Genome-Wide Survey and Expression Analysis of the KT/HAK/KUP Family in Brassica napus and Its Potential Roles in the Response to K+ Deficiency



Abstract
:1. Introduction
2. Results
2.1. Identification of HAK Family Members in the B. napus Genome
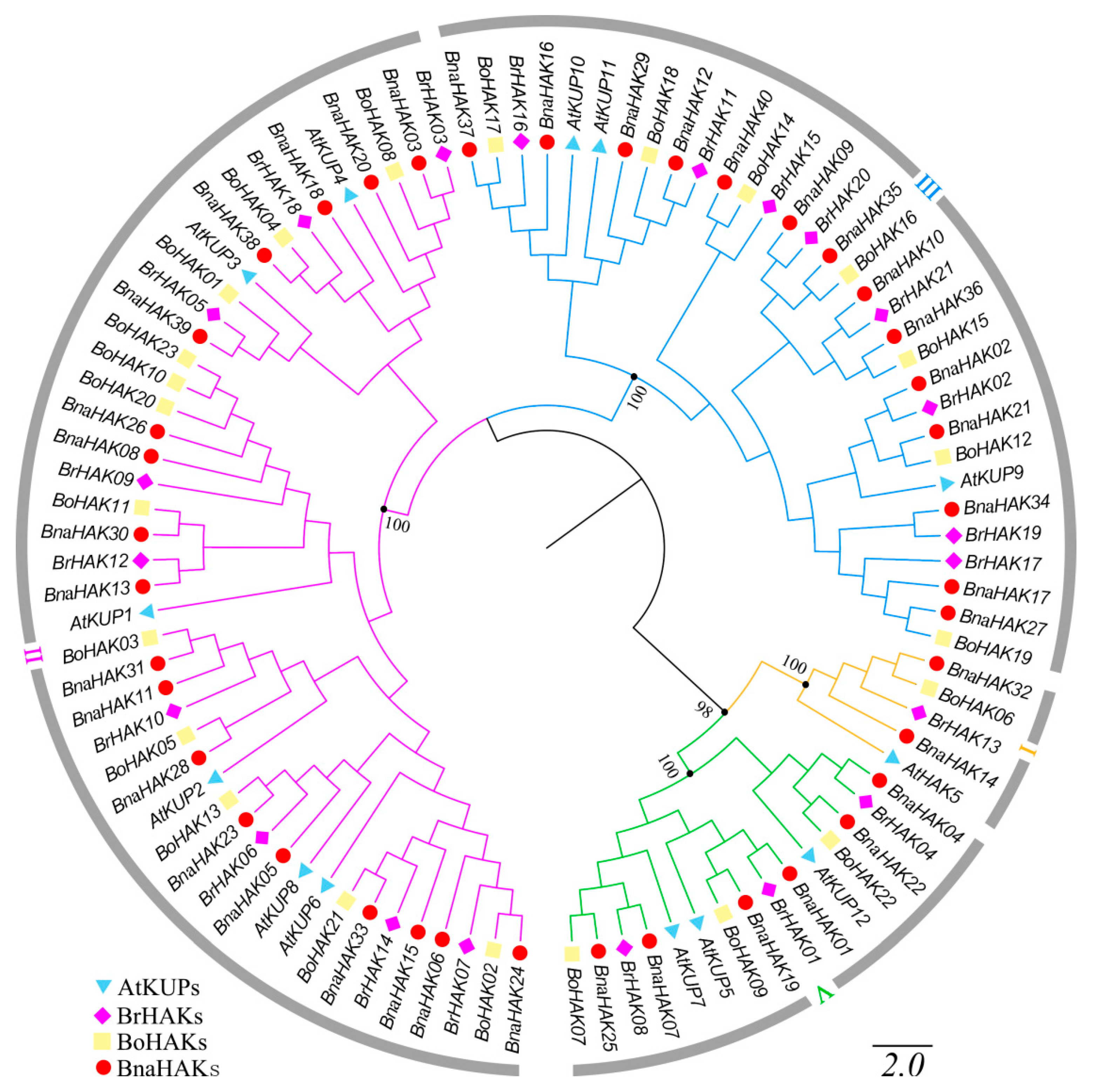
2.2. Phylogenetic Analysis of the BnaHAK Family
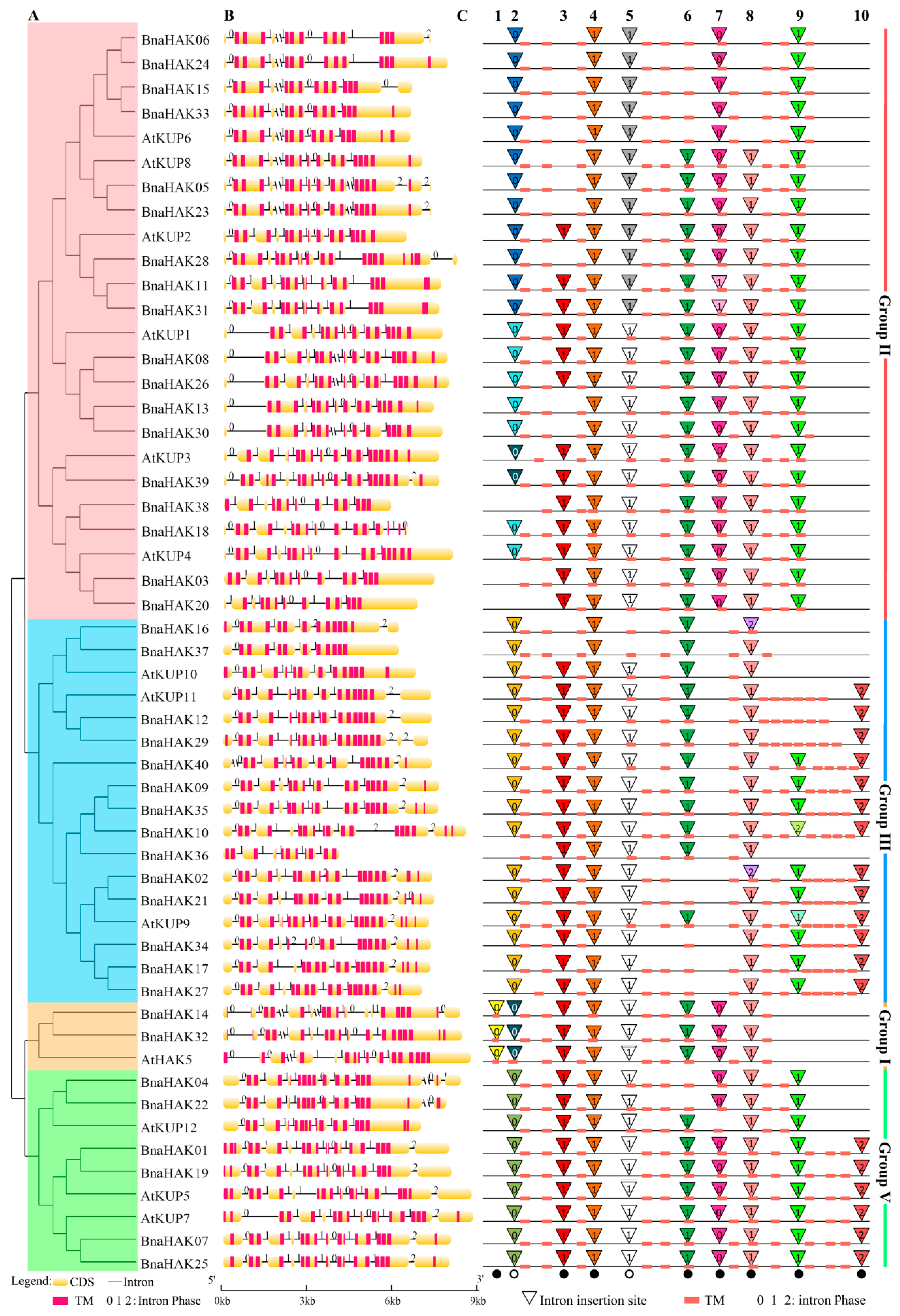
2.3. Gene Structure and Intron Pattern of BnaHAKs
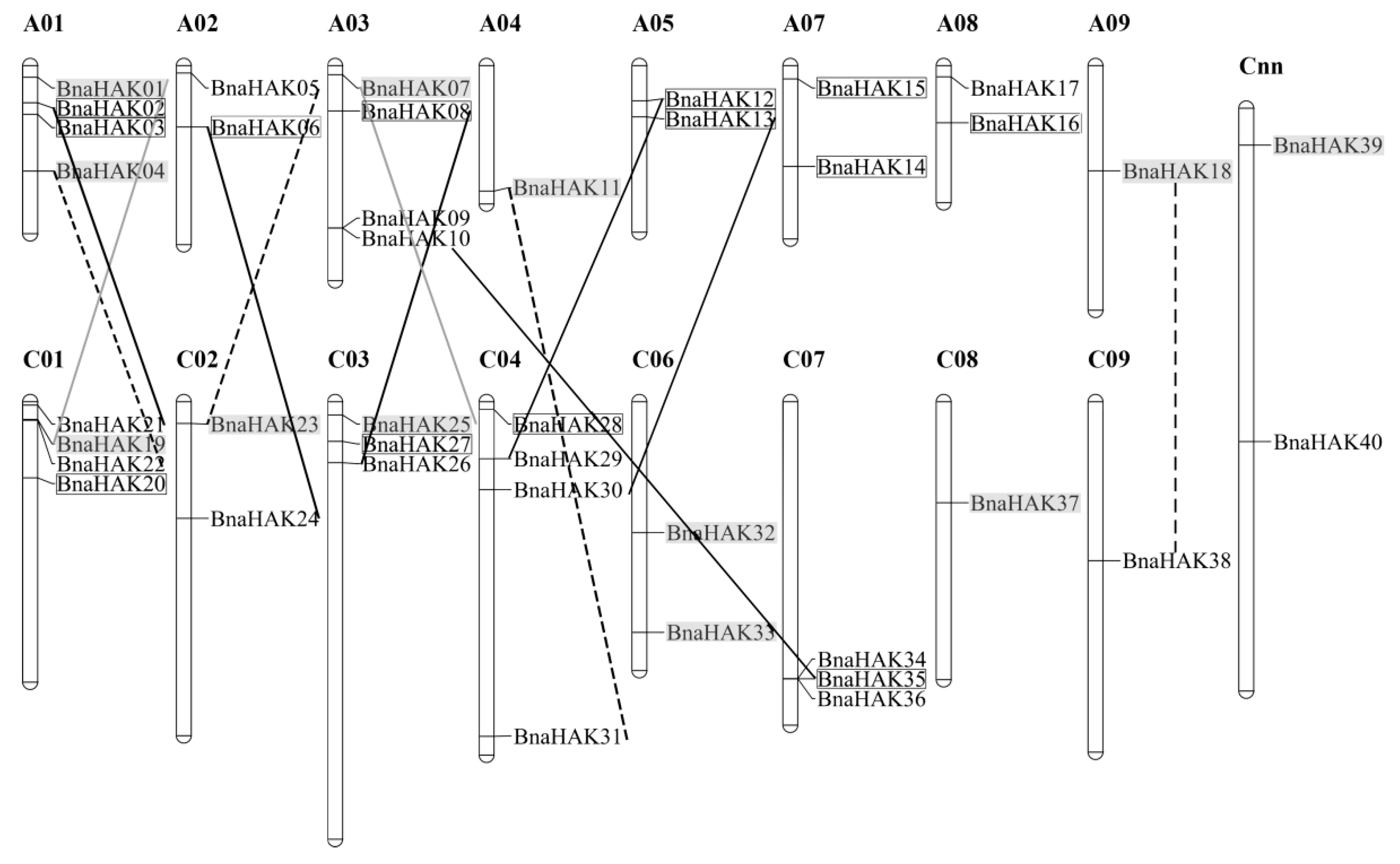
2.4. Chromosomal Distribution and Duplication of BnaHAKs
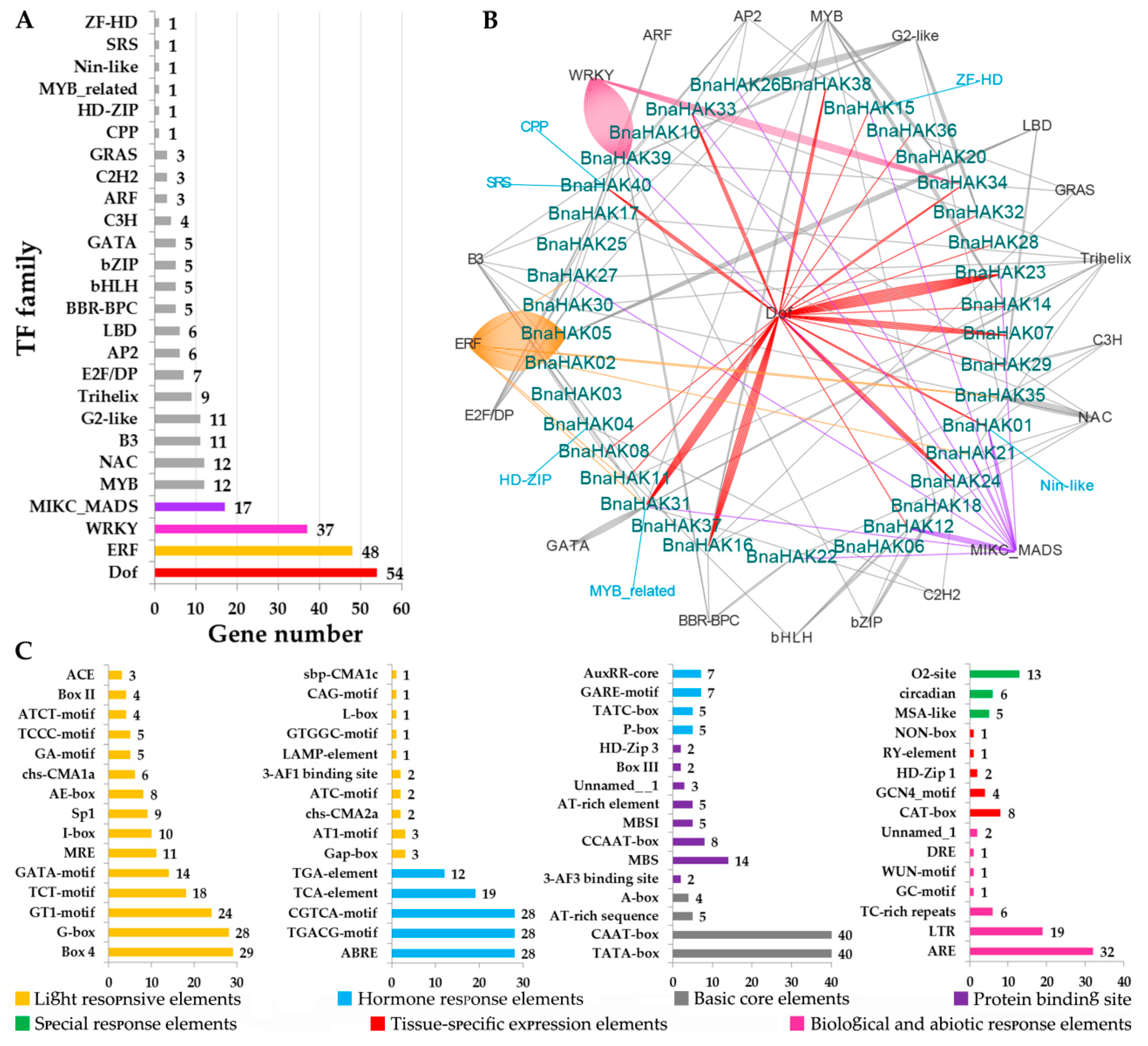
2.5. Regulation Mechanism in the Promoter Regions of BnaHAKs
2.6. Comprehensive Analysis of miRNAs Targeting BnaHAKs
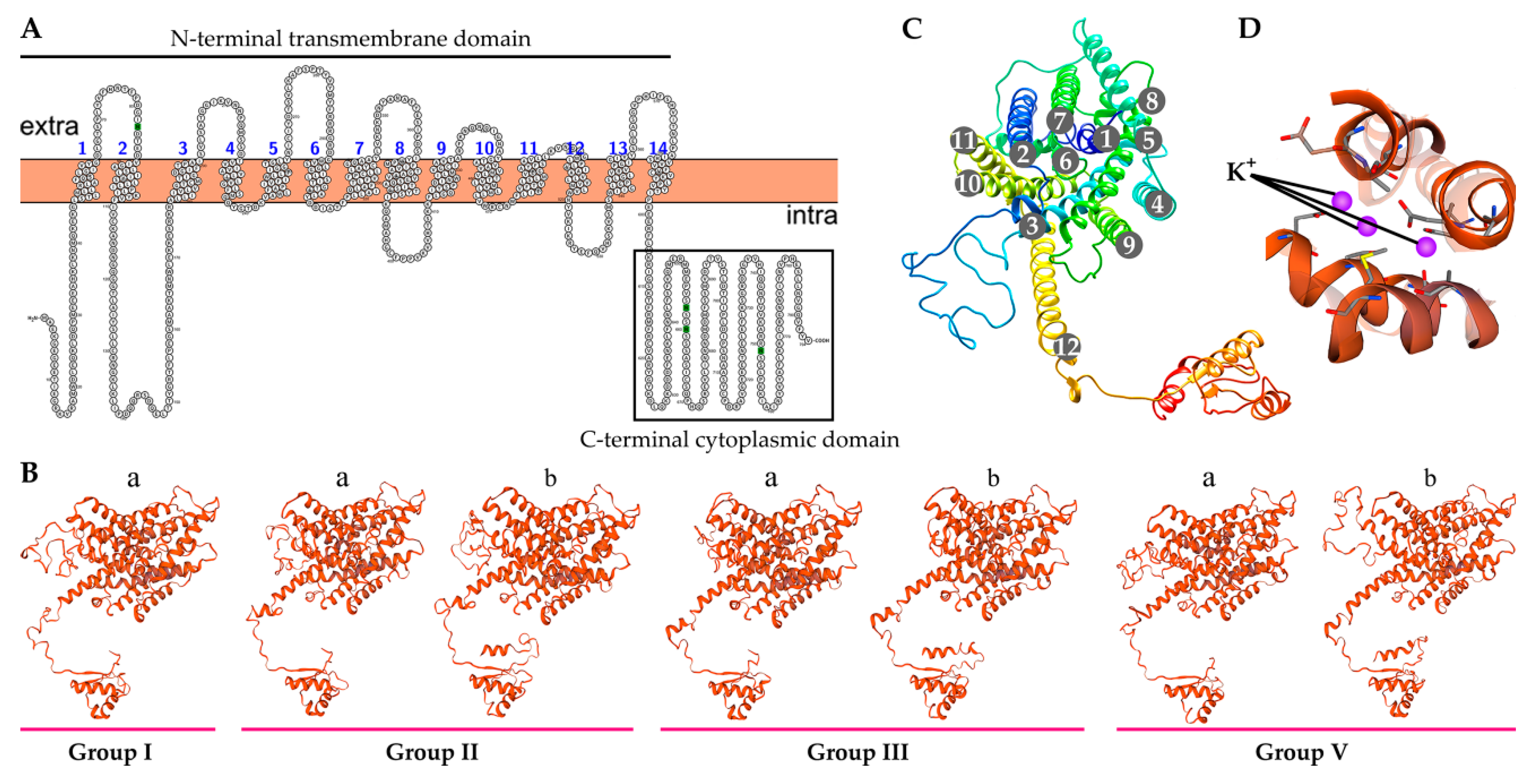
2.7. Characteristics of the Predicted BnaHAK Proteins
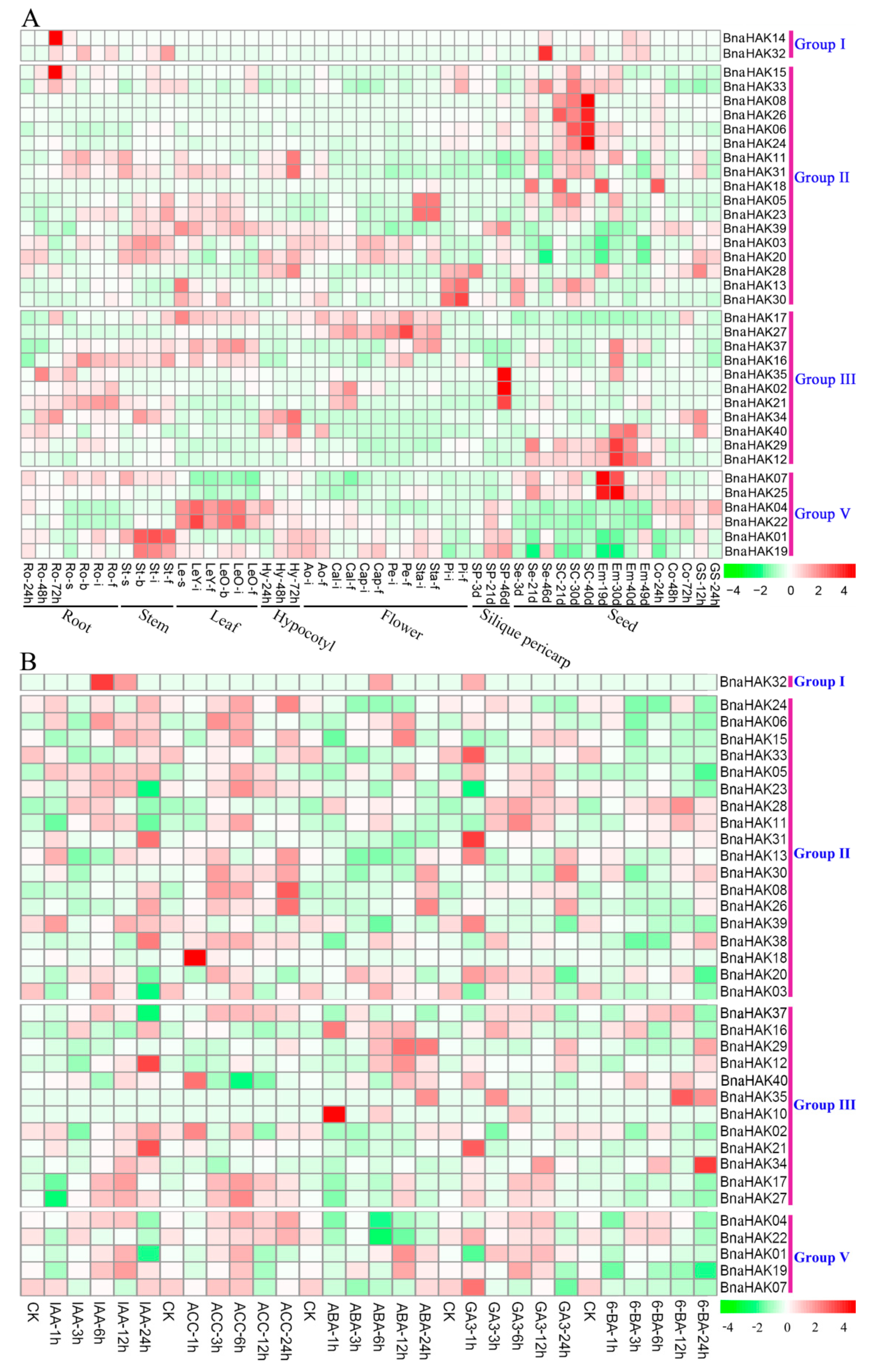
2.8. Spatiotemporal Expression Profiles of BnaHAKs across Different Developmental Stages
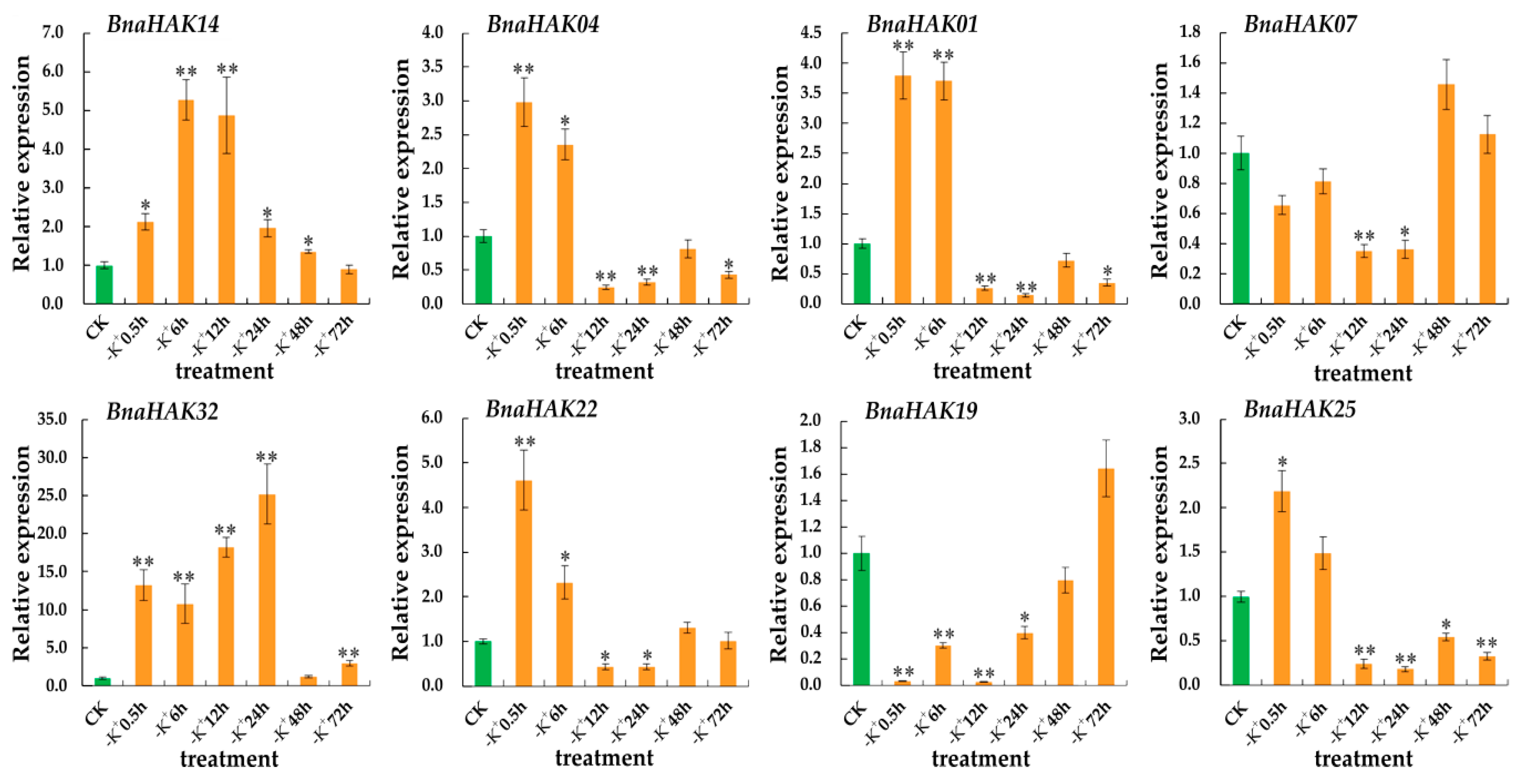
2.9. Expression Levels of BnaHAKs under Low-K+ Conditions
3. Discussion
4. Materials and Methods
4.1. Identification and Phylogenetic Analysis of the HAK Family in B. napus
4.2. Gene Structure Analysis of BnaHAKs
4.3. Chromosomal Location and Collinearity Analysis of BnaHAKs
4.4. TF-Binding Network, Cis-Elements, and MiRNA Target Analysis
4.5. Structure Prediction Analysis of BnaHAK Proteins
4.6. Gene Expression Analysis
4.7. Plant Materials and Growth Condition
4.8. RT-qPCR Analysis of BnaHAKs under Low-K+ Conditions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Véry, A.A.; Sentenac, H. Molecular mechanisms and regulation of K+ transport in higher plants. Annu. Rev. Plant Biol. 2003, 54, 575–603. [Google Scholar] [CrossRef] [PubMed]
- Marschner, P. Marschner’s Mineral Nutrition of Higher Plants, 3rd ed.; Academic Press: London, UK, 2012; pp. 178–189. [Google Scholar]
- Zörb, C.; Senbayram, M.; Peiter, E. Potassium in agriculture--status and perspectives. J. Plant Physiol. 2014, 171, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Osakabe, Y.; Arinaga, N.; Umezawa, T.; Katsura, S.; Nagamachi, K.; Tanaka, H.; Ohiraki, H.; Yamada, K.; Seo, S.U.; Abo, M.; et al. Osmotic stress responses and plant growth controlled by potassium transporters in Arabidopsis. Plant Cell 2013, 25, 609–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebaudy, A.; Véry, A.A.; Sentenac, H. K+ channel activity in plants: Genes, regulations and functions. FEBS Lett. 2007, 581, 2357–2366. [Google Scholar] [CrossRef]
