Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells

 ,
,  ,
,  ,
,
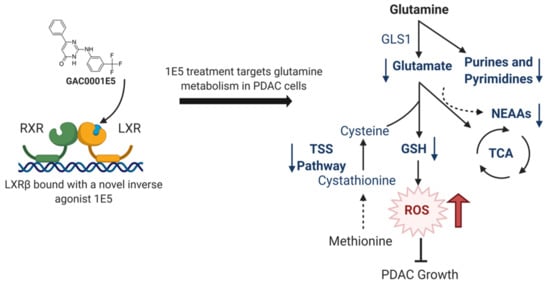
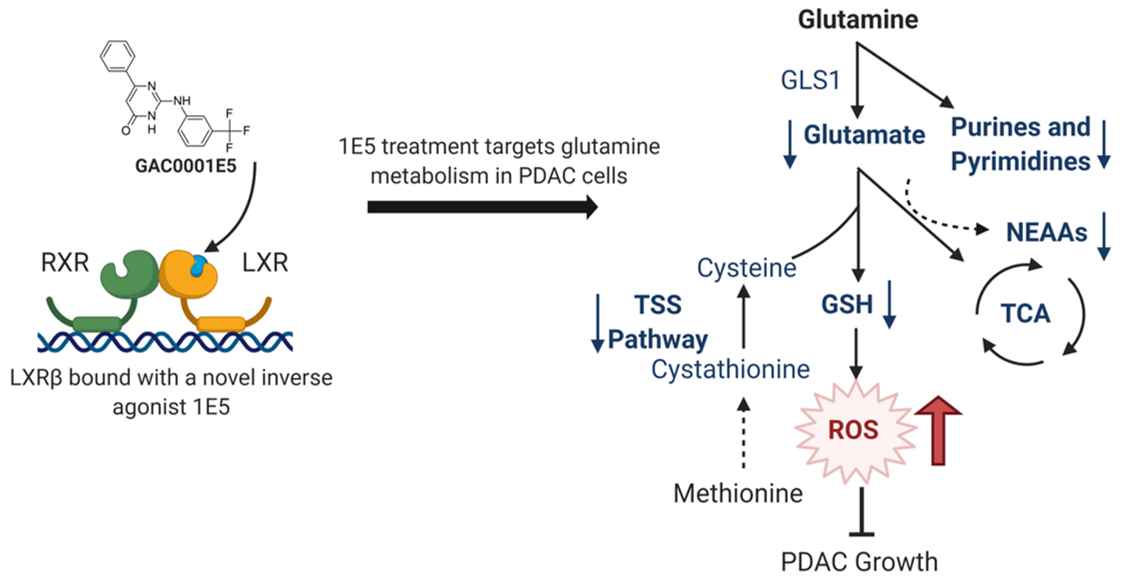
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
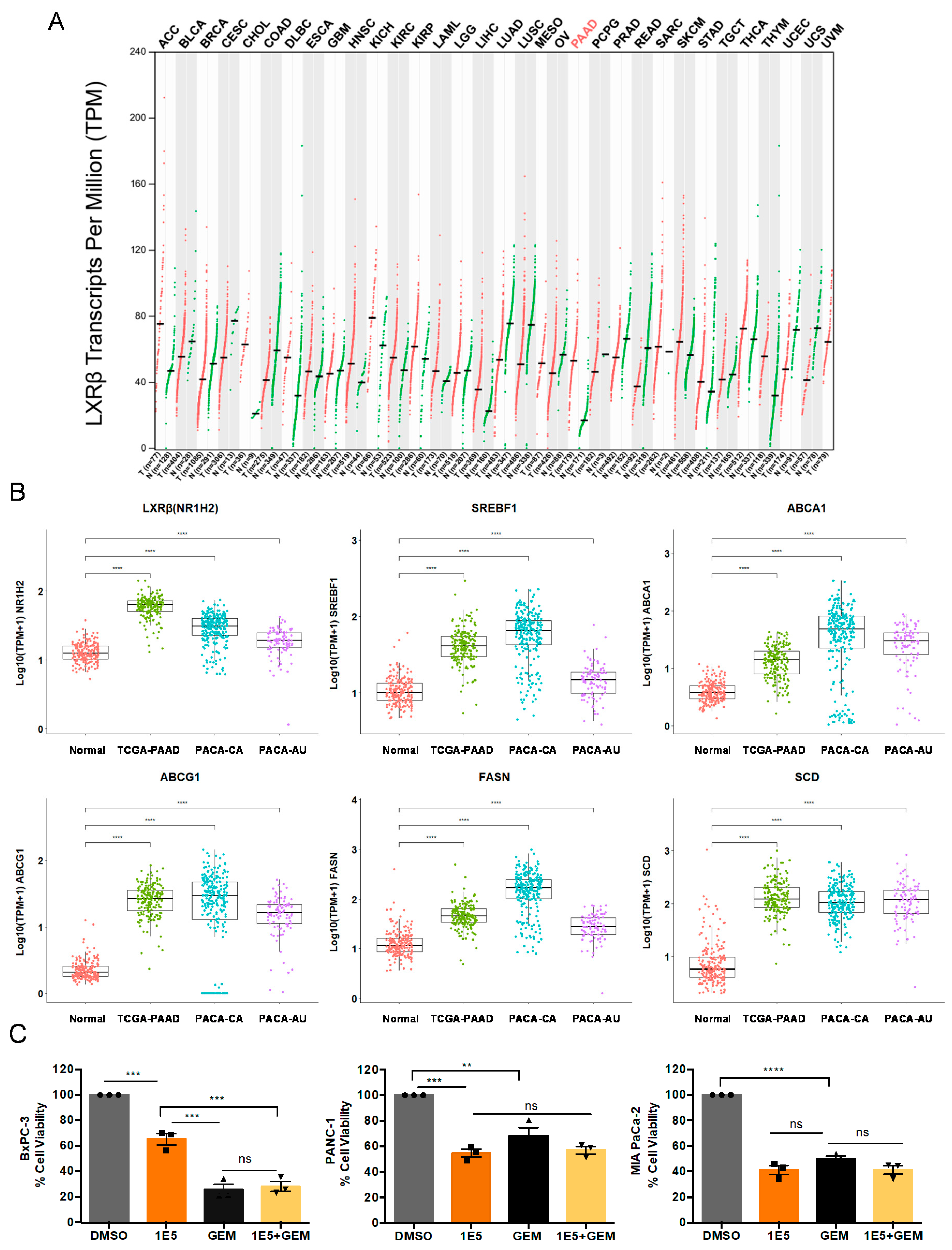
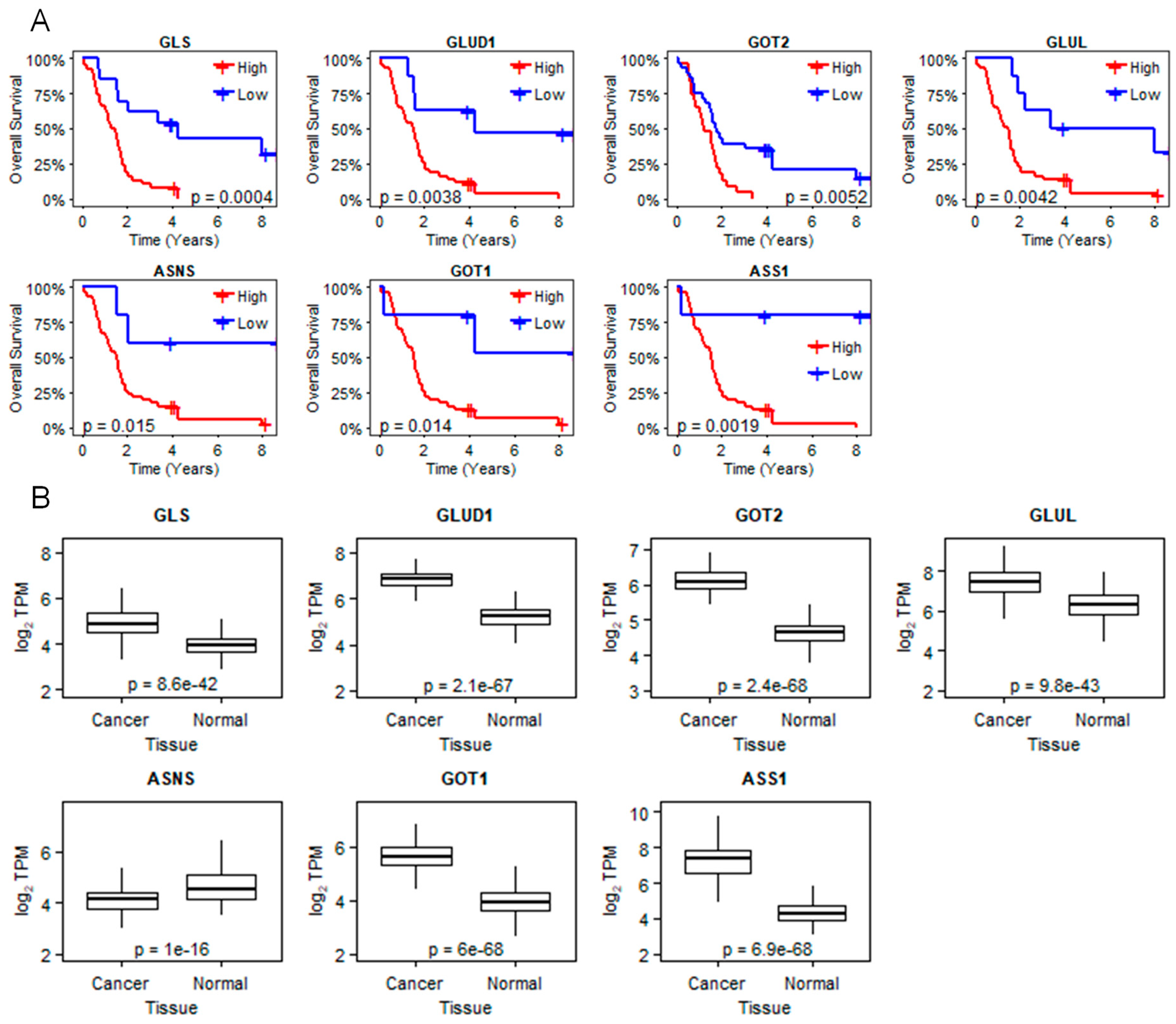
2.1. LXRβ Is Overexpressed in Pancreatic Cancer and LXR Ligand 1E5 Inhibits PDAC Cell Proliferation
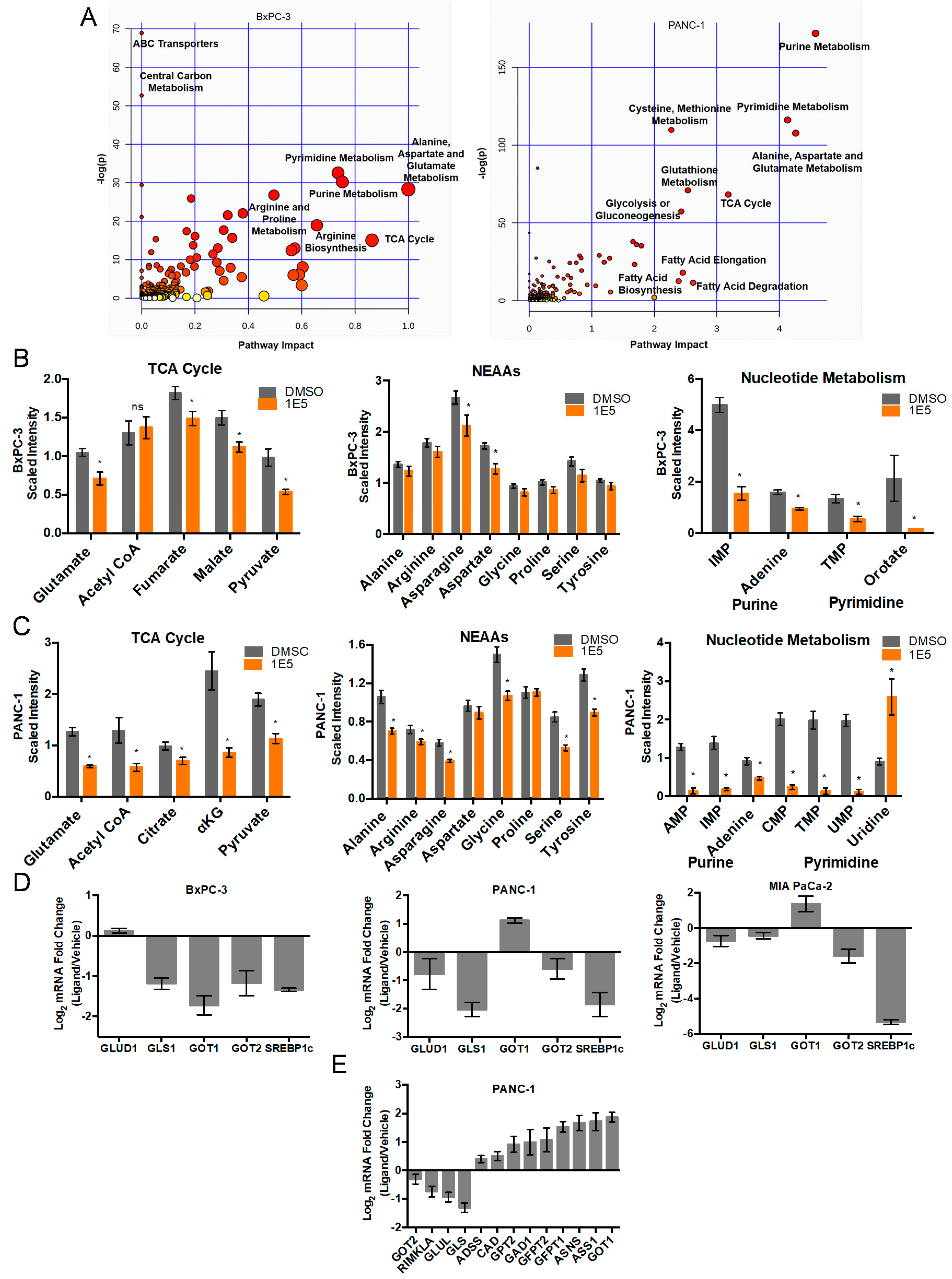
2.2. LXR Inverse Agonist and Degrader 1E5 Is a Potent Inhibitor of Glutamine Metabolism
2.3. Combination Treatment of 1E5 with GLS Inhibitor BPTES Synergistically Decreases Glutaminolysis and Growth of PDAC Cells
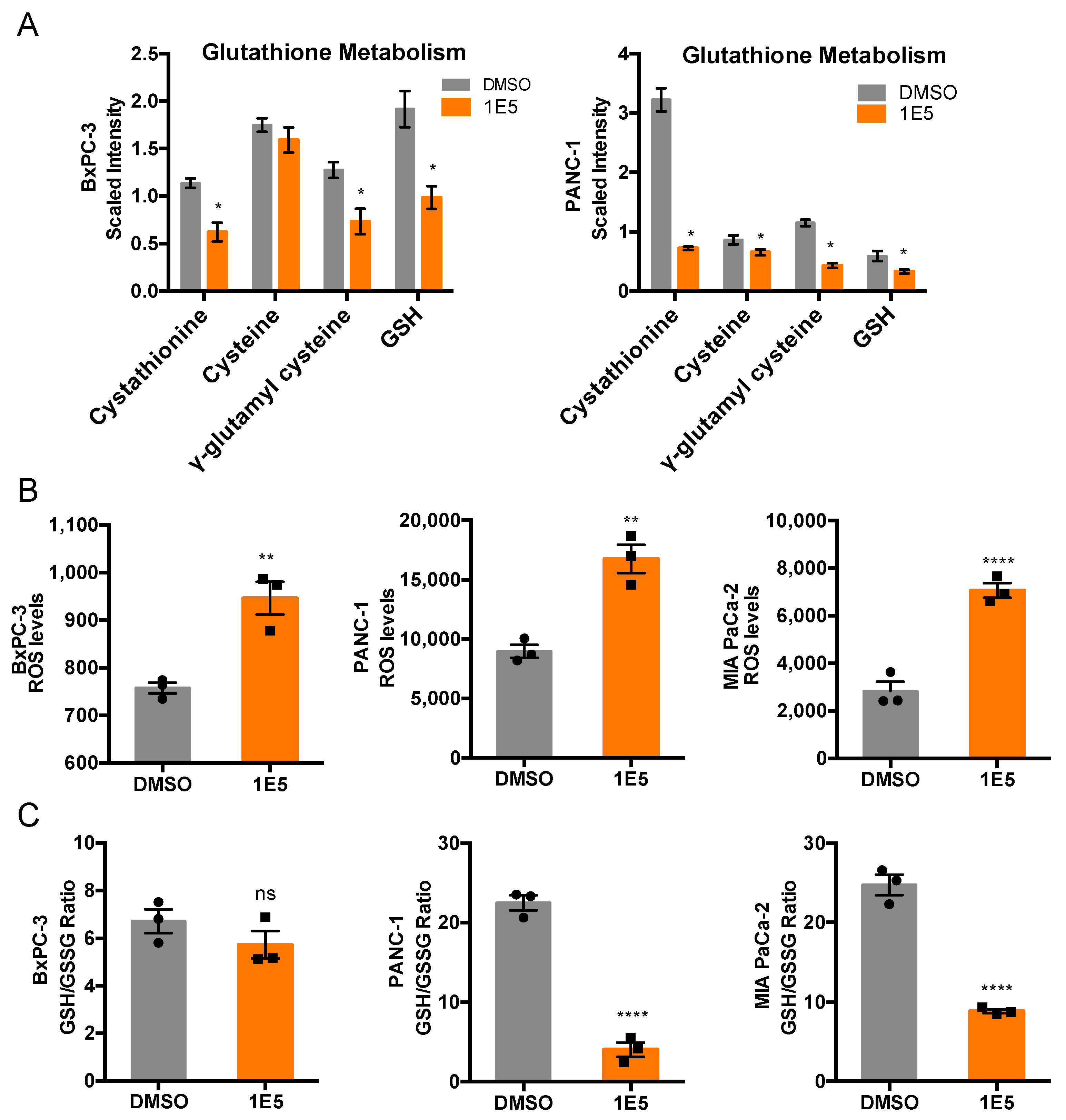
2.4. 1E5 Treatment Induces Oxidative Stress in PDAC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Cell Proliferation Assay
4.3. Real-Time Quantitative PCR
- 36B4-Forward: 5′-GTGTTCGACAATGGCAGCAT-3′
- 36B4-Reverse: 5′-GACACCCTCCAGGAAGCGA-3′
- GLUD1-Forward: 5′-AGGAATGACACCAGGGTTTG-3′
- GLUD1-Reverse: 5′-GAGGGTTGGAATACATGGGAC-3′
- GLS1-Forward: 5′-TTCCAGAAGGCACAGACATG-3′
- GLS1-Reverse: 5′-GGCTCAGTACTCTTTCACCAG-3′
- GOT1-Forward: 5′-CAACTGGGATTGACCCAACT-3′
- GOT1-Reverse: 5′-GGAACAGAAACCGGTGCTT-3′
