Microfluidic Tumor-on-a-Chip Model to Study Tumor Metabolic Vulnerability

 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
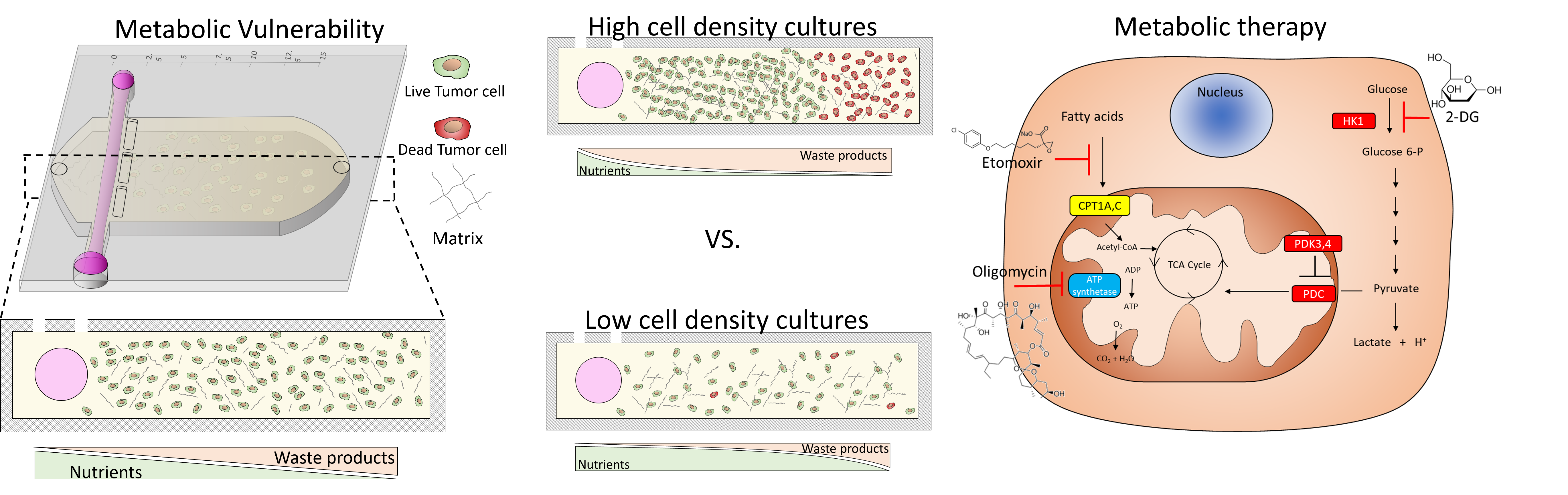
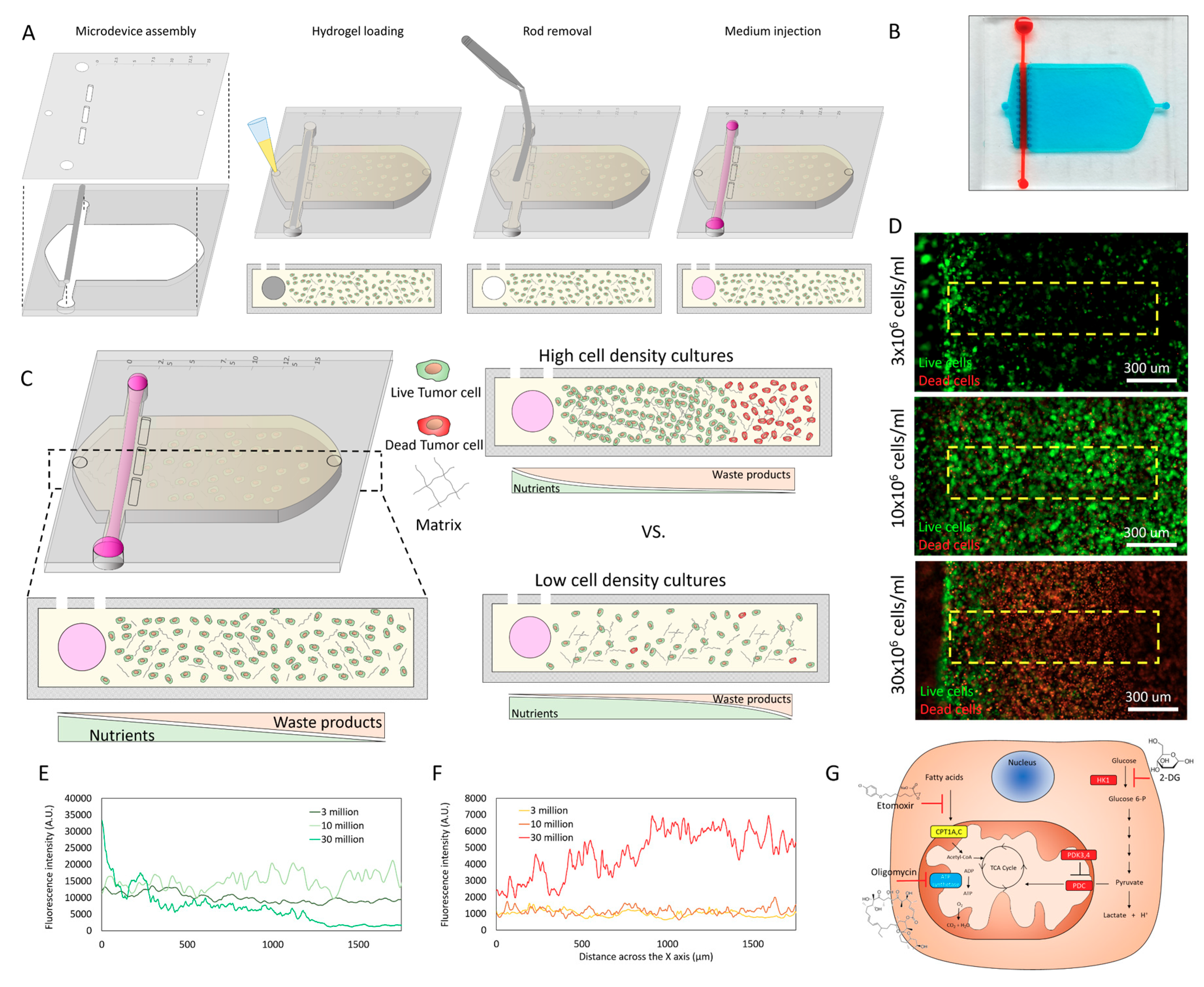
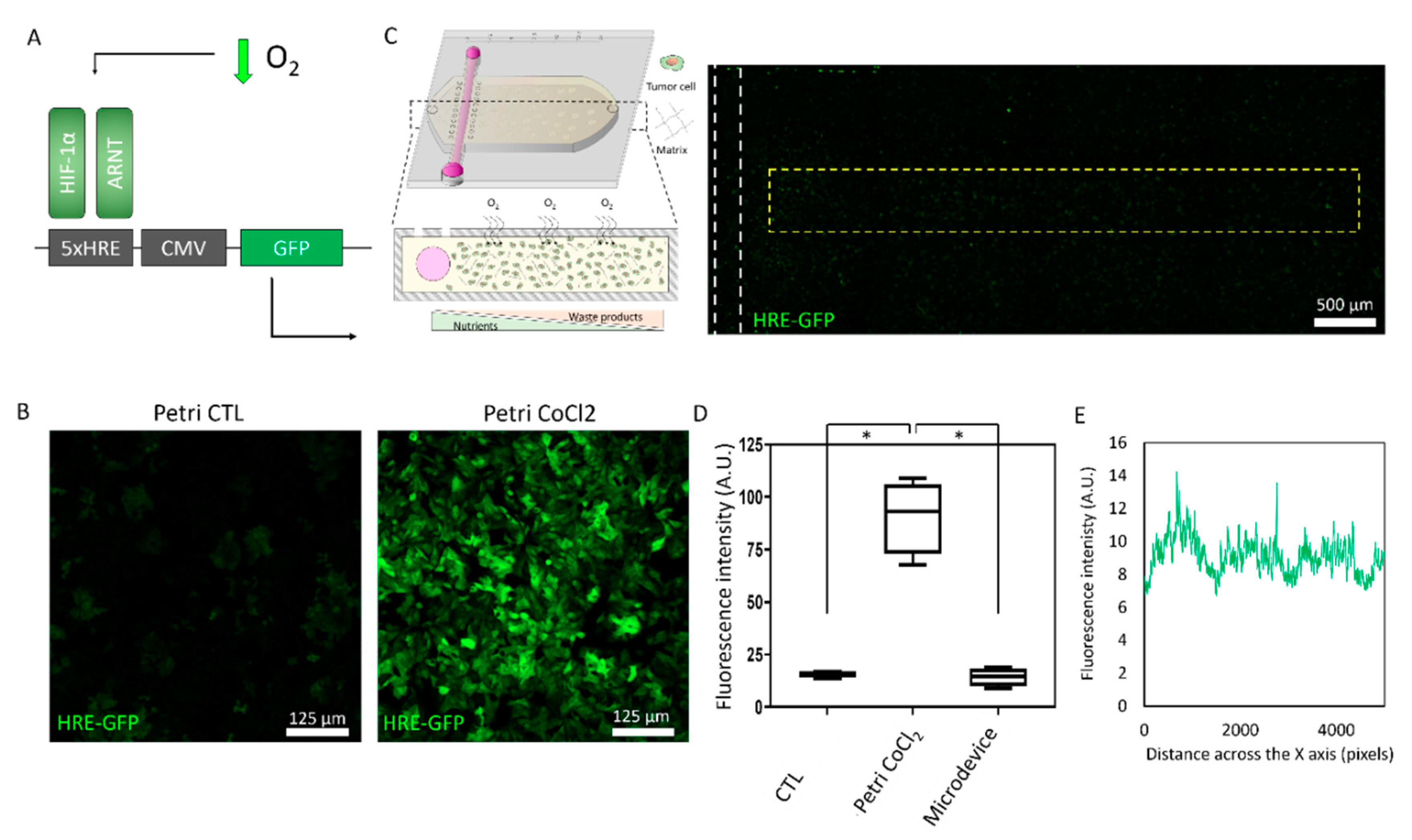
2.1. Cell Density Modulates Necrotic Core Generation
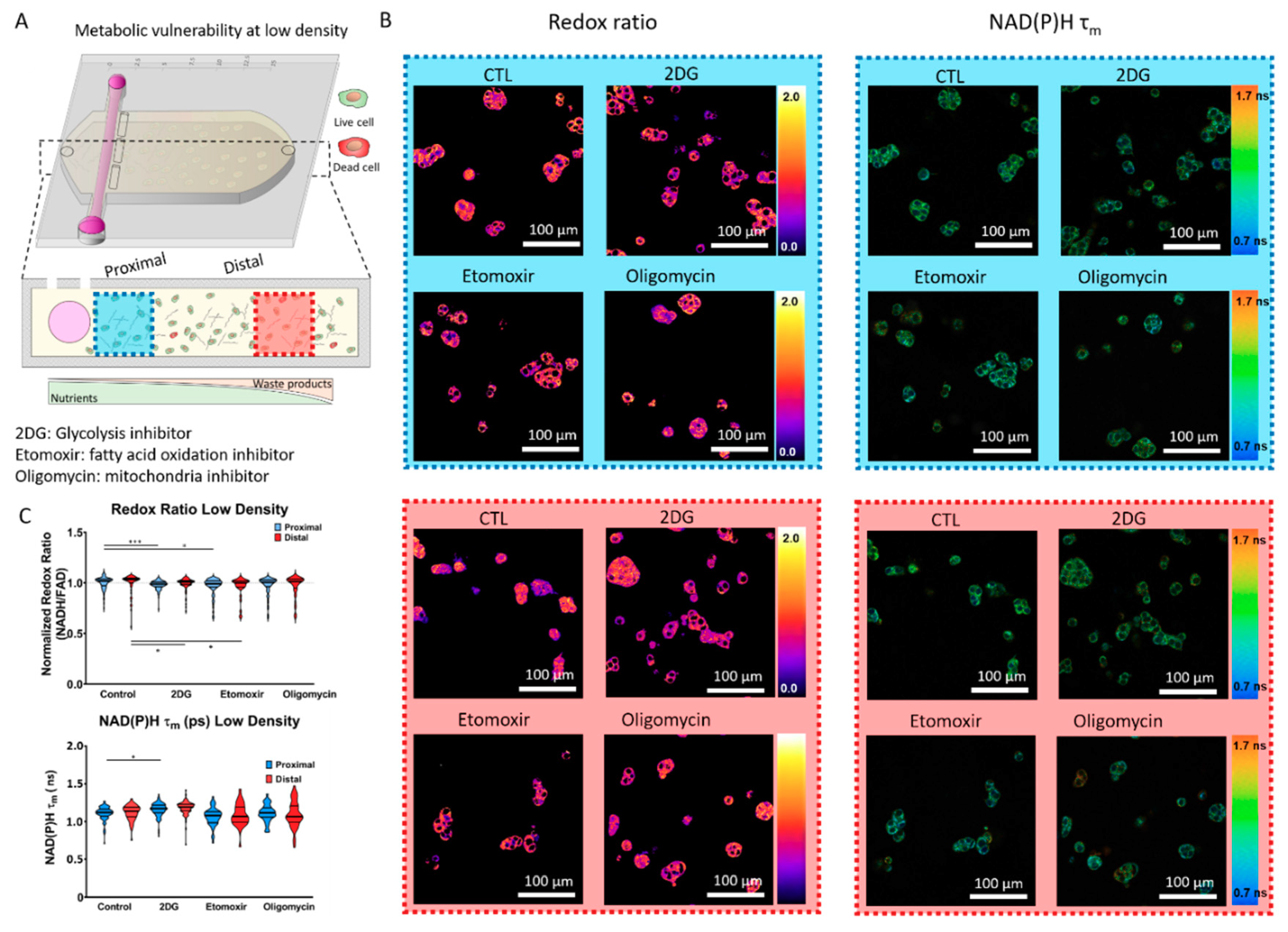
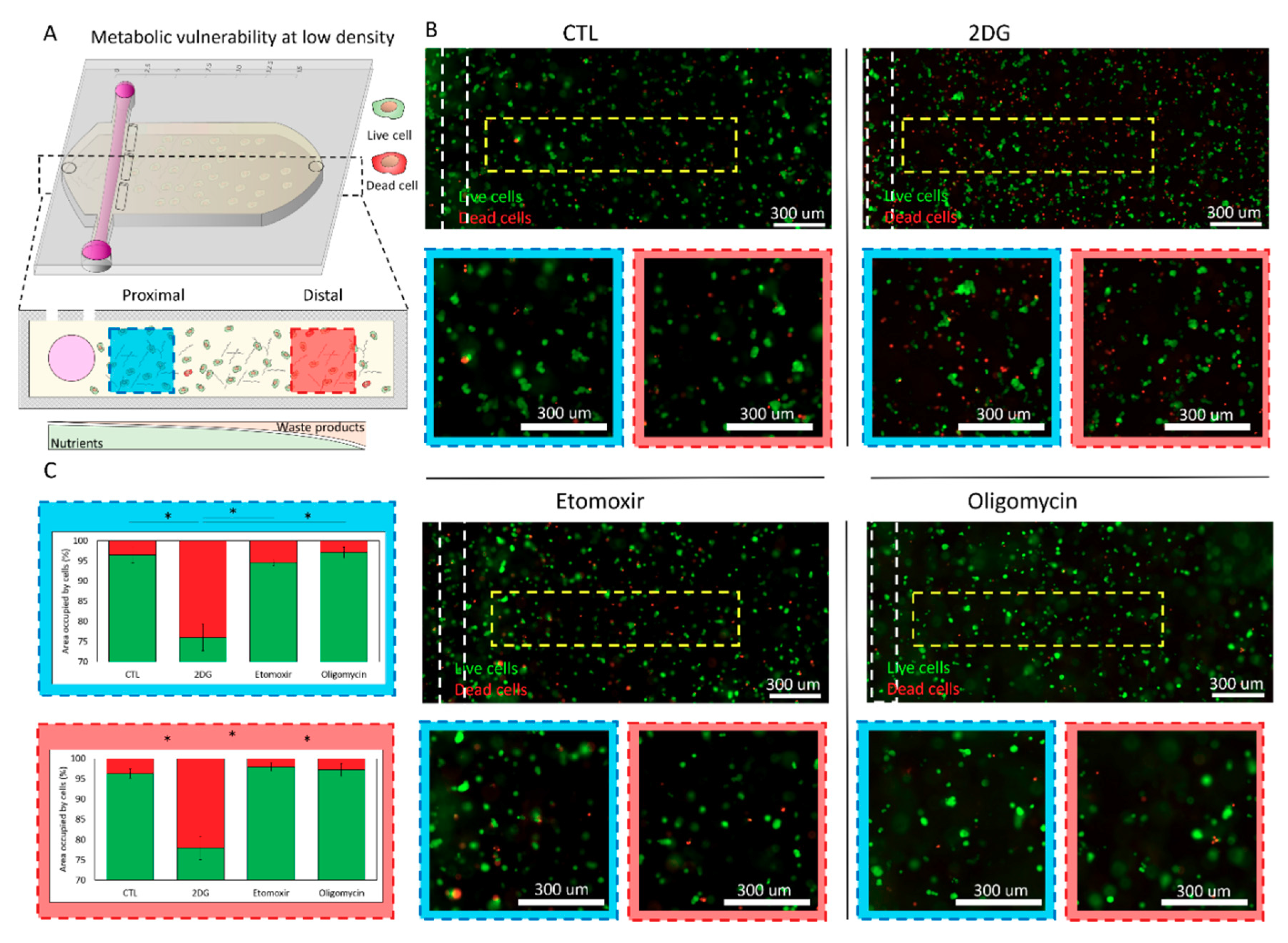
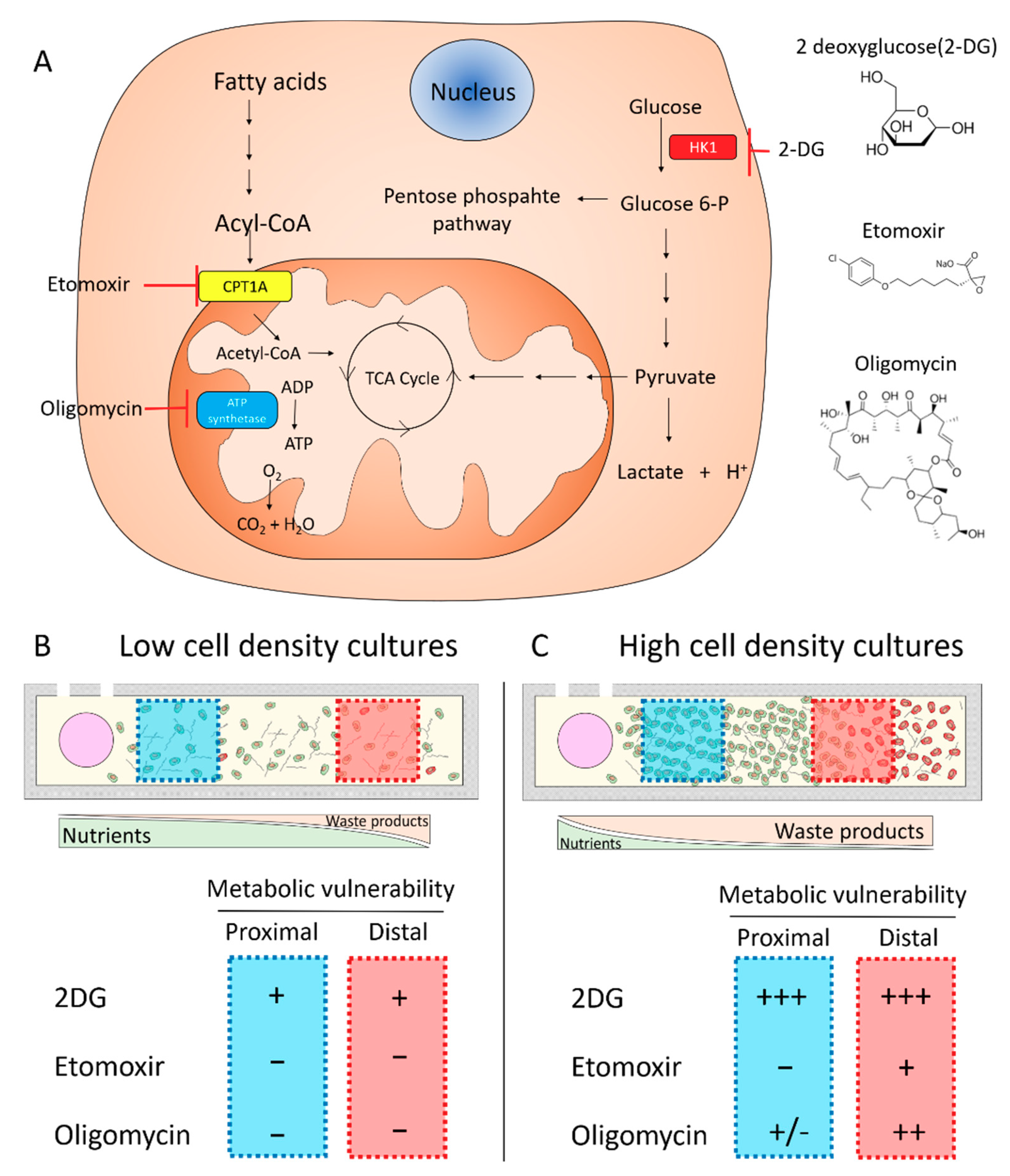
2.2. Metabolic Vulnerability in Low Cell Density Cultures
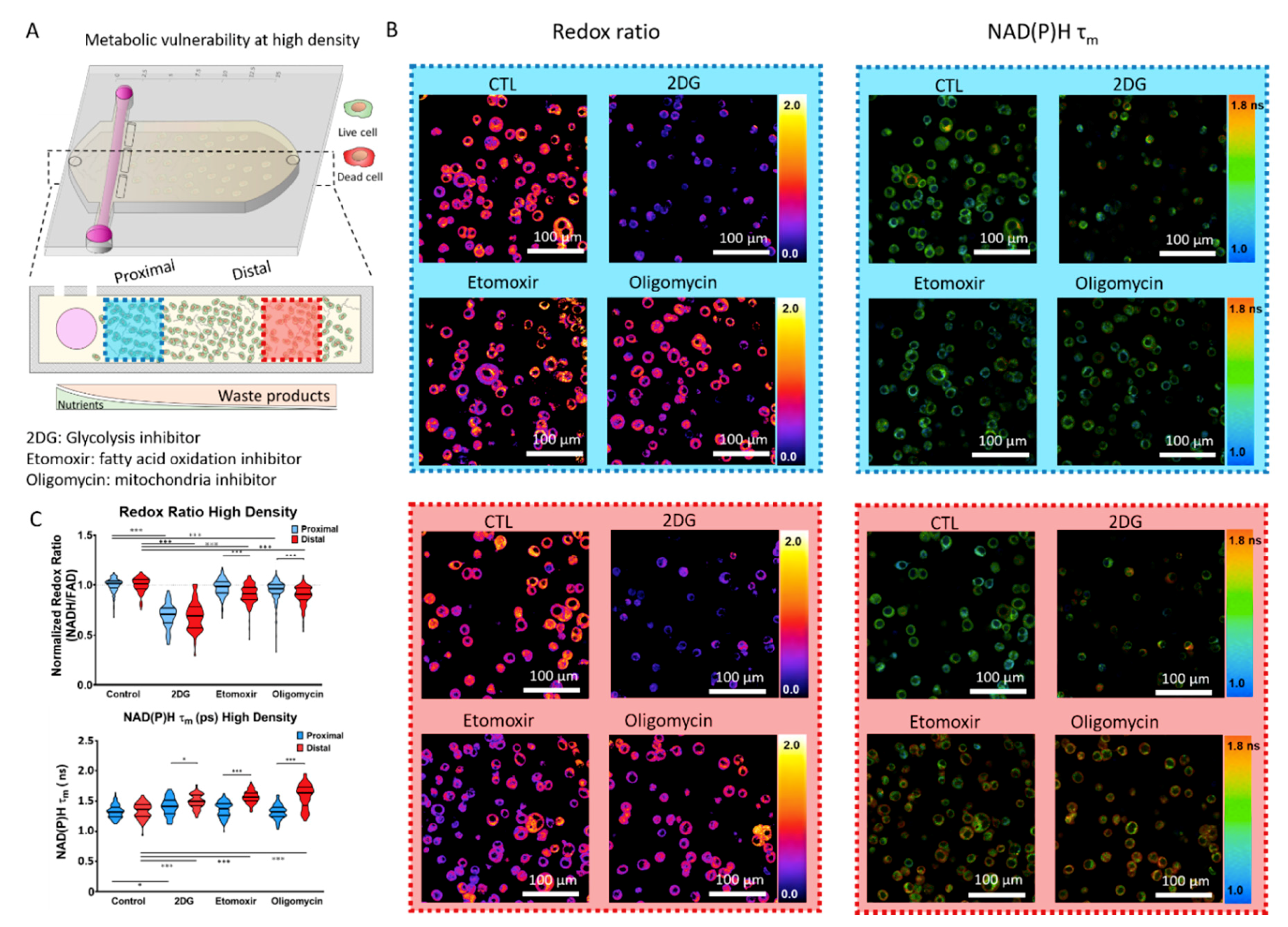
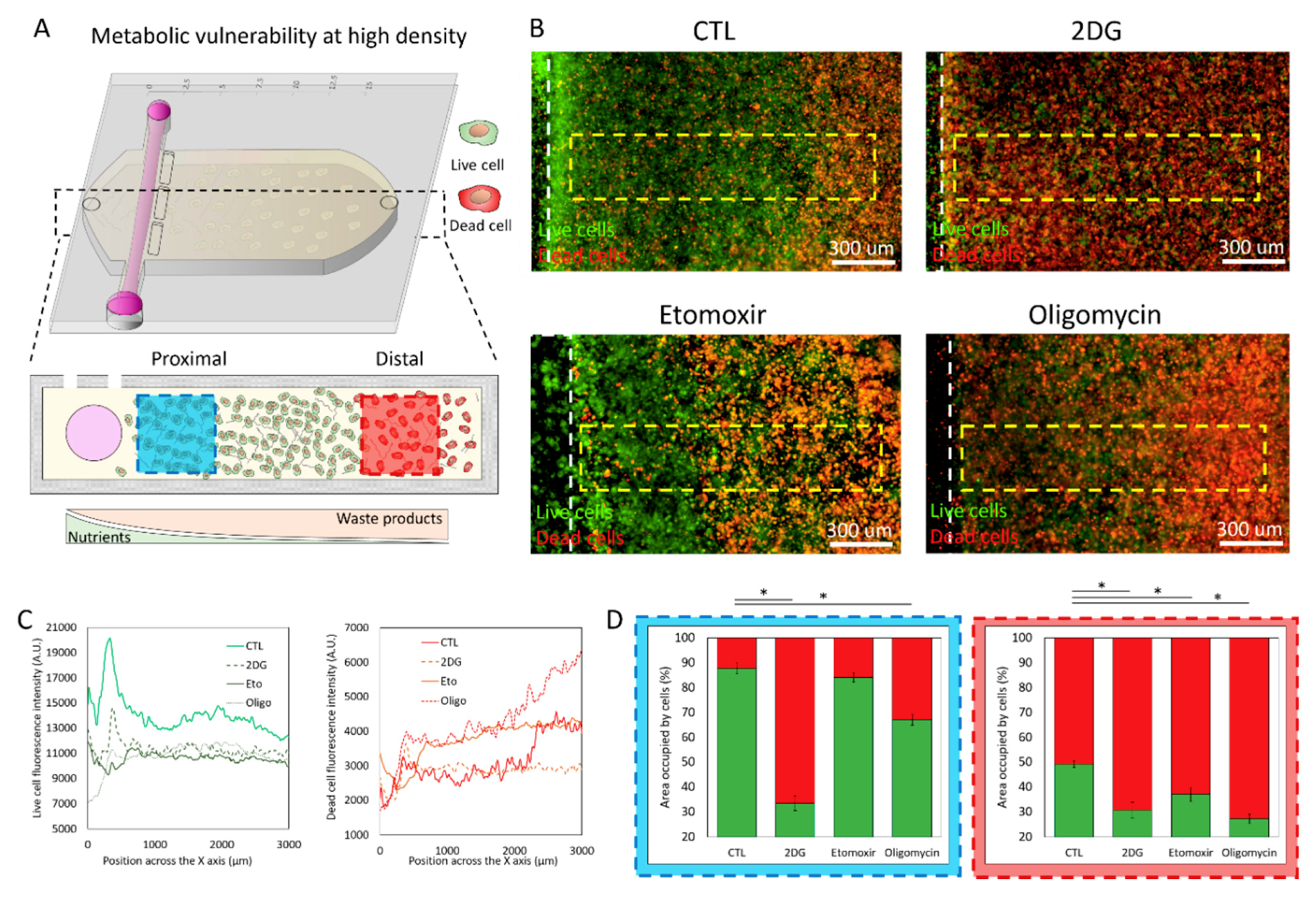
2.3. Metabolic Vulnerability in High Cell Density Cultures
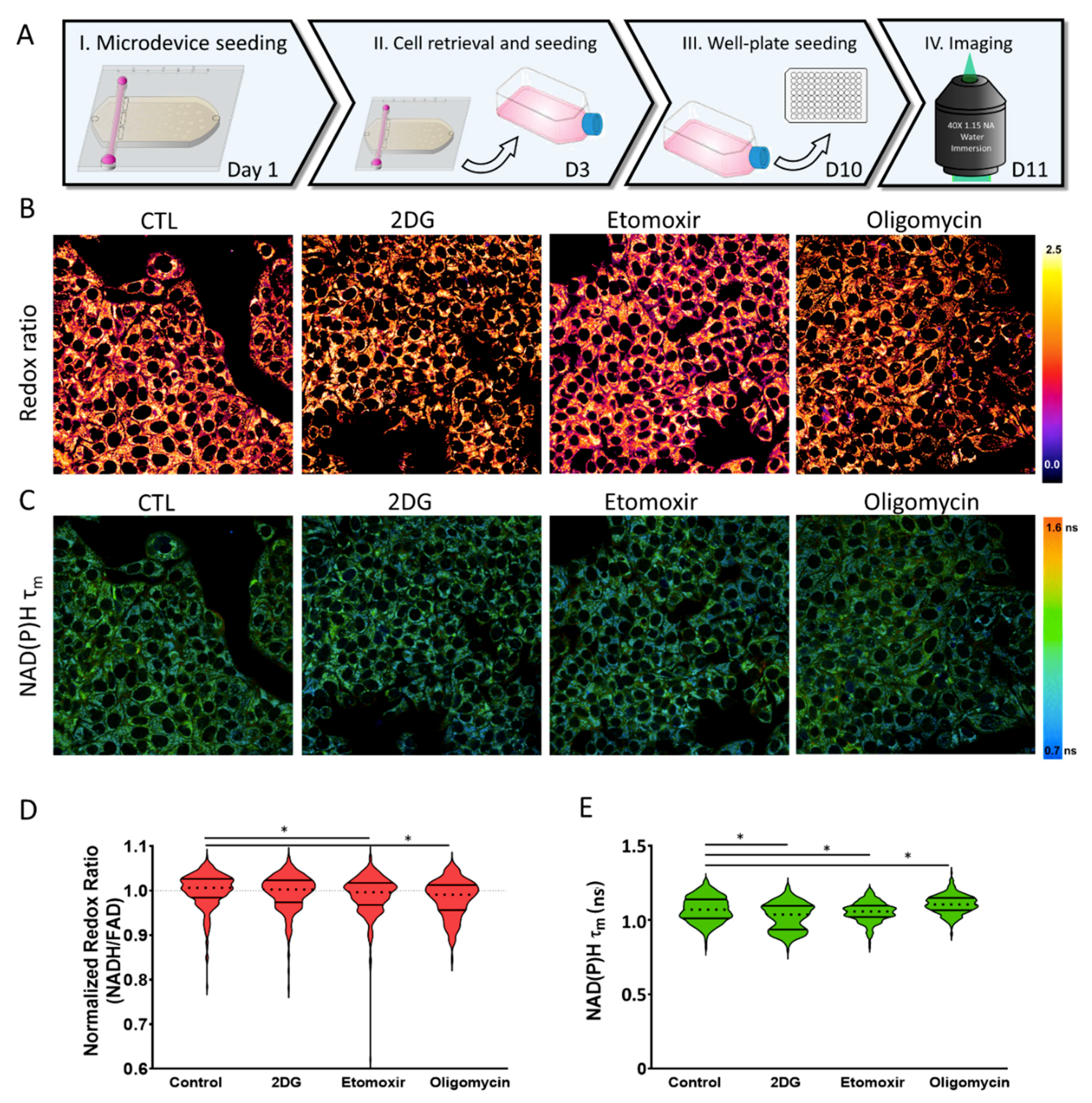
2.4. Metabolic Plasticity
3. Conclusions
4. Materials and Methods
4.1. Tumor-on-a-Chip Microdevice Fabrication
4.2. Cell Culture
4.3. HRE-GFP Transfection