- Lu, Z.; Pan, Y.; Hu, W.; Cong, R.; Ren, T.; Guo, S.; Lu, J. The photosynthetic and structural differences between leaves and siliques of Brassica napus exposed to potassium deficiency. BMC Plant Biol. 2017, 17, 240. [Google Scholar] [CrossRef] [Green Version]
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Nahar, K.; Hossain, M.S.; Mahmud, J.A.; Hossen, M.S.; Masud, A.A.C.; Moumita; Fujita, M. Potassium: A Vital Regulator of Plant Responses and Tolerance to Abiotic Stresses. Agronomy 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Maathuis, F.J. Physiological functions of mineral macronutrients. Curr. Opin. Plant Biol. 2009, 12, 250–258. [Google Scholar] [CrossRef]
- Rengel, Z.; Damon, P.M. Crops and genotypes differ in efficiency of potassium uptake and use. Physiol. Plant 2008, 133, 624–636. [Google Scholar] [CrossRef]
- Römheld, V.; Kirkby, E.A. Research on potassium in agriculture: Needs and prospects. Plant Soil. 2010, 335, 155–180. [Google Scholar] [CrossRef]
- Lu, Z.; Ren, T.; Pan, Y.; Li, X.; Cong, R.; Lu, J. Differences on photosynthetic limitations between leaf margins and leaf centers under potassium deficiency for Brassica napus L. Sci. Rep. 2016, 6, 21725. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Lu, J.; Li, H.; Zou, J.; Xu, H.; Liu, X.; Li, X. Potassium-fertilizer management in winter oilseed-rape production in China†. J. Plant Nutr. Soil Sci. 2013, 176, 429–440. [Google Scholar] [CrossRef]
- Mäser, P.; Thomine, S.; Schroeder, J.I.; Ward, J.M.; Hirschi, K.; Sze, H.; Talke, I.N.; Amtmann, A.; Maathuis, F.J.; Sanders, D.; et al. Phylogenetic relationships within cation transporter families of Arabidopsis. Plant Physiol. 2001, 126, 1646–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Qiu, X.; Wang, L.; Xie, W.; Zhang, C.; Xiong, L.; Lian, X.; Zhang, Q. KT/HAK/KUP potassium transporters gene family and their whole-life cycle expression profile in rice (Oryza sativa). Mol. Genet. Genom. 2008, 280, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Santa-María, G.E.; Rubio, F.; Dubcovsky, J.; Rodríguez-Navarro, A. The HAK1 gene of barley is a member of a large gene family and encodes a high-affinity potassium transporter. Plant Cell 1997, 9, 2281–2289. [Google Scholar] [CrossRef] [Green Version]