- GOT2-Forward: 5′-GTTTGCCTCTGCCAATCATATG-3′
- GOT2-Reverse: 5′-GAGGGTTGGAATACATGGGAC-3′
- SREBP-1c-Forward: 5′-GGAGGGGTAGGGCCAACGGCCT-3′
- SREBP-1c-Reverse: 5′-CATGTCTTCGAAAGTGCAATCC-3′
4.4. Metabolomics
4.5. Intracellular Glutamate Assay
4.6. ROS Measurement
4.7. GSH/GSSG Assay
4.8. RNAseq Library Preparation and Sequencing
4.9. RNAseq Analysis
4.10. Survival Analysis
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| LXR | Liver X receptor |
| RXR | Retinoid X receptor |
| GEM | Gemcitabine |
| GAC0001E5 | 1E5 |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| TCGA-PAAD | The Cancer Genome Atlas-Pancreatic Adenocarcinoma |
| PACA-CA | Pancreatic Cancer-Canada |
| PACA-AU | Pancreatic Cancer-Australia |
| GSEA | Gene set enrichment analysis |
| GLS | Glutaminase |
| NEAAs | Non-essential amino acids |
| GSH | Glutathione |
| ROS | Reactive oxygen species |
| TSS | Transsulfuration |
| TPM | Transcripts per million |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [PubMed]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F.; et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res. 2020, 80, 3101–3115. [Google Scholar] [PubMed] [Green Version]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharm. Sci. 2012, 33, 394–404. [Google Scholar]