4.4. Metabolic Inhibitors
4.5. Cell Viability
4.6. Hydrogel Degradation and Cell Recovery
4.7. Optical Metabolic Imaging
4.8. Image and Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walsh, A.J.; Cook, R.S.; Manning, H.C.; Hicks, D.J.; Lafontant, A.; Arteaga, C.L.; Skala, M.C. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res. 2013, 73, 6164–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the tca cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Br. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef]
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose regulates apoptosis in ovarian cancer cells via p38 mapk/jnk signaling pathway. Am. J. Otransl. Res. 2016, 8, 4812–4821. [Google Scholar]
- Chen, M.; Huang, J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis. Clin. Med. 2019, 2, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase i: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The cpt1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [Green Version]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. Metabolic fitness and plasticity in cancer progression. Trends Cancer 2020, 6, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, C.A.; Gallez, B. Metabolic plasticity of tumor cells: How they do adapt to food deprivation. Tumor Microenvir. 2020, 1219, 109–123. [Google Scholar]
- Wek, R.C.; Staschke, K.A. How do tumours adapt to nutrient stress? EMBO J. 2010, 29, 1946–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve cancer cells of glutamine: Break the spell or make a hungry monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, T.; Shibuya, M. Targeting cancer cells resistant to hypoxia and nutrient starvation to improve anti-angiogeneic therapy. Cell Cycle 2013, 12, 2519–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef]
- Virumbrales-Munoz, M.; Ayuso, J.M.; Gong, M.M.; Humayun, M.; Livingston, M.K.; Lugo-Cintron, K.M.; McMinn, P.; Alvarez-Garcia, Y.R.; Beebe, D.J. Microfluidic lumen-based systems for advancing tubular organ modeling. Chem. Soc. Rev. 2020, 49, 6402–6442. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Virumbrales-Munoz, M.; McMinn, P.H.; Rehman, S.; Gomez, I.; Karim, M.R.; Trusttchel, R.; Wisinski, K.B.; Beebe, D.J.; Skala, M.C. Tumor-on-a-chip: A microfluidic model to study cell response to environmental gradients. Lab. Chip. 2019, 19, 3461–3471. [Google Scholar] [CrossRef]