- Gierth, M.; Mäser, P.; Schroeder, J.I. The potassium transporter AtHAK5 functions in K(+) deprivation-induced high-affinity K(+) uptake and AKT1 K(+) channel contribution to K(+) uptake kinetics in Arabidopsis roots. Plant Physiol. 2005, 137, 1105–1114. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Garvin, D.F.; Kochian, L.V. Rapid induction of regulatory and transporter genes in response to phosphorus, potassium, and iron deficiencies in tomato roots. Evidence for cross talk and root/rhizosphere-mediated signals. Plant Physiol. 2002, 130, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- Bañuelos, M.A.; Garciadeblas, B.; Cubero, B.; Rodríguez-Navarro, A. Inventory and functional characterization of the HAK potassium transporters of rice. Plant Physiol. 2002, 130, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Wu, W.; Wu, W.H.; Wang, Y. Potassium Transporter KUP7 Is Involved in K(+) Acquisition and Translocation in Arabidopsis Root under K(+)-Limited Conditions. Mol. Plant 2016, 9, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Armengaud, P.; Breitling, R.; Amtmann, A. The potassium-dependent transcriptome of Arabidopsis reveals a prominent role of jasmonic acid in nutrient signaling. Plant Physiol. 2004, 136, 2556–2576. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Nakayama, H.; Shinmyo, A.; Yoshida, K. Expression of OsHAK genes encoding potassium ion transporters in rice. Plant Biotechnol. 2008, 25, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Senn, M.E.; Rubio, F.; Bañuelos, M.A.; Rodríguez-Navarro, A. Comparative functional features of plant potassium HvHAK1 and HvHAK2 transporters. J. Biol. Chem. 2001, 276, 44563–44569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garciadeblas, B.; Benito, B.; Rodríguez-Navarro, A. Molecular cloning and functional expression in bacteria of the potassium transporters CnHAK1 and CnHAK2 of the seagrass Cymodocea nodosa. Plant Mol. Biol. 2002, 50, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, C.; Gao, Z.; Zhang, Y.; Jiang, H.; Zhu, L.; Ren, D.; Yu, L.; Xu, G.; Qian, Q. OsHAK1, a High-Affinity Potassium Transporter, Positively Regulates Responses to Drought Stress in Rice. Front. Plant Sci. 2017, 8, 1885. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Tang, Q.; Cai, J.; Xu, B.; Xu, G.; Yu, L. Rice OsHAK16 functions in potassium uptake and translocation in shoot, maintaining potassium homeostasis and salt tolerance. Planta 2019, 250, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liang, X.; Wang, L.; Cao, Y.; Song, W.; Shi, J.; Lai, J.; Jiang, C. A HAK family Na+ transporter confers natural variation of salt tolerance in maize. Nat. Plants 2019, 5, 1297–1308. [Google Scholar] [CrossRef]
