An In Vitro Model to Investigate the Role of Helicobacter pylori in Type 2 Diabetes, Obesity, Alzheimer’s Disease and Cardiometabolic Disease

 , and
, and
Abstract
:1. Introduction
2. Results
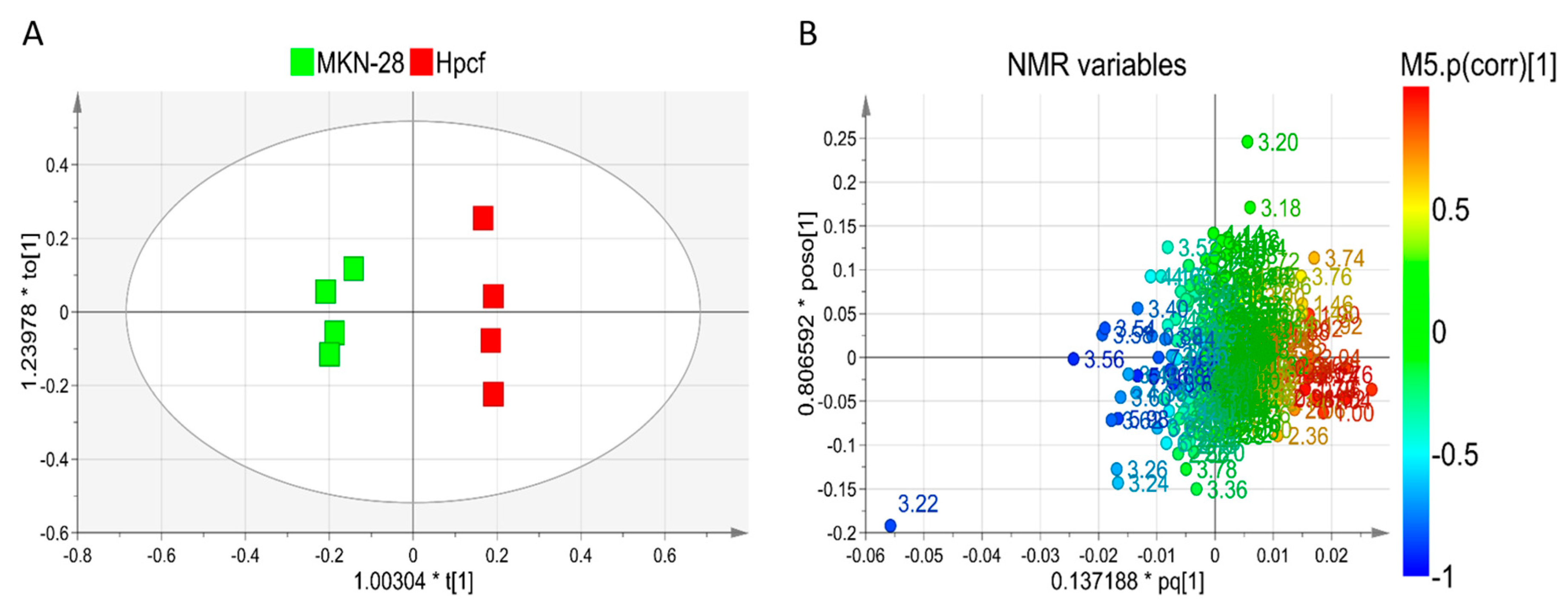
2.1. NMR-Based Metabolomics Analysis
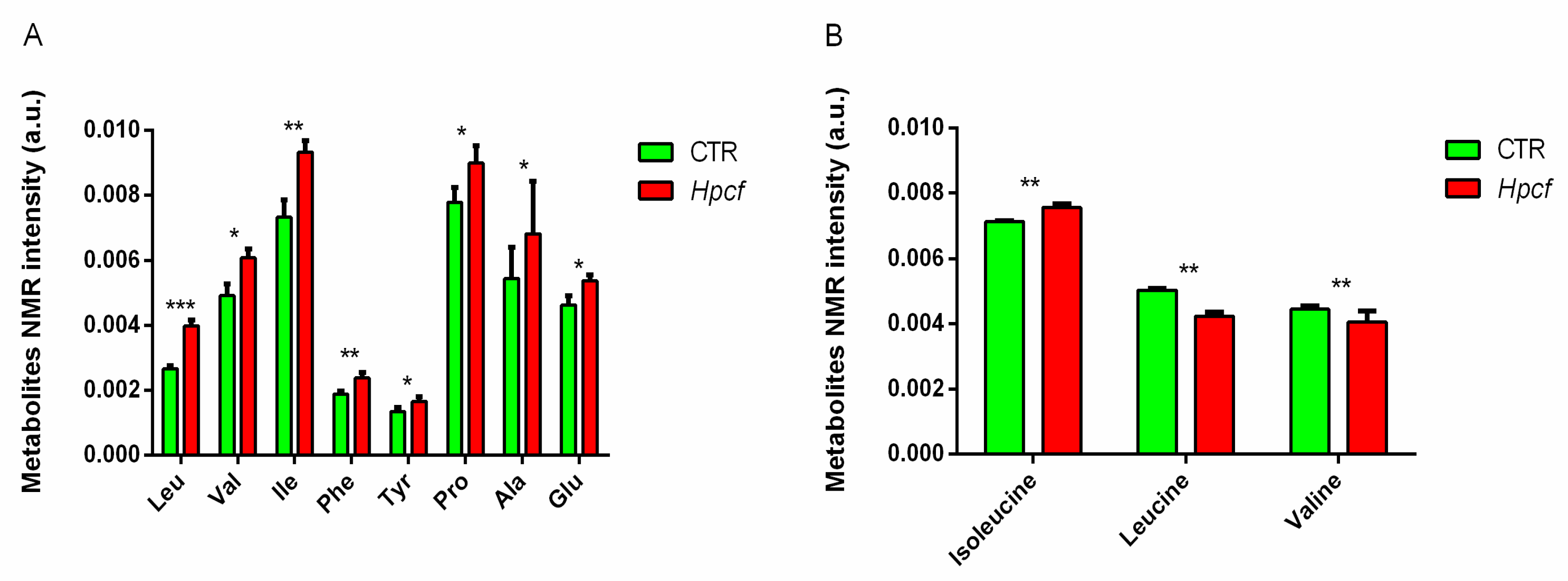
2.2. Helicobacter Pylori Alters the Amino Acids Metabolism
2.3. MKN-28 Cells Uptake BCAA from Culture Medium
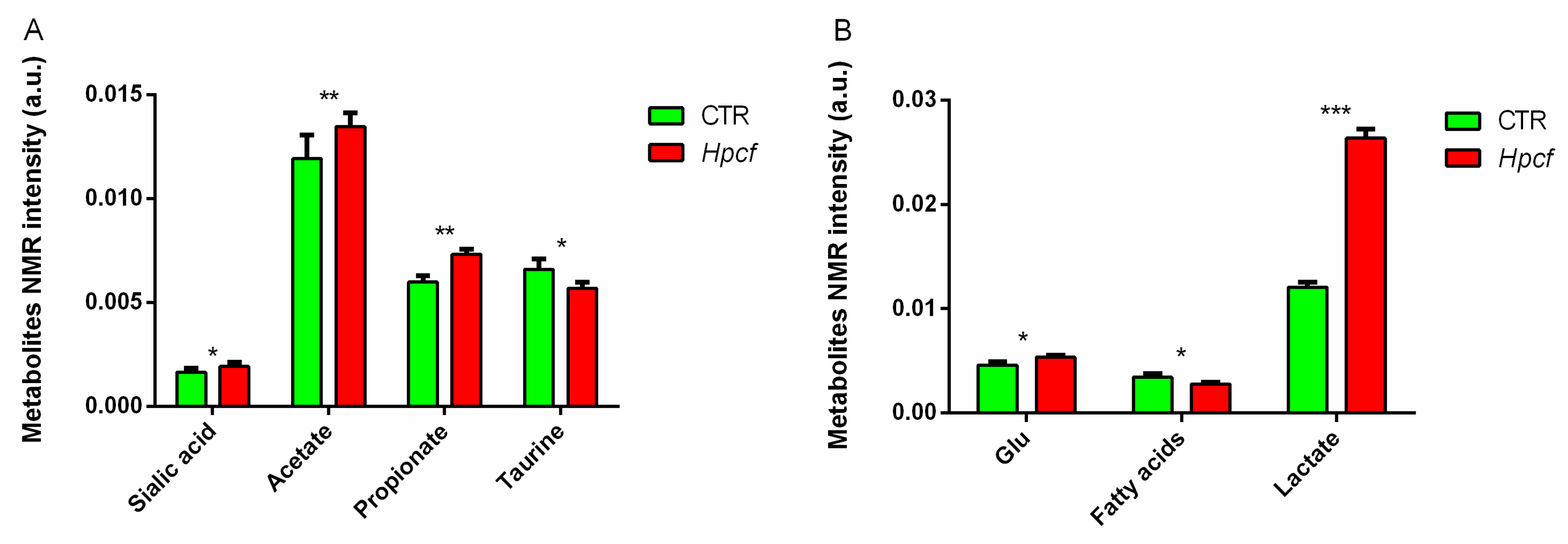
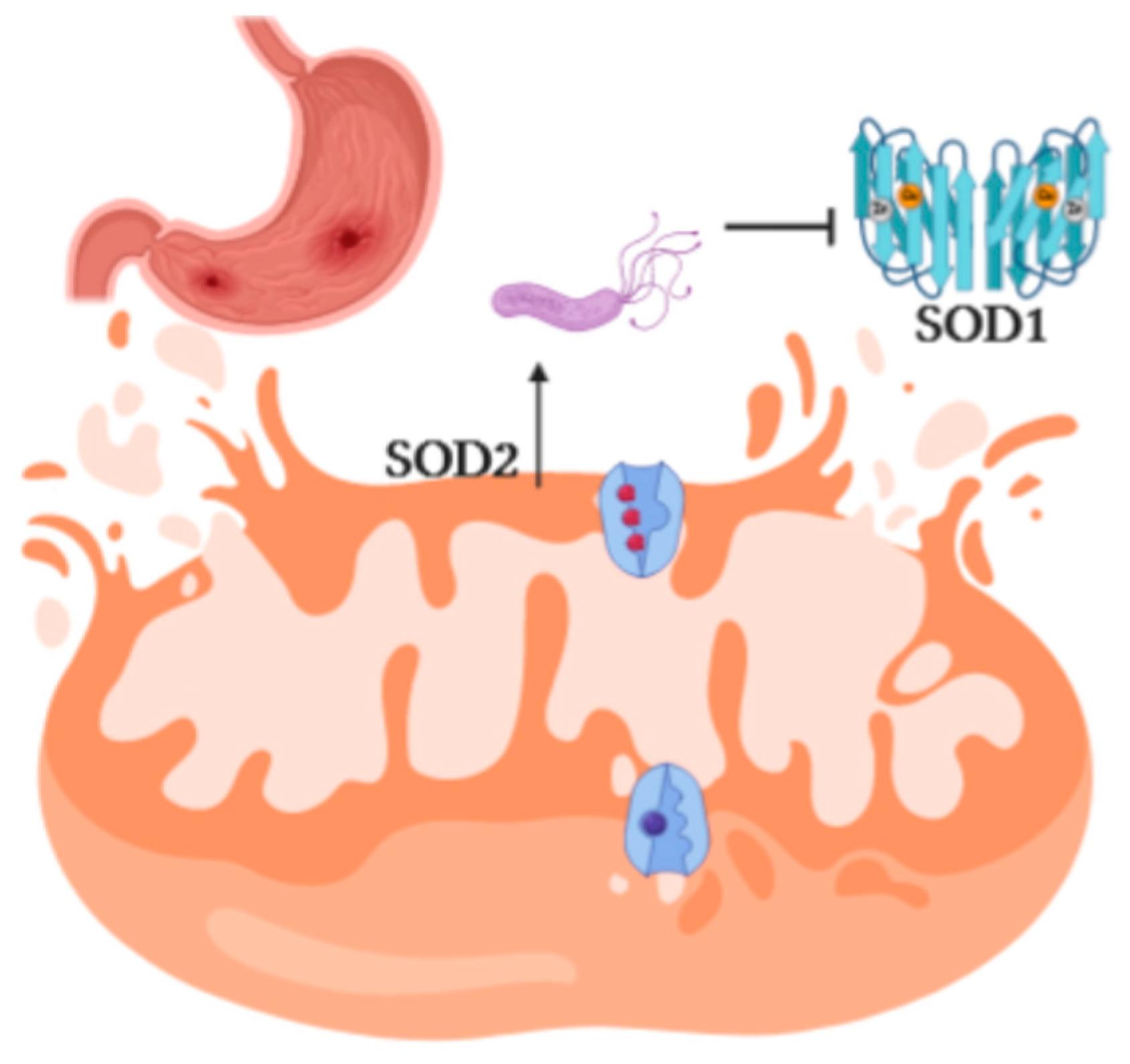
2.4. Helicobacter Pylori Induces Inflammation and Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Helicobacter Pylori
4.2. Cell Culture Conditions
4.3. Cell Culture for Metabolomics Analysis
4.4. Metabolites Extraction and NMR Samples Preparation
4.5. NMR Spectroscopy
4.6. RNA Extraction and Real-Time PCR
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Hp | Helicobacter pylori |
| NMR | Nuclear Magnetic Resonance |
| Hpcf | Hp cell filtrate |
| BCAA | Branched chain amino acids |
| mTORC1 | mammalian Target Of Rapamycin Complex 1 |
| TLRs | Tool like receptors |
| NF-AT | nuclear translocation of the T transcription factor |
| T2D | Type 2 diabetes |
| OB | Obesity |
| AD | Alzheimer’s disease |
| CMD | Cardiometabolic disease |
| PCA | Principal Component Analysis |
| OPLS- | Orthogonal partial least squares discriminant analysis |
| AAA | Aromatic amino acids |
| ROS | Reactive oxygen species |
| LAT1 | Large neutral amino acid transporter |
| BCAT | Branched chain amino acid transaminase |
| KLF15 | Kruppel-like factor 15 |
| FBS | Fetal Bovine Serum |
| BHI | Brain Heart Infusion medium |
| PBS | Phosphate-Buffered Saline |
References
