Inhibition of AKT-Signaling Sensitizes Soft Tissue Sarcomas (STS) and Gastrointestinal Stromal Tumors (GIST) to Doxorubicin via Targeting of Homology-Mediated DNA Repair

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
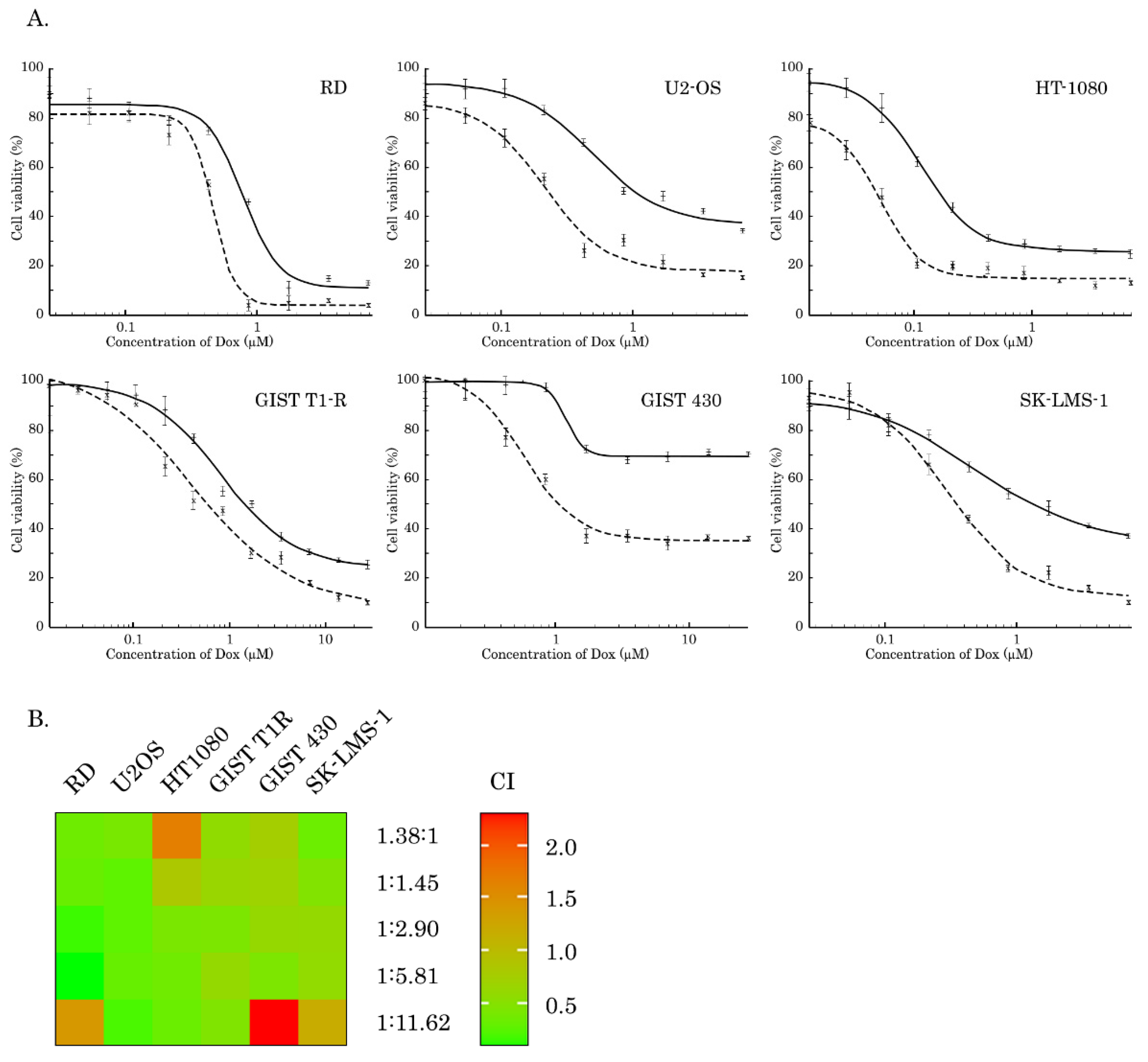
2.1. Inhibition of AKT-Signaling Enhances Cytotoxicity of Topo II Inhibitors in STS and GIST
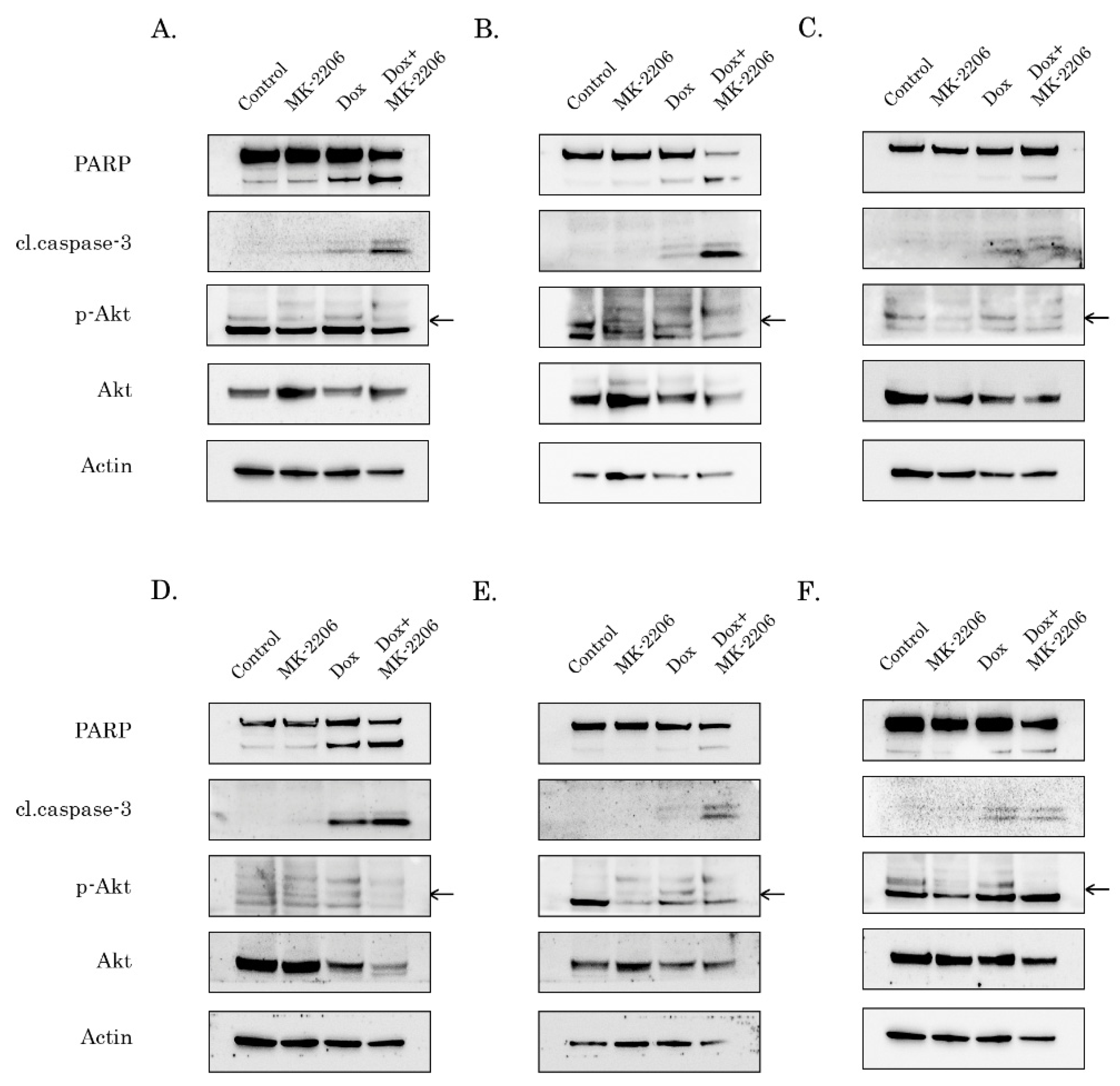
2.2. Inhibition of AKT-Signaling Enhances Doxorubicin-Induced Apoptosis of STS and GIST
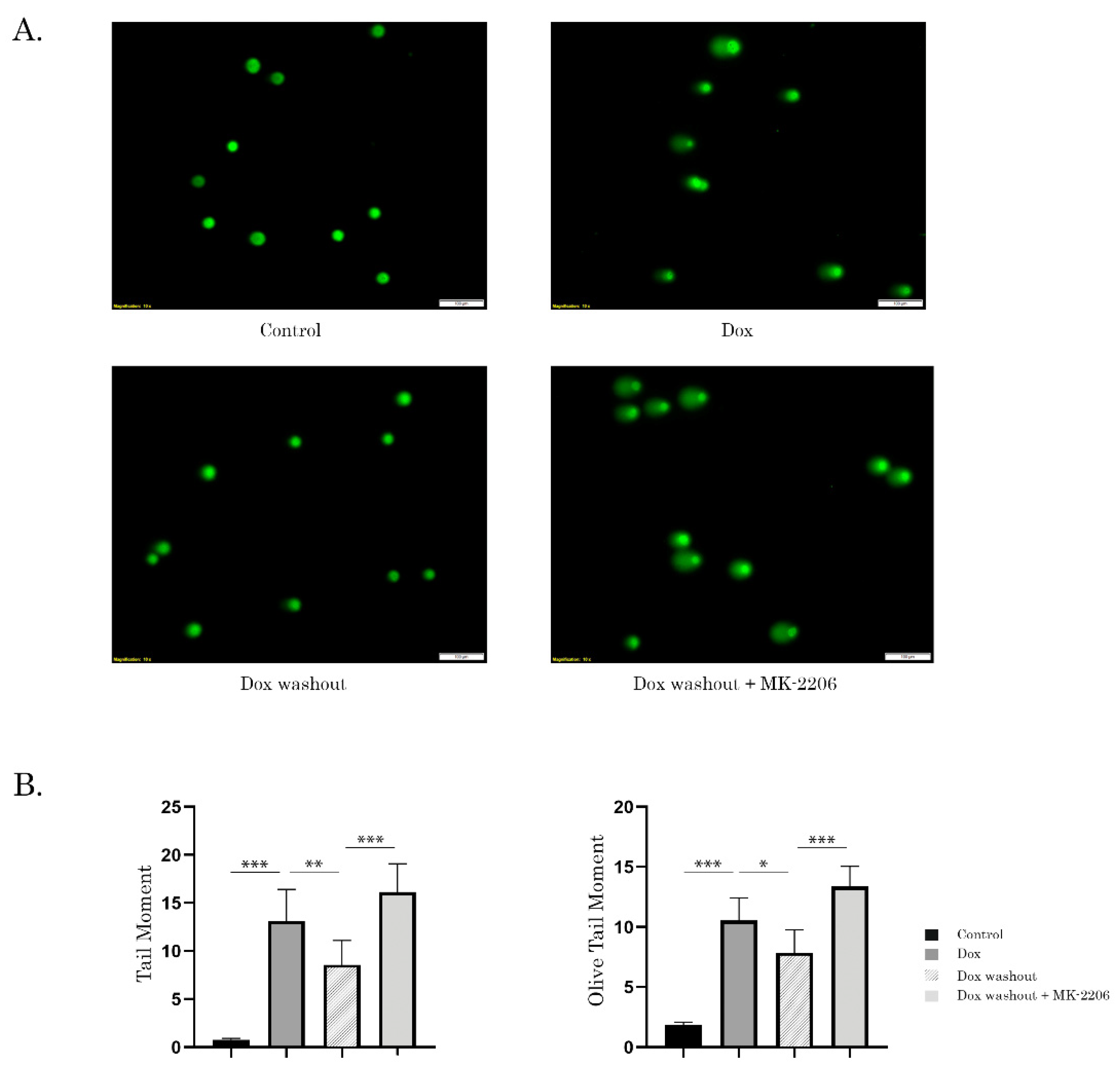
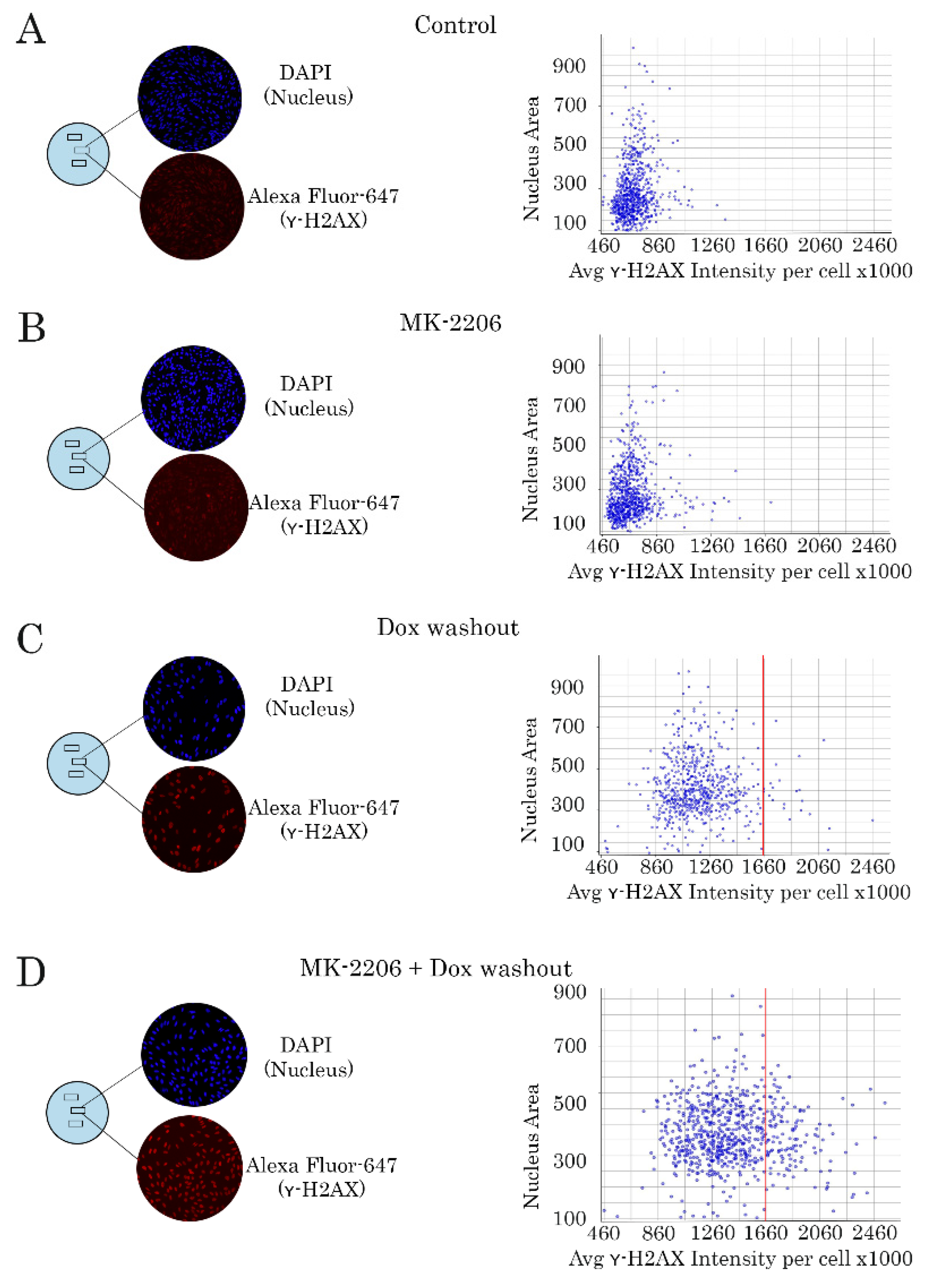
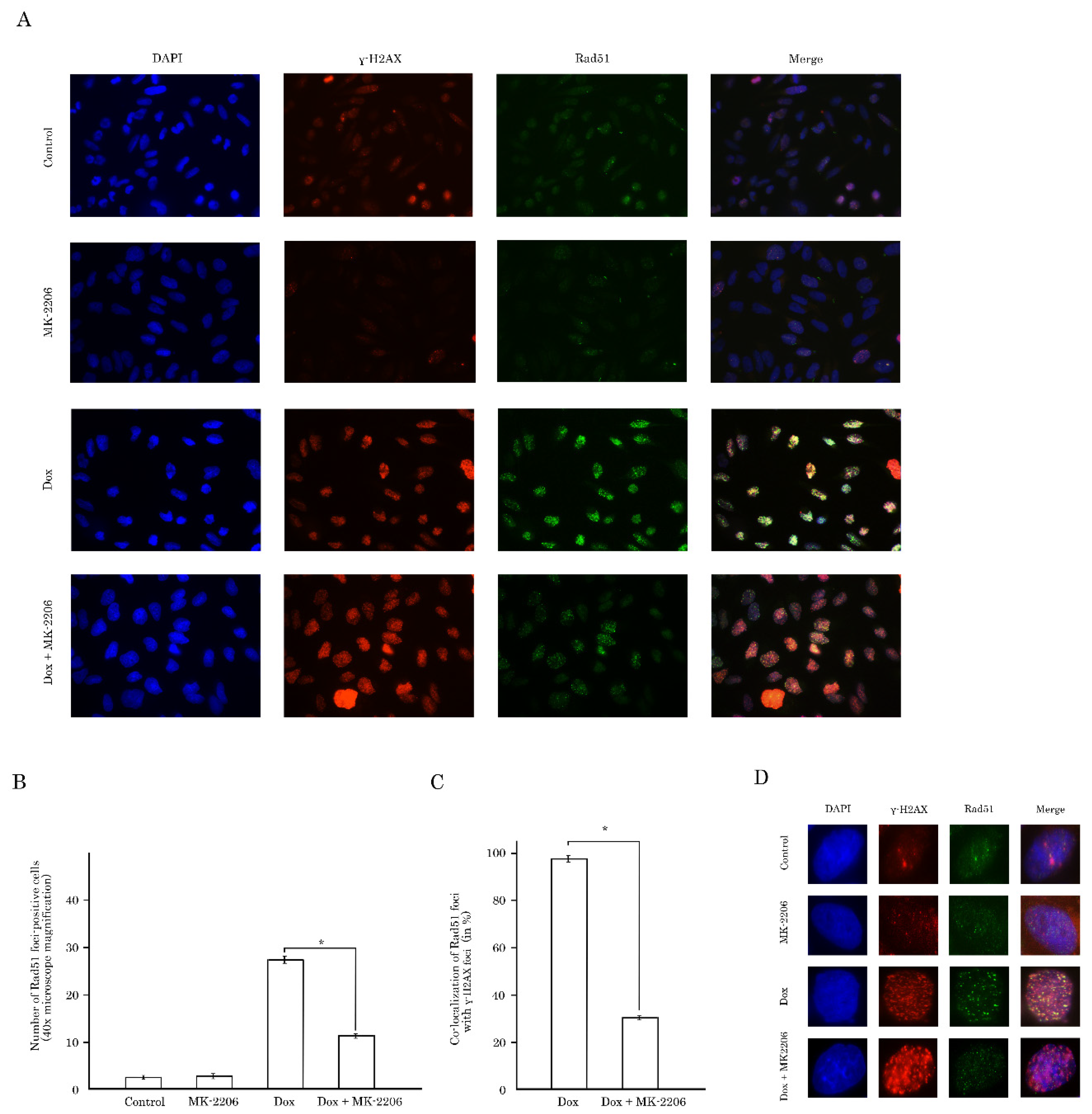
2.3. Inhibition of AKT Signaling Attenuates Repair of Dox-Induced DNA Damage
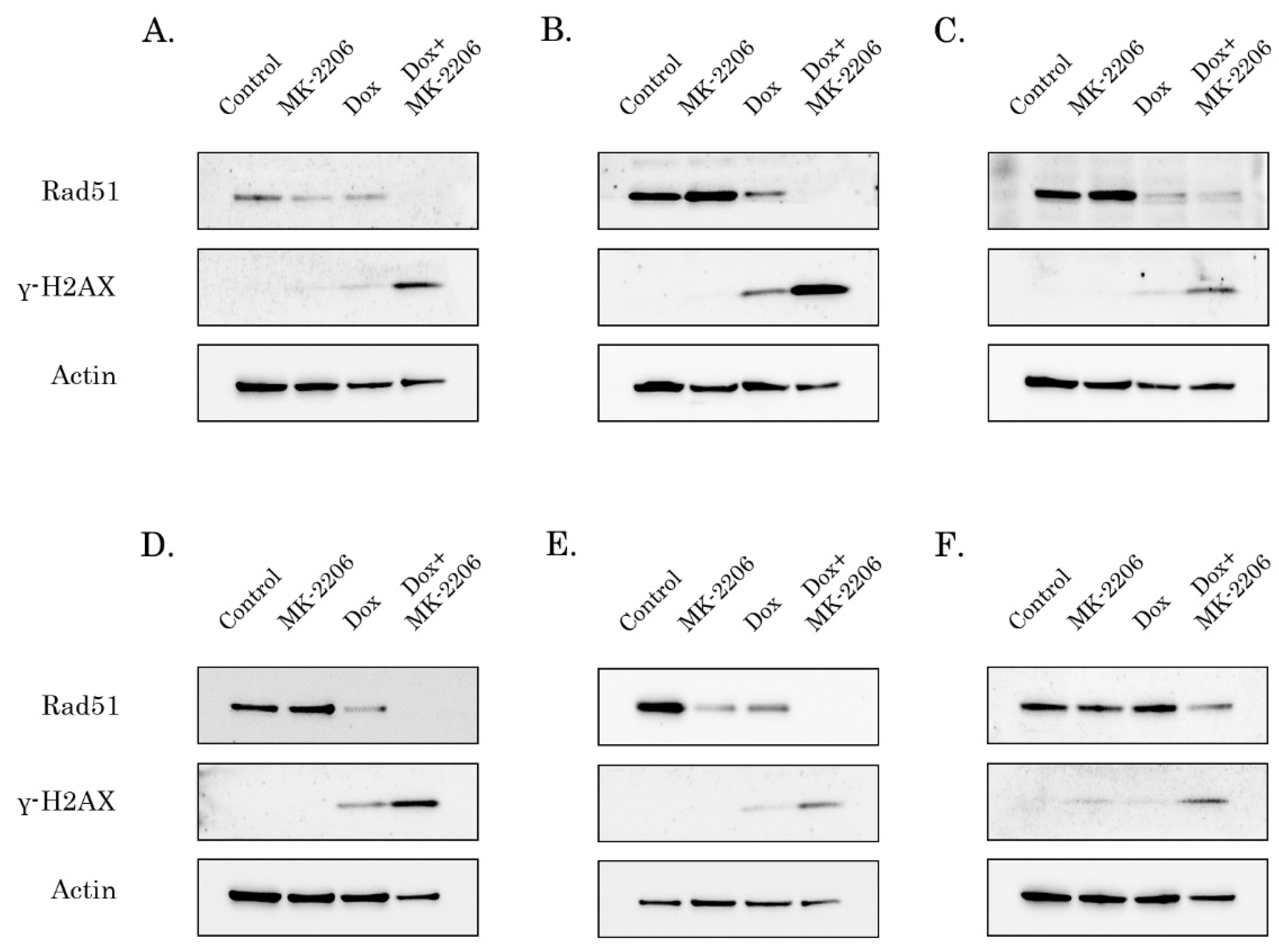
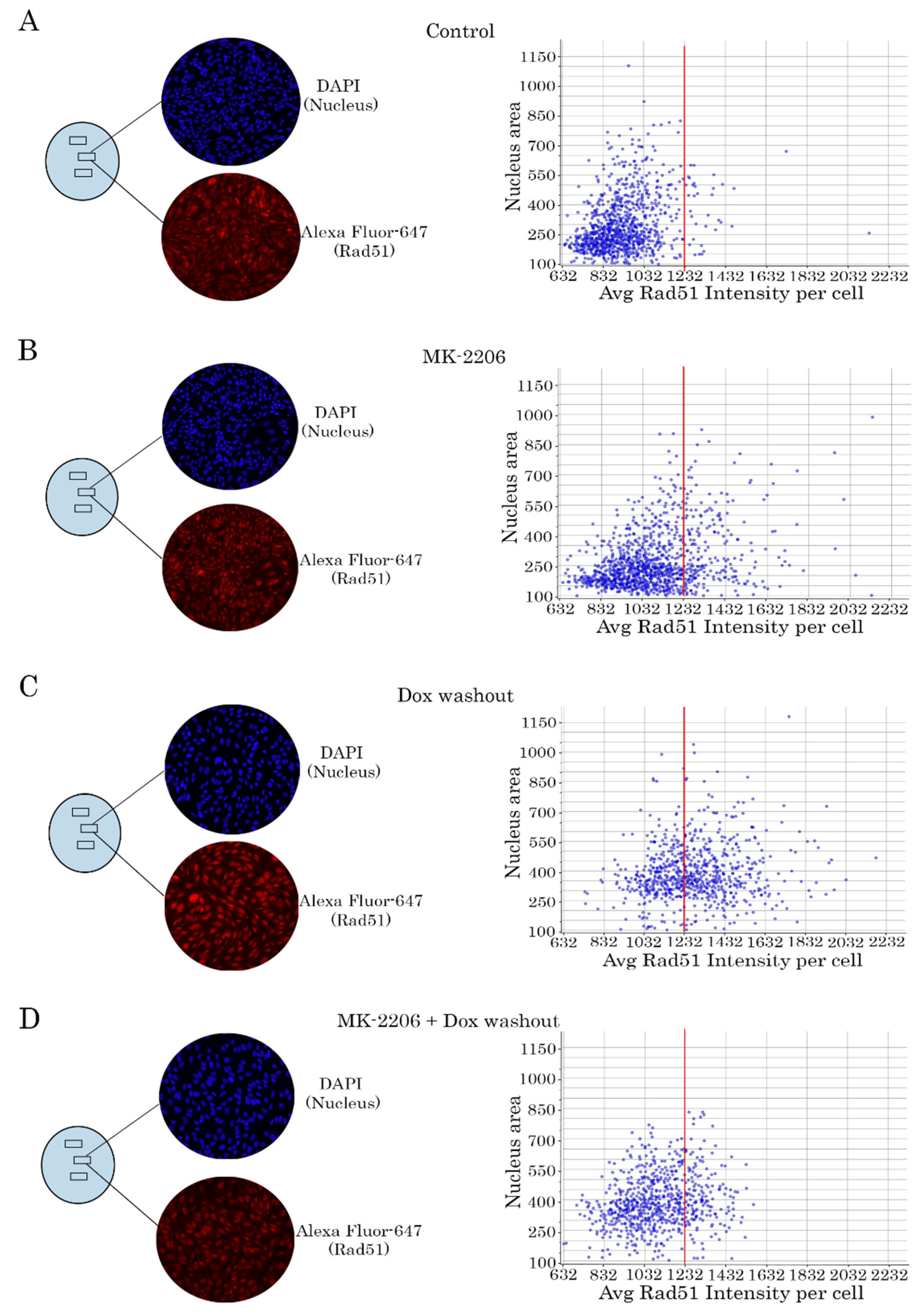
2.4. Rad51 Expression Is Substantially Reduced in AKT-Inhibited Cancer Cells
2.5. AKT Impacts Rad51 Stability in GIST and STS Cell Lines
3. Discussion
4. Materials and Methods
4.1. Chemical Compounds
4.2. Antibodies
4.3. Cell lines and Culture Conditions
4.4. Cellular Survival MTS-Based Assay
4.5. Western Blotting and Co-Immunoprecipitation (Co-IP)
4.6. Immunofluorescence Staining
4.7. RNA Extraction and Real-Time Quantitative PCR
4.8. Single-Cell Electrophoresis (Comet Assay)
4.9. Sub-Cellular Fractionation
4.10. Images Quantification and Registration
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.; McDougall, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Huang, K.; Zhou, Y.; Du, C.Y.; Ye, Y.W.; Fu, H.; Zhou, X.Y.; Shi, Y.Q. Molecular mechanisms of secondary imatinib resistance in patients with gastrointestinal stromal tumors. J. Cancer Res. Clin. Oncol. 