- Zhang, M.L.; Huang, P.P.; Ji, Y.; Wang, S.; Wang, S.S.; Li, Z.; Guo, Y.; Ding, Z.; Wu, W.H.; Wang, Y. KUP9 maintains root meristem activity by regulating K+ and auxin homeostasis in response to low K. EMBO Rep. 2020, 21, e50164. [Google Scholar] [CrossRef]
- Davies, C.; Shin, R.; Liu, W.; Thomas, M.R.; Schachtman, D.P. Transporters expressed during grape berry (Vitis vinifera L.) development are associated with an increase in berry size and berry potassium accumulation. J. Exp. Bot. 2006, 57, 3209–3216. [Google Scholar] [CrossRef] [Green Version]
- Elumalai, R.P.; Nagpal, P.; Reed, J.W. A mutation in the Arabidopsis KT2/KUP2 potassium transporter gene affects shoot cell expansion. Plant Cell 2002, 14, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Rigas, S.; Debrosses, G.; Haralampidis, K.; Vicente-Agullo, F.; Feldmann, K.A.; Grabov, A.; Dolan, L.; Hatzopoulos, P. TRH1 encodes a potassium transporter required for tip growth in Arabidopsis root hairs. Plant Cell 2001, 13, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.J.; Shin, R.; Schachtman, D.P. Expression of KT/KUP genes in Arabidopsis and the role of root hairs in K+ uptake. Plant Physiol. 2004, 134, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Liu, X.; Mao, W.; Zhang, X.; Chen, S.; Zhan, K.; Bi, H.; Xu, H. Genome-Wide Identification and Analysis of HAK/KUP/KT Potassium Transporters Gene Family in Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2018, 19, 3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Wang, Y.; Zhang, N.; Wu, Z.; Zeng, Q.; Wu, J.; Wu, X.; Wang, L.; Zhang, J.; Qi, Y. Genome-wide systematic characterization of the HAK/KUP/KT gene family and its expression profile during plant growth and in response to low-K+ stress in Saccharum. BMC Plant Biol. 2020, 20, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivetta, A.; Allen, K.E.; Slayman, C.W.; Slayman, C.L. Coordination of K+ transporters in neurospora: TRK1 is scarce and constitutive, while HAK1 is abundant and highly regulated. Eukaryot. Cell 2013, 12, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tascón, I.; Sousa, J.S.; Corey, R.A.; Mills, D.J.; Griwatz, D.; Aumüller, N.; Mikusevic, V.; Stansfeld, P.J.; Vonck, J.; Hänelt, I. Structural basis of proton-coupled potassium transport in the KUP family. Nat. Commun. 2020, 11, 626. [Google Scholar] [CrossRef]