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Cuomo, P.; Papaianni, M.; Fulgione, A.; Guerra, F.; Capparelli, R.; Medaglia, C. An innovative approach to control H. Pylori-induced persistent inflammation and colonization. Microorganisms 2020, 8, 1214. [Google Scholar] [CrossRef]
- Cooke, C.L.; Huff, J.L.; Solnick, J.V. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol. Med. Microbiol. 2005, 45, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Cullen, T.W.; Giles, D.K.; Wolf, L.N.; Ecobichon, C.; Boneca, I.G.; Trent, M.S. Helicobacter pylori versus the Host: Remodeling of the Bacterial Outer Membrane Is Required for Survival in the Gastric Mucosa. PLoS Pathog. 2011, 7, e1002454. [Google Scholar] [CrossRef] [Green Version]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, F.; Gasbarrini, A.; Polyzos, S.A.; Kountouras, J. Extragastric Diseases and Helicobacter pylori. Helicobacter 2015, 20, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.; Yamaoka, Y. Helicobacter pylori virulence factors exploiting gastric colonization and its pathogenicity. Toxins 2019, 11, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.-J.; Lee, J.; Oh, S.J.; Chung, H.J.; Chen, J.; Blanke Correspondence, S.R. Helicobacter pylori Infection Modulates Host Cell Metabolism through VacA-Dependent Inhibition of mTORC1. Cell Host Microbe 2018, 23, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.W.; Luo, R.H.; Zhao, Q.; Shen, Z.Z.; Huang, L.L.; An, X.Y.; Zhao, L.J.; Wang, J.; Huang, Y.Z. Helicobacter pylori induces mitochondrial DNA mutation and reactive oxygen species level in AGS cells. Int. J. Med. Sci. 2011, 8, 56–67. [Google Scholar] [CrossRef] [Green Version]
- White, J.R.; Winter, J.A.; Robinson, K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J. Inflamm. Res. 2015, 8, 137–147. [Google Scholar]
- White, P.J.; Newgard, C.B. Branched-chain amino acids in disease. Science 2019, 363, 582–583. [Google Scholar] [CrossRef]