2010, 136, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ikezoe, T.; Nishioka, C.; Takezaki, Y.; Hanazaki, K.; Taguchi, T.; Yokoyama, A. Long-term exposure of gastrointestinal stromal tumor cells to sunitinib induces epigenetic silencing of the PTEN gene. Int. J. Cancer 2012, 130, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Ricci, R.; Maggiano, N.; Castri, F.; Rinelli, A.; Murazio, M.; Pacelli, F.; Potenza, A.E.; Vecchio, F.M.; Larocca, L.M. Role of PTEN in gastrointestinal stromal tumor progression. Arch. Pathol. Lab. Med. 2004, 128, 421–425. [Google Scholar]
- Patel, S. Exploring novel therapeutic targets in GIST: Focus on the PI3K/Akt/mTOR pathway. Curr. Oncol. Rep. 2013, 15, 386–395. [Google Scholar] [CrossRef]
- Van Looy, T.; Wozniak, A.; Floris, G.; Sciot, R.; Li, H.; Wellens, J.; Vanleeuw, U.; Fletcher, J.A.; Manley, P.W.; Schöffski, P.; et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: Rationale and efficacy. Clin. Cancer Res. 2014, 20, 6071–6082. [Google Scholar] [CrossRef] [Green Version]
- Zook, P.; Pathak, H.B.; Belinsky, M.G.; Gersz, L.; Devarajan, K.; Zhou, Y.; Godwin, A.K.; von Mehren, M.; Rink, L. Combination of imatinibmesylate and AKT inhibitor provides synergistic effects in preclinical study of gastrointestinal stromal tumor. Clin. Cancer Res. 2016, 23, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Conley, A.P.; Araujo, D.; Ludwig, J.; Ravi, V.; Samuels, B.L.; Choi, H.; Thall, P.F.; Patel, S.; Benjamin, R.; Trent, J. A randomized phase II study of perifosine (P) plus imatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST). J. Clin. Oncol. 2009, 27, 10563. [Google Scholar] [CrossRef]