- Gierth, M.; Mäser, P. Potassium transporters in plants--involvement in K+ acquisition, redistribution and homeostasis. FEBS Lett. 2007, 581, 2348–2356. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Porras, J.L.; Riaño-Pachón, D.M.; Benito, B.; Haro, R.; Sklodowski, K.; Rodríguez-Navarro, A.; Dreyer, I. Phylogenetic analysis of k(+) transporters in bryophytes, lycophytes, and flowering plants indicates a specialization of vascular plants. Front. Plant Sci. 2012, 3, 167. [Google Scholar] [CrossRef] [Green Version]
- Nieves-Cordones, M.; Ródenas, R.; Chavanieu, A.; Rivero, R.M.; Martinez, V.; Gaillard, I.; Rubio, F. Uneven HAK/KUP/KT Protein Diversity among Angiosperms: Species Distribution and Perspectives. Front. Plant Sci. 2016, 7, 127. [Google Scholar] [CrossRef] [Green Version]
- Santa-María, G.E.; Oliferuk, S.; Moriconi, J.I. KT-HAK-KUP transporters in major terrestrial photosynthetic organisms: A twenty years tale. J. Plant Physiol. 2018, 226, 77–90. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Chen, Y.; Li, R.; Wang, H.; Wei, J. Genome-wide analysis and identification of HAK potassium transporter gene family in maize (Zea mays L.). Mol. Biol. Rep. 2012, 39, 8465–8473. [Google Scholar] [CrossRef]
- He, C.; Cui, K.; Duan, A.; Zeng, Y.; Zhang, J. Genome-wide and molecular evolution analysis of the Poplar KT/HAK/KUP potassium transporter gene family. Ecol. Evol. 2012, 2, 1996–2004. [Google Scholar] [CrossRef]
- Rehman, H.M.; Nawaz, M.A.; Shah, Z.H.; Daur, I.; Khatoon, S.; Yang, S.H.; Chung, G. In-Depth Genomic and Transcriptomic Analysis of Five K+ Transporter Gene Families in Soybean Confirm Their Differential Expression for Nodulation. Front. Plant Sci. 2017, 8, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lü, J.; Chen, D.; Zhang, J.; Qi, K.; Cheng, R.; Zhang, H.; Zhang, S. Genome-wide identification, evolution, and expression analysis of the KT/HAK/KUP family in pear. Genome 2018, 61, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Hampton, C.R.; Shin, R.; Barkla, B.J.; White, P.J.; Schachtman, D.P. The high affinity K+ transporter AtHAK5 plays a physiological role in planta at very low K+ concentrations and provides a caesium uptake pathway in Arabidopsis. J. Exp. Bot. 2008, 59, 595–607. [Google Scholar] [CrossRef] [Green Version]