- Shao, D.; Villet, O.; Zhang, Z.; Choi, S.W.; Yan, J.; Ritterhoff, J.; Gu, H.; Djukovic, D.; Christodoulou, D.; Kolwicz, S.C.; et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.I.; Romano, M.; Basarali, M.K.; Elzagallaai, A.; Karaman, M.; Demir, Z.; Demir, M.F.; Akcay, F.; Seyrek, M.; Haksever, N.; et al. The Effect of Corrected Inflammation, Oxidative Stress and Endothelial Dysfunction on Fmd Levels in Patients with Selected Chronic Diseases: A Quasi-Experimental Study. Sci. Rep. 2020, 10, 9018. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Sul, J.H.; Martin, L.S.; Eskin, E. Population structure in genetic studies: Confounding factors and mixed models. PLoS Genet. 2018, 14, e1007309. [Google Scholar] [CrossRef]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar] [CrossRef]
- Shaham, O.; Slate, N.G.; Goldberger, O.; Xu, Q.; Ramanathan, A.; Souza, A.L.; Clish, C.B.; Sims, K.B.; Mootha, V.K. A plasma signature of human mitochondrial disease revealed through metabolic profiling of spent media from cultured muscle cells. Proc. Natl. Acad. Sci. USA 2010, 107, 1571–1575. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef]
- Frühbeck, G. Obesity: Screening for the evident in obesity. Nat. Rev. Endocrinol. 2012, 8, 570–572. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Yamakado, M.; Nagao, K.; Imaizumi, A.; Tani, M.; Toda, A.; Tanaka, T.; Jinzu, H.; Miyano, H.; Yamamoto, H.; Daimon, T.; et al. Plasma Free Amino Acid Profiles Predict Four-Year Risk of Developing Diabetes, Metabolic Syndrome, Dyslipidemia, and Hypertension in Japanese Population. Sci. Rep. 2015, 5, 11918. [Google Scholar] [CrossRef]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 2018, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Toledo, J.B.; Arnold, M.; Kastenmüller, G.; Chang, R.; Baillie, R.A.; Han, X.; Thambisetty, M.; Tenenbaum, J.D.; Suhre, K.; Thompson, J.W.; et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 2017, 13, 965–984. [Google Scholar] [CrossRef]