- Schoffski, P.; Reichardt, P.; Blay, J.Y.; Dumez, H.; Morgan, J.A.; Ray-Coquard, I.; Hollaender, N.; Jappe, A.; Demetri, G.D. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann. Oncol. 2010, 21, 1990–1998. [Google Scholar] [CrossRef]
- Yang, J.; Du, X.; Chen, K.; Ylipää, A.; Lazar, A.J.; Trent, J.; Lev, D.; Pollock, R.; Hao, X.; Hunt, K.; et al. Genetic aberrations in soft tissue leiomyosarcoma. Cancer Lett. 2009, 275, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando, E.; Charytonowicz, E.; Dudas, M.E.; Mendez, S.; Matushansky, I.; Mills, J.; Socci, N.D.; Behrendt, N.; Ma, L.; Maki, R.G.; et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat. Med. 2007, 13, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Bathan, A.J.; Constantinidou, A.; Pollack, S.M.; Jones, R.J. Diagnosis, prognosis, and management of leiomyosarcoma: Recognition of anatomic variants. Curr. Opin. 2013, 25, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Mateo-Lozano, S.; Tirado, O.M.; Notario, V. Rapamycin induces the fusion-type independent downregulation of the EWS/FLI-1 proteins and inhibits Ewing’s sarcoma cell proliferation. Oncogene 2003, 22, 9282–9287. [Google Scholar] [CrossRef] [PubMed]
- Manara, M.C.; Nicoletti, G.; Zambelli, D.; Ventura, S.; Guerzoni, C.; Landuzzi, L.; Lollini, P.L.; Maira, S.M.; García-Echeverría, C.; Mercuri, M.; et al. NVP-BEZ235 as a new therapeutic option for sarcomas. Clin. Cancer Res. 2010, 16, 530–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petricoin, E.F., 3rd; Espina, V.; Araujo, R.P.; Midura, B.; Yeung, C.; Wan, X.; Eichler, G.S.; Johann, D.J., Jr.; Qualman, S.; Tsokos, M.; et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007, 67, 3431–3440. [Google Scholar] [PubMed] [Green Version]