- Nieves-Cordones, M.; Mohamed, S.; Tanoi, K.; Kobayashi, N.I.; Takagi, K.; Vernet, A.; Guiderdoni, E.; Périn, C.; Sentenac, H.; Véry, A.A. Production of low-Cs+ rice plants by inactivation of the K+ transporter OsHAK1 with the CRISPR-Cas system. Plant J. 2017, 92, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Ródenas, R.; Nieves-Cordones, M.; Rivero, R.M.; Martinez, V.; Rubio, F. Pharmacological and gene regulation properties point to the SlHAK5 K+ transporter as a system for high-affinity Cs+ uptake in tomato plants. Physiol. Plant 2018, 162, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Liu, W.; Qiu, C.W.; Zeng, F.; Wang, Y.; Zhang, G.; Chen, Z.H.; Wu, F. HvAKT2 and HvHAK1 confer drought tolerance in barley through enhanced leaf mesophyll H+ homoeostasis. Plant Biotechnol. J. 2020, 18, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- Nieves-Cordones, M.; Alemán, F.; Martínez, V.; Rubio, F. The Arabidopsis thaliana HAK5 K+ transporter is required for plant growth and K+ acquisition from low K+ solutions under saline conditions. Mol. Plant 2010, 3, 326–333. [Google Scholar] [CrossRef]
- Benito, B.; Garciadeblas, B.; Rodriguez-Navarro, A. HAK transporters from Physcomitrella patens and Yarrowia lipolytica mediate sodium uptake. Plant Cell Physiol. 2012, 53, 1117–1123. [Google Scholar] [CrossRef]
- Takahashi, R.; Nishio, T.; Ichizen, N.; Takano, T. High-affinity K+ transporter PhaHAK5 is expressed only in salt-sensitive reed plants and shows Na+ permeability under NaCl stress. Plant Cell Rep. 2007, 26, 1673–1679. [Google Scholar] [CrossRef]
- Tenorio-Berrío, R.; Pérez-Alonso, M.M.; Vicente-Carbajosa, J.; Martín-Torres, L.; Dreyer, I.; Pollmann, S. Identification of Two Auxin-Regulated Potassium Transporters Involved in Seed Maturation. Int. J. Mol. Sci. 2018, 19, 2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.L.; Llewellyn, D.J.; Furbank, R.T. The control of single-celled cotton fiber elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansin. Plant Cell 2001, 13, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbrosses, G.; Kopka, C.; Ott, T.; Udvardi, M.K. Lotus japonicus LjKUP is induced late during nodule development and encodes a potassium transporter of the plasma membrane. Mol. Plant-Microbe Interact. MPMI 2004, 17, 789–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [Green Version]
- Galili, T.; O’Callaghan, A.; Sidi, J.; Sievert, C. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 2018, 34, 1600–1602. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Genome ID | Chromosome | Protein Length (aa) | CDS Length | DNA Length | pI | Molecular Weight | Subcellular Localization |