- Boado, R.J.; Li, J.Y.; Nagaya, M.; Zhang, C.; Pardridge, W.M. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 12079–12084. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.K.; Bischoff, D.S.; Song, A.M.; Uyemura, K.; Yamaguchi, D.T. Metabolic relationship between diabetes and Alzheimer’s Disease affected by Cyclo(His-Pro) plus zinc treatment. BBA Clin. 2017, 7, 41–54. [Google Scholar] [CrossRef]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-Induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Koelman, L.; Pivovarova-Ramich, O.; Pfeiffer, A.F.H.; Grune, T.; Aleksandrova, K. Cytokines for evaluation of chronic inflammatory status in ageing research: Reliability and phenotypic characterisation. Immun. Ageing 2019, 16, 11. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex i Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Zhao, H.; Zhu, R.; Chen, C.; Cao, H.; Han, J.; Han, S. On the use of abiotic sialic acids to attenuate cell inflammation. Sci. Rep. 2018, 8, 17320. [Google Scholar] [CrossRef]
- Paglia, G.; Stocchero, M.; Cacciatore, S.; Lai, S.; Angel, P.; Alam, M.T.; Keller, M.; Ralser, M.; Astarita, G. Unbiased Metabolomic Investigation of Alzheimer’s Disease Brain Points to Dysregulation of Mitochondrial Aspartate Metabolism. J. Proteome Res. 2016, 15, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B. The hypoxia–lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Bond, P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr. 2016, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar]