- Zhang, J.; Yu, X.H.; Yan, Y.G.; Wang, C.; Wang, W.J. PI3K/Akt signaling in osteosarcoma. Clin. Chim. Acta 2015, 444, 182–192. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, L.B.; Peng, A.F.; Wang, T.F.; Long, X.H.; Gao, S.; Zhou, R.P.; Liu, Z.L. LY294002 inhibits the malignant phenotype of osteosarcoma cells by modulating the phosphatidylinositol 3-kinase/Akt/fatty acid synthase signaling pathway in vitro. Mol. Med. Rep. 2015, 11, 1352–1357. [Google Scholar] [CrossRef] [Green Version]
- Schöffski, P.; Cornillie, J.; Wozniak, A.; Li, H.; Hompes, D. Soft tissue sarcoma: An update on systemic treatment options for patients with advanced disease. Oncol. Res. Treat. 2014, 37, 355–362. [Google Scholar] [CrossRef]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Dickson, M.A.; D’Adamo, D.R.; Keohan, M.L.; D’Angelo, S.P.; Carvajal, R.D.; Gounder, M.M.; Maki, R.G.; Qin, L.X.; Lefkowitz, R.A.; McKennon, O.R.; et al. Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015, 2015, 532478. [Google Scholar] [CrossRef] [PubMed]
- García-Del-Muro, X.; López-Pousa, A.; Maurel, J.; Martín, J.; Martínez-Trufero, J.; Casado, A.; Gómez-España, A.; Fra, J.; Cruz, J.; Poveda, A.; et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: A Spanish Group for Research on Sarcomas study. J. Clin. Oncol. 2011, 29, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Lee, D.J.; Mehalek, K.R.; Makielski, K.R.; Wozniak, A.; Seneviratne, D.S.; Korzeniewski, N.; Cuevas, R.; Parry, J.A.; Brown, M.F.; et al. Unbiased compound screening identifies unexpected drug sensitivities and novel treatment options for gastrointestinal stromal tumors. Cancer Res. 2014, 74, 1200–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessetto, Z.Y.; Ma, Y.; Hirst, J.J.; von Mehren, M.; Weir, S.J.; Godwin, A.K. Drug repurposing identifies a synergistic combination therapy with imatinibmesylate for gastrointestinal stromal tumor. Mol. Cancer Ther. 2014, 13, 2276–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, G.H. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymonowicz, K.; Oeck, S.; Krysztofiak, A.; van der Linden, J.; Iliakis, G.; Jendrossek, V. Restraining AKT1 phosphorylation attenuates the repair of radiation-induced DNA double-strand breaks and reduces the survival of irradiated cancer cells. Int. J. Mol. Sci. 2018, 19, 2233. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Kehlbach, R.; Florczak, U.; Sak, A.; Wang, S.; Chen, J.; Lobrich, M.; Rodemann, H.P. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Ther. 2008, 7, 1772–1781. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Maier, J.; Iida, M.; Rebholz, S.; Holler, M.; Grottke, A.; Juker, M.; Wheeler, D.L.; Rothbauer, U.; Rodemann, H.P. Akt1 and Akt3 but not Akt2 through interaction with DNA-PKcs stimulate proliferation and post-irradiation cell survival of K-RAS-mutated cancer cells. Cell Death Discov. 2017, 3, 17072. [Google Scholar] [CrossRef]
- Mueck, K.; Rebholz, S.; Harati, M.; Rodemann, H.; Toulany, M. AKT1 stimulates homologous recombination repair of DNA double-strand breaks in a Rad51-dependent manner. Int. J. Mol. Sci. 2017, 18, 2473. [Google Scholar] [CrossRef] [Green Version]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene 2013, 32, 1943–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkov, A.; Kilvaer, T.K.; Sorbye, S.W.; Donnem, T.; Smeland, E.; Bremnes, R.M.; Busund, L.T. The prognostic impact of Akt isoforms, PI3K and PTEN related to female