|---|---|---|---|---|---|---|---|---|
| BnaHAK01 | BnaA01g03380D | chrA01 | 856 | 2571 | 3630 | 5.22 | 94.66 | Vacuole |
| BnaHAK02 | BnaA01g10310D | chrA01 | 812 | 2439 | 3238 | 6.78 | 90.71 | Vacuole |
| BnaHAK03 | BnaA01g13320D | chrA01 | 782 | 2349 | 3779 | 9.37 | 87.82 | Cell membrane, Vacuole |
| BnaHAK04 | BnaA01g22220D | chrA01 | 834 | 2868 | 5005 | 5.61 | 106.49 | Cell membrane, Vacuole |
| BnaHAK05 | BnaA02g02340D | chrA02 | 710 | 4299 | 8919 | 8.48 | 161.23 | Vacuole |
| BnaHAK06 | BnaA02g14820D | chrA02 | 665 | 1998 | 3831 | 8.75 | 74.33 | Cell membrane, Vacuole |
| BnaHAK07 | BnaA03g02700D | chrA03 | 864 | 2595 | 3806 | 5.30 | 96.22 | Vacuole |
| BnaHAK08 | BnaA03g13690D | chrA03 | 711 | 2136 | 5533 | 8.03 | 78.97 | Cell membrane, Vacuole |
| BnaHAK09 | BnaA03g44320D | chrA03 | 808 | 2427 | 3476 | 7.04 | 91.09 | Vacuole |
| BnaHAK10 | BnaA03g44330D | chrA03 | 810 | 2433 | 3761 | 6.28 | 91.26 | Cell membrane, Vacuole |
| BnaHAK11 | BnaA04g23330D | chrA04 | 776 | 2331 | 3847 | 6.97 | 87.06 | Vacuole |
| BnaHAK12 | BnaA05g08850D | chrA05 | 787 | 2364 | 3752 | 8.65 | 88.09 | Vacuole |
| BnaHAK13 | BnaA05g12300D | chrA05 | 714 | 2145 | 3338 | 7.54 | 79.24 | Cell membrane, Vacuole |
| BnaHAK14 | BnaA07g16500D | chrA07 | 747 | 2244 | 7046 | 8.00 | 84.14 | Cell membrane, Vacuole |
| BnaHAK15 | BnaA07g38760D | chrA07 | 700 | 2103 | 3392 | 8.86 | 78.23 | Cell membrane, Vacuole |
| BnaHAK16 | BnaA08g08020D | chrA08 | 757 | 2274 | 2724 | 8.38 | 85.19 | Cell membrane, Vacuole |
| BnaHAK17 | BnaA08g30510D | chrA08 | 808 | 2427 | 3259 | 6.61 | 90.57 | Vacuole |
| BnaHAK18 | BnaA09g21950D | chrA09 | 489 | 1476 | 2821 | 9.25 | 55.07 | Cell membrane, Vacuole |
| BnaHAK19 | BnaC01g04660D | chrC01 | 850 | 2553 | 3831 | 5.27 | 94.31 | Vacuole |
| BnaHAK20 | BnaC01g15360D | chrC01 | 705 | 2118 | 3625 | 9.34 | 78.82 | Cell membrane, Vacuole |
| BnaHAK21 | BnaC01g41320D | chrC01 | 789 | 2373 | 3584 | 7.59 | 88.43 | Vacuole |
| BnaHAK22 | BnaC01g43090D | chrC01 | 832 | 2700 | 4041 | 5.75 | 99.91 | Cell membrane, Vacuole |
| BnaHAK23 | BnaC02g05800D | chrC02 | 775 | 4581 | 10,988 | 8.37 | 171.55 | Vacuole |
| BnaHAK24 | BnaC02g19780D | chrC02 | 777 | 2334 | 4163 | 8.68 | 86.67 | Vacuole |
| BnaHAK25 | BnaC03g03790D | chrC03 | 864 | 2595 | 3755 | 5.30 | 96.21 | Vacuole |