- Lin, C.Y.; Gustafsson, J.A. Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 2015, 15, 216–224. [Google Scholar]
- Lin, C.Y.; Vedin, L.L.; Steffensen, K.R. The emerging roles of liver X receptors and their ligands in cancer. Expert Opin. Ther. Targets 2016, 20, 61–71. [Google Scholar]
- Candelaria, N.R.; Addanki, S.; Zheng, J.; Nguyen-Vu, T.; Karaboga, H.; Dey, P.; Gabbi, C.; Vedin, L.L.; Liu, K.; Wu, W.; et al. Antiproliferative effects and mechanisms of liver X receptor ligands in pancreatic ductal adenocarcinoma cells. PLoS ONE 2014, 9, e106289. [Google Scholar]
- Karaboga, H.; Huang, W.; Srivastava, S.; Widmann, S.; Addanki, S.; Gamage, K.T.; Mazhar, Z.; Ebalunode, J.O.; Briggs, J.M.; Gustafsson, J.-Å.; et al. Screening of focused compound library targeting liver X receptors in pancreatic cancer identified ligands with inverse agonist and degrader activity. ACS Chem. Biol. 2020, 15, 2916–2928. [Google Scholar]
- Pencheva, N.; Buss, C.G.; Posada, J.; Merghoub, T.; Tavazoie, S.F. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell 2014, 156, 986–1001. [Google Scholar]