steroid hormone receptors in soft tissue sarcomas. J. Transl. Med. 2011, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Morooka, T.; Hoshida, Y.; Zhang, B.; Qiu, Y.; Nakamichi, I.; Hamada, K.; Ueda, T.; Naka, N.; Kudawara, I.; et al. Prognostic significance of activated AKT expression in soft-tissue sarcoma. Clin. Cancer Res. 2006, 12, 3070–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Bikinieva, F.; Nurgatina, I.; Mustafin, I.; Aukhadieva, A.; Kurtasanov, R.; Andriutsa, N.; Shagimardanova, E.; et al. Inhibition of FGFR2-Signaling Attenuates a Homology-Mediated DNA Repair in GIST and Sensitizes Them to DNA-Topoisomerase II Inhibitors. Int. J. Mol. Sci. 2020, 21, 352. [Google Scholar] [CrossRef] [Green Version]
- Sahlberg, S.H.; Gustafsson, A.S.; Pendekanti, P.N.; Glimelius, B.; Stenerlow, B. The influence of akt isoforms on radiation sensitivity and DNA repair in colon cancer cell lines. Tumour. Biol. 2014, 35, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Babichev, Y.; Kabaroff, L.; Datti, A.; Uehling, D.; Isaac, M.; Al-Awar, R.; Prakesch, M.; Sun, R.; Boutros, P.; Venier, R.; et al. PI3K/AKT/mTOR inhibition in combination with doxorubicin is an effective therapy for leiomyosarcoma. J. Transl. Med. 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, V.; Vögler, O.; Barceló, F.; Martín-Broto, J.; Martínez-Serra, J.; Ruiz-Gutiérrez, V.; Alemany, R. Down-regulation of AKT signaling by ursolic acid induces intrinsic apoptosis and sensitization to doxorubicin in soft tissue sarcoma. PLoS ONE 2016, 11, e0155946. [Google Scholar] [CrossRef]
