.06 | −33.52 | −35.69 | 2.13 | 202.61 |
| Meloxicam | −34.02 | −37.74 | −37.32 | −0.42 | 1.09 |
| Compound | Concentration [µM] | ROS | RNS | DNA Damage |
|---|---|---|---|---|
| 10 | −33.1 (0.04) * | −3.7 (0.03) | −68 (0.1) * | |
| 5a | 50 | −34.2 (0.06) * | −5.9 (0.02) | −47 (0.08) * |
| 100 | −28.7 (0.03) * | 12.1 (0.02) * | −38 (0.1) * | |
| 10 | −29.0 (0.02) * | −6.7 (0.03) * | −59 (0.08) * | |
| 5b | 50 | −9.7 (0.02) * | 3.4 (0.04) | −12 (0.07) * |
| 100 | −0.5 (0.04) | 9.8 (0.04) | 5.7 (0.2) | |
| 10 | −7.4 (0.03) | −11.0 (0.07) * | −2.4 (0.1) | |
| 6a | 50 | 12.3 (0.05) | −6.9 (0.05) | 26 (0.06) * |
| 100 | 18.9 (0.06) * | −4.1 (0.04) | 24 (0.08) * | |
| 10 | −24.1 (0.05) * | −7.9 (0.05) * | −70 (0.06) * | |
| 6b | 50 | −12.6 (0.04) * | −1.4 (0.04) | −22 (0.09) * |
| 100 | −1.8 (0.02) | 4.6 (0.02) | −9.7 (0.1) * |
| Compound | ROS Level vs. DNA Damage | NO Level vs. DNA Damage |
|---|---|---|
| 5a | 0.589 | 0.650 |
| 5b | 0.998 | 0.991 |
| 6a | 0.953 | 0.888 |
| 6b | 0.951 | 0.950 |
| Quenching | Binding | Thermodynamic | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T [K] | Ksv × 105 [dm3·mol−1] | kq × 1013 [dm3·mol−1·s−1] | logKb | Kb × 104 [dm3·mol−1] | n | ∆G° [kJmol−1] | ∆H° [kJmol−1] | ∆S° [Jmol−1K−1] | |
| 5a | 294 | 4.44 | 4.44 | 5.09 ± 0.14 | 12.30 | 0.91 ± 0.02 | −29.31 | −133.03 | −352.78 |
| 301 | 2.45 | 4.45 | 4.90 ± 0.30 | 7.94 | 0.92 ± 0.05 | ||||
| 308 | 2.09 | 2.09 | 4.01 ± 0.13 | 1.02 | 0.77 ± 0.03 | ||||
| 5b | 294 | 4.44 | 4.44 | 5.09 ± 0.14 | 12.30 | 0.91 ± 0.02 | −29.31 | −133.03 | −352.78 |
| 301 | 2.45 | 4.45 | 4.90 ± 0.30 | 7.94 | 0.92 ± 0.05 | ||||
| 308 | 2.09 | 2.09 | 4.01 ± 0.13 | 1.02 | 0.77 ± 0.03 | ||||
| 6a | 294 | 2.12 | 2.12 | 4.86 ± 0.05 | 7.24 | 0.92 ± 0.01 | −27.51 | −108.79 | −276.43 |
| 301 | 1.27 | 1.17 | 4.50 ± 0.13 | 3.16 | 0.89 ± 0.02 | ||||
| 308 | 1.13 | 2.74 | 3.98 ± 0.27 | 0.96 | 0.81 ± 0.04 | ||||
| 6b | 294 | 2.90 | 2.90 | 4.93 ± 0.07 | 8.51 | 0.91 ± 0.01 | −27.37 | −77.15 | −169.33 |
| 301 | 1.80 | 1.80 | 4.41 ± 0.29 | 2.57 | 0.85 ± 0.05 | ||||
| 308 | 1.74 | 4.43 | 4.31 ± 0.26 | 2.04 | 0.84 ± 0.04 | ||||
| Binding Site | ∆G° [kJmol−1] | ∆E1 [kJmol−1] | ∆E2 [kJmol−1] | ∆E3 [kJmol−1] | |
|---|---|---|---|---|---|
| 5a | site I | −31.43 | −41.42 | −40.88 | −0.54 |
| site II | −38.96 | −48.94 | −48.44 | −0.46 | |
| 5b | site I | −27.38 | −39.83 | −38.70 | −1.13 |
| site II | −38.87 | −51.37 | −51.03 | −0.33 | |
| 6a | site I | −34.73 | −44.72 | −40.34 | −4.39 |
| site II | −36.94 | −47.10 | −44.51 | −2.59 | |
| 6b | site I | −30.55 | −43.01 | −38.70 | −4.31 |
| site II | −39.12 | −51.62 | −49.49 | −0.46 |
| BSA/Analyzed Compound Molar Ratio | α-helix(%) | |||
|---|---|---|---|---|
| 5a | 5b | 6a | 6b | |
| 1:0 | 54.05 | 53.55 | 53.78 | 53.46 |
| 1:0.5 | 51.08 | 52.15 | 52.77 | 52.45 |
| 1:1 | 50.51 | 51.11 | 51.72 | 51.83 |
| 1:2 | 49.94 | 50.50 | 51.70 | 51.26 |
| 1:3 | 49.70 | 50.47 | 51.45 | 50.91 |
| 1:4 | 49.62 | 49.81 | 51.49 | 50.47 |
| 1:5 | 49.48 | 49.78 | 51.40 | 50.23 |
| 1:6 | 48.85 | 49.51 | 50.28 | 50.15 |
| 1:7 | 48.57 | 49.21 | 50.29 | 50.10 |
| 1:8 | 48.23 | 48.85 | 50.31 | 49.97 |
| 1:9 | 48.11 | 48.06 | 50.24 | 49.82 |
| 1:10 | 47.59 | 47.76 | 50.11 | 49.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczukowski, Ł.; Krzyżak, E.; Zborowska, A.; Zając, P.; Potyrak, K.; Peregrym, K.; Wiatrak, B.; Marciniak, A.; Świątek, P. Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-d]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors. Int. J. Mol. Sci. 2020, 21, 9623. https://doi.org/10.3390/ijms21249623
Szczukowski Ł, Krzyżak E, Zborowska A, Zając P, Potyrak K, Peregrym K, Wiatrak B, Marciniak A, Świątek P. Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-d]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors. International Journal of Molecular Sciences. 2020; 21(24):9623. https://doi.org/10.3390/ijms21249623
Chicago/Turabian StyleSzczukowski, Łukasz, Edward Krzyżak, Adrianna Zborowska, Patrycja Zając, Katarzyna Potyrak, Krzysztof Peregrym, Benita Wiatrak, Aleksandra Marciniak, and Piotr Świątek. 2020. "Design, Synthesis and Comprehensive Investigations of Pyrrolo[3,4-d]pyridazinone-Based 1,3,4-Oxadiazole as New Class of Selective COX-2 Inhibitors" International Journal of Molecular Sciences 21, no. 24: 9623. https://doi.org/10.3390/ijms21249623