- Carpenter, K.J.; Valfort, A.C.; Steinauer, N.; Chatterjee, A.; Abuirqeba, S.; Majidi, S.; Sengupta, M.; Di Paolo, R.J.; Shornick, L.P.; Zhang, J.; et al. LXR-inverse agonism stimulates immune-mediated tumor destruction by enhancing CD8 T-cell activity in triple negative breast cancer. Sci. Rep. 2019, 9, 19530. [Google Scholar]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [PubMed] [Green Version]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 2019, 29, 1287–1298. [Google Scholar] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A hallmark of cancer metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar]
- Vučetić, M.; Cormerais, Y.; Parks, S.K.; Pouysségur, J. The Central Role of Amino Acids in Cancer Redox Homeostasis: Vulnerability Points of the Cancer Redox Code. Front. Oncol. 2017, 7, 309. [Google Scholar]
- Biancur, D.E.; Paulo, J.A.; Malachowska, B.; Quiles Del Rey, M.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar]
- Biancur, D.E.; Kimmelman, A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 67–75. [Google Scholar]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, M.K.; Ramaker, R.C.; Gertz, J.; Davis, N.S.; Johnston, B.E.; Oliver, P.G.; Sexton, K.C.; Greeno, E.W.; Christein, J.D.; Heslin, M.J.; et