- Hayasaka, N.; Takada, K.; Nakamura, H.; Arihara, Y.; Kawano, Y.; Osuga, T.; Murase, K.; Kikuchi, S.; Iyama, S.; Emori, M.; et al. Combination of eribulin plus AKT inhibitor evokes synergistic cytotoxicity in soft tissue sarcoma cells. Sci. Rep. 2019, 9, 5759. [Google Scholar] [CrossRef]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef] [Green Version]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A novel receptor tyrosine kinase switch promotes gastrointestinal stromal tumor drug resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mladenov, E.; Anachkova, B.; Tsaneva, I. Sub-nuclear localization of Rad51 in response to DNA damage. Genes Cells 2006, 11, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chang, T.T.; Chou, T.C. Rational approach to the clinical protocol design for drug combinations: A review. Acta Paediatr. Taiwan 2000, 41, 294–302. [Google Scholar] [PubMed]
- Chou, T.C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 | MK-2206 (µM) | Dox (µM) | Dox + MK-2206 (µM) | Fold Increase |
|---|---|---|---|---|
| GIST T1-R | 32.9 ± 4.3 | 0.77 ± 0.13 | 0.42 ± 0.1 | 1.8 |
| GIST 430 | 17 ± 0.2 | 1.21 ± 0.08 | 0.59 ± 0.07 | 2.1 |
| SK-LMS-1 | 13.5 ± 1.02 | 0.49 ± 0.05 | 0.3 ± 0.03 | 1.6 |
| RD | 26.4 ± 1 | 0.79 ± 0.08 | 0.47 ± 0.03 | 1.7 |
| U2-OS | 27.8 ± 1.2 | 0.49 ± 0.07 | 0.22 ± 0.04 | 2.2 |
| HT-1080 | 19.7 ± 0.4 | 0.116 ± 0.005 | 0.05 ± 0.006 | 2.3 |
| Cell Line | Molar Ratio of DOX: MK-2206 | CI Value ± SD |
|---|---|---|
| RD | 1:11.62 | 1.41 ± 0.23 |
| 1:5.81 | 0.07 ± 0.01 | |
| 1:2.90 | 0.15 ± 0.02 | |
| 1:1.45 | 0.31 ± 0.04 | |
| 1.38:1 | 0.34 ± 0.04 | |
| SK-LMS-1 | 1:11.62 | 1.18 ± 0.10 |
| 1:5.81 | 0.58 ± 0.06 | |
| 1:2.90 | 0.60 ± 0.02 | |
| 1:1.45 | 0.46 ± 0.05 | |
| 1.38:1 | 0.33 ± 0.03 | |
| HT-1080 | 1:11.62 | 0.32 ± 0.06 |
| 1:5.81 | 0.35 ± 0.05 | |