- Rexius-Hall, M.L.; Rehman, J.; Eddington, D.T. A microfluidic oxygen gradient demonstrates differential activation of the hypoxia-regulated transcription factors hif-1alpha and hif-2alpha. Integr. Biol. 2017, 9, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Ayuso, J.M.; Gillette, A.; Lugo-Cintron, K.; Acevedo-Acevedo, S.; Gomez, I.; Morgan, M.; Heaster, T.; Wisinski, K.B.; Palecek, S.P.; Skala, M.C.; et al. Organotypic microfluidic breast cancer model reveals starvation-induced spatial-temporal metabolic adaptations. EBioMedicine 2018, 37, 144–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayuso, J.M.; Truttschel, R.; Gong, M.M.; Humayun, M.; Virumbrales-Munoz, M.; Vitek, R.; Felder, M.; Gillies, S.D.; Sondel, P.; Wisinski, K.B.; et al. Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology 2019, 8, 1553477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacio-Castaneda, V.; Kooijman, L.; Venzac, B.; Verdurmen, W.P.R.; Le Gac, S. Metabolic switching of tumor cells under hypoxic conditions in a tumor-on-a-chip model. Micromachines 2020, 11, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br. J. Cancer 2017, 116, 277–286. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Seminars. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Vitek, R.; Swick, A.D.; Skala, M.C.; Wisinski, K.B.; Kimple, R.J.; Lambert, P.F.; Beebe, D.J. Effects of culture method on response to egfr therapy in head and neck squamous cell carcinoma cells. Sci. Rep. 2019, 9, 12480. [Google Scholar] [CrossRef]
- Lamb, R.; Harrison, H.; Hulit, J.; Smith, D.L.; Lisanti, M.P.; Sotgia, F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via mct1/2 inhibition. Oncotarget 2014, 5, 11029–11037. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Virumbrales-Munoz, M.; Ayuso, J.M.; Lacueva, A.; Randelovic, T.; Livingston, M.K.; Beebe, D.J.; Olivan, S.; Pereboom, D.; Doblare, M.; Fernandez, L.; et al. Enabling cell recovery from 3d cell culture microfluidic devices for tumour microenvironment biomarker profiling. Sci. Rep. 2019, 9, 6199. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.J.; Skala, M.C. An automated image processing routine for segmentation of cell cytoplasms in high-resolution autofluorescence images. In Proceedings of the Multiphoton Microscopy in the Biomedical Sciences XIV, San Francisco, CA, USA, 1–6 February 2014. [Google Scholar]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayuso, J.M.; Rehman, S.; Farooqui, M.; Virumbrales-Muñoz, M.; Setaluri, V.; Skala, M.C.; Beebe, D.J. Microfluidic Tumor-on-a-Chip Model to Study Tumor Metabolic Vulnerability. Int. J. Mol. Sci. 2020, 21, 9075. https://doi.org/10.3390/ijms21239075
Ayuso JM, Rehman S, Farooqui M, Virumbrales-Muñoz M, Setaluri V, Skala MC, Beebe DJ. Microfluidic Tumor-on-a-Chip Model to Study Tumor Metabolic Vulnerability. International Journal of Molecular Sciences. 2020; 21(23):9075. https://doi.org/10.3390/ijms21239075
Chicago/Turabian StyleAyuso, Jose M, Shujah Rehman, Mehtab Farooqui, María Virumbrales-Muñoz, Vijayasaradhi Setaluri, Melissa C Skala, and David J Beebe. 2020. "Microfluidic Tumor-on-a-Chip Model to Study Tumor Metabolic Vulnerability" International Journal of Molecular Sciences 21, no. 23: 9075. https://doi.org/10.3390/ijms21239075