al. RNA sequencing of pancreatic adenocarcinoma tumors yields novel expression patterns associated with long-term survival and reveals a role for ANGPTL4. Mol. Oncol. 2016, 10, 1169–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tataranni, T.; Agriesti, F.; Ruggieri, V.; Mazzoccoli, C.; Simeon, V.; Laurenzana, I.; Scrima, R.; Pazienza, V.; Capitanio, N.; Piccoli, C. Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles. Oncotarget 2017, 8, 41265–41281. [Google Scholar] [CrossRef] [Green Version]
- Roux, C.; Riganti, C.; Borgogno, S.F.; Curto, R.; Curcio, C.; Catanzaro, V.; Digilio, G.; Padovan, S.; Puccinelli, M.P.; Isabello, M.; et al. Endogenous glutamine decrease is associated with pancreatic cancer progression. Oncotarget 2017, 8, 95361–95376. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinina, E.V.; Gavriliuk, L.A. Glutathione synthesis in cancer cells. Biochemistry 2020, 85, 895–907. [Google Scholar] [CrossRef]
- Chakrabarti, G.; Moore, Z.R.; Luo, X.; Ilcheva, M.; Ali, A.; Padanad, M.; Zhou, Y.; Xie, Y.; Burma, S.; Scaglioni, P.P.; et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.T.; Ishida, C.T.; Shang, E.; Shu, C.; Torrini, C.; Zhang, Y.; Bianchetti, E.; Sanchez-Quintero, M.J.; Kleiner, G.; Quinzii, C.M.; et al. Activation of LXRβ inhibits tumor respiration and is synthetically lethal with Bcl-xL inhibition. EMBO Mol. Med. 2019, 11, e10769. [Google Scholar] [CrossRef]
- Pehkonen, P.; Welter-Stahl, L.; Diwo, J.; Ryynänen, J.; Wienecke-Baldacchino, A.; Heikkinen, S.; Treuter, E.; Steffensen, K.R.; Carlberg, C. Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages. BMC Genomics 2012, 13, 50. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Berisa, M.; Schwörer, S.; Qin, W.; Cross, J.R.; Thompson, C.B. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019, 30, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Hichor, M.; Sundaram, V.K.; Eid, S.A.; Abdel-Rassoul, R.; Petit, P.X.; Borderie, D.; Bastin, J.; Eid, A.A.; Manuel, M.; Grenier, J.; et al. Liver X Receptor exerts a protective effect against the oxidative stress in the peripheral nerve. Sci. Rep. 2018, 8, 2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.M.; Bridgewater, B.; Liu, Q.; Mitchell, M.; Robinson, R.; Dai, H.; Stewart, S.; Dehaven, C.D.; Miller, L. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in highthroughput profiling metabolomics. Metab. Open Access. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef] [PubMed]
- Dehaven, C.D.; Evans, A.M.; Dai, H.; Lawton, K.A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2010, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010, 26, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Widmann, S.; Ho, C.; Nguyen, D.; Nguyen, A.; Premaratne, A.; Gustafsson, J.-Å.; Lin, C.-Y. Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9622. https://doi.org/10.3390/ijms21249622
Srivastava S, Widmann S, Ho C, Nguyen D, Nguyen A, Premaratne A, Gustafsson J-Å, Lin C-Y. Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2020; 21(24):9622. https://doi.org/10.3390/ijms21249622
Chicago/Turabian StyleSrivastava, Shivangi, Scott Widmann, Charles Ho, Donovan Nguyen, Alexis Nguyen, Asitha Premaratne, Jan-Åke Gustafsson, and Chin-Yo Lin. 2020. "Novel Liver X Receptor Ligand GAC0001E5 Disrupts Glutamine Metabolism and Induces Oxidative Stress in Pancreatic Cancer Cells" International Journal of Molecular Sciences 21, no. 24: 9622. https://doi.org/10.3390/ijms21249622