| 1:2.90 | 0.41 ± 0.06 | |
| 1:1.45 | 0.83 ± 0.09 | |
| 1.38:1 | 1.68 ± 0.60 | |
| U2-OS | 1:11.62 | 0.19 ± 0.02 |
| 1:5.81 | 0.30 ± 0.02 | |
| 1:2.90 | 0.26 ± 0.01 | |
| 1:1.45 | 0.26 ± 0.02 | |
| 1.38:1 | 0.40 ± 0.10 | |
| GIST T1-R | 1:11.62 | 0.45 ± 0.05 |
| 1:5.81 | 0.60 ± 0.06 | |
| 1:2.90 | 0.42 ± 0.05 | |
| 1:1.45 | 0.64 ± 0.04 | |
| 1.38:1 | 0.57 ± 0.06 | |
| GIST 430 | 1:11.62 | 2.32 ± 0.42 |
| 1:5.81 | 0.43 ± 0.07 | |
| 1:2.90 | 0.61 ± 0.09 | |
| 1:1.45 | 0.69 ± 0.11 | |
| 1.38:1 | 0.75 ± 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichuk, S.; Bikinieva, F.; Nurgatina, I.; Dunaev, P.; Valeeva, E.; Aukhadieva, A.; Sabirov, A.; Galembikova, A. Inhibition of AKT-Signaling Sensitizes Soft Tissue Sarcomas (STS) and Gastrointestinal Stromal Tumors (GIST) to Doxorubicin via Targeting of Homology-Mediated DNA Repair. Int. J. Mol. Sci. 2020, 21, 8842. https://doi.org/10.3390/ijms21228842
Boichuk S, Bikinieva F, Nurgatina I, Dunaev P, Valeeva E, Aukhadieva A, Sabirov A, Galembikova A. Inhibition of AKT-Signaling Sensitizes Soft Tissue Sarcomas (STS) and Gastrointestinal Stromal Tumors (GIST) to Doxorubicin via Targeting of Homology-Mediated DNA Repair. International Journal of Molecular Sciences. 2020; 21(22):8842. https://doi.org/10.3390/ijms21228842
Chicago/Turabian StyleBoichuk, Sergei, Firuza Bikinieva, Ilmira Nurgatina, Pavel Dunaev, Elena Valeeva, Aida Aukhadieva, Alexey Sabirov, and Aigul Galembikova. 2020. "Inhibition of AKT-Signaling Sensitizes Soft Tissue Sarcomas (STS) and Gastrointestinal Stromal Tumors (GIST) to Doxorubicin via Targeting of Homology-Mediated DNA Repair" International Journal of Molecular Sciences 21, no. 22: 8842. https://doi.org/10.3390/ijms21228842