| BnaHAK26 | BnaC03g16580D | chrC03 | 710 | 2133 | 3984 | 8.21 | 78.89 | Vacuole |
| BnaHAK27 | BnaC03g76940D | chrC03 | 804 | 2415 | 3188 | 6.89 | 90.16 | Vacuole |
| BnaHAK28 | BnaC04g01430D | chrC04 | 784 | 3825 | 6298 | 6.23 | 142.58 | Vacuole |
| BnaHAK29 | BnaC04g10260D | chrC04 | 728 | 2187 | 3978 | 9.03 | 81.62 | Cell membrane, Vacuole |
| BnaHAK30 | BnaC04g14750D | chrC04 | 712 | 2139 | 4392 | 7.81 | 79.02 | Cell membrane, Vacuole |
| BnaHAK31 | BnaC04g47240D | chrC04 | 777 | 2334 | 3898 | 6.83 | 87.16 | Vacuole |
| BnaHAK32 | BnaC06g15440D | chrC06 | 784 | 2355 | 8981 | 7.64 | 88.06 | Cell membrane, Vacuole |
| BnaHAK33 | BnaC06g31400D | chrC06 | 785 | 2358 | 3467 | 8.47 | 87.48 | Vacuole |
| BnaHAK34 | BnaC07g36080D | chrC07 | 709 | 2130 | 3221 | 6.61 | 79.36 | Cell membrane, Vacuole |
| BnaHAK35 | BnaC07g36130D | chrC07 | 798 | 2397 | 3462 | 6.51 | 89.55 | Vacuole |
| BnaHAK36 | BnaC07g36140D | chrC07 | 402 | 1209 | 2867 | 8.98 | 44.40 | Vacuole |
| BnaHAK37 | BnaC08g09300D | chrC08 | 794 | 2385 | 3563 | 8.65 | 89.18 | Vacuole |
| BnaHAK38 | BnaC09g24170D | chrC09 | 539 | 1620 | 5134 | 9.44 | 75.90 | Vacuole |
| BnaHAK39 | BnaCnng05490D | chrCnn_random | 770 | 2313 | 3546 | 9.19 | 85.60 | Vacuole |
| BnaHAK40 | BnaCnng46720D | chrCnn_random | 794 | 2385 | 4156 | 8.53 | 88.49 | Cell membrane, Vacuole |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Zhou, H.-J.; Chen, P.; Zhang, L.-L.; Zhu, J.-T.; Li, P.-F.; Yang, J.; Ke, Y.-Z.; Zhou, Y.-H.; Li, J.-N.; et al. Genome-Wide Survey and Expression Analysis of the KT/HAK/KUP Family in Brassica napus and Its Potential Roles in the Response to K+ Deficiency. Int. J. Mol. Sci. 2020, 21, 9487. https://doi.org/10.3390/ijms21249487
Zhou J, Zhou H-J, Chen P, Zhang L-L, Zhu J-T, Li P-F, Yang J, Ke Y-Z, Zhou Y-H, Li J-N, et al. Genome-Wide Survey and Expression Analysis of the KT/HAK/KUP Family in Brassica napus and Its Potential Roles in the Response to K+ Deficiency. International Journal of Molecular Sciences. 2020; 21(24):9487. https://doi.org/10.3390/ijms21249487
Chicago/Turabian StyleZhou, Jie, Hong-Jun Zhou, Ping Chen, Lan-Lan Zhang, Jia-Tian Zhu, Peng-Feng Li, Jin Yang, Yun-Zhuo Ke, Yong-Hong Zhou, Jia-Na Li, and et al. 2020. "Genome-Wide Survey and Expression Analysis of the KT/HAK/KUP Family in Brassica napus and Its Potential Roles in the Response to K+ Deficiency" International Journal of Molecular Sciences 21, no. 24: 9487. https://doi.org/10.3390/ijms21249487