- Matsunaga, S.; Nishiumi, S.; Tagawa, R.; Yoshida, M. Alterations in metabolic pathways in gastric epithelial cells infected with Helicobacter pylori. Microb. Pathog. 2018, 124, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Luc, Y.; Martinez, J.; Bi, Y.; Lian, G.; Wang, T.; Milasta, S.; Wang, J.; Yang, M.; Liu, G.; et al. Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. USA 2016, 113, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5′-Monophosphate-Activated Protein Kinase Promotes Macrophage Polarization to an Anti-Inflammatory Functional Phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Ricci, V. Relationship between vaca toxin and host cell autophagy in Helicobacter pylori infection of the human stomach: A few answers, Many questions. Toxins 2016, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- D’Souza, G.; Waschina, S.; Pande, S.; Bohl, K.; Kaleta, C.; Kost, C. Less is more: Selective advantages can explain the prevalent loss of biosynthetic genes in bacteria. Evolution 2014, 68, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Bayram, T.; Van De Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, P.; Peters, R.; Hans, P.J.C. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Contaldi, F.; Capuano, F.; Fulgione, A.; Aiese Cigliano, R.; Sanseverino, W.; Iannelli, D.; Medaglia, C.; Capparelli, R. The hypothesis that Helicobacter pylori predisposes to Alzheimer’s disease is biologically plausible. Sci. Rep. 2017, 7, 7817. [Google Scholar] [CrossRef]
- Papaianni, M.; Paris, D.; Woo, S.L.; Fulgione, A.; Rigano, M.M.; Parrilli, E.; Tutino, M.L.; Marra, R.; Manganiello, G.; Casillo, A.; et al. Plant dynamic metabolic response to bacteriophage treatment after Xanthomonas campestris pv campestris infection. Front. Microbiol. 2020, 11, 732. [Google Scholar] [CrossRef]
- Fulgione, A.; Papaianni, M.; Cuomo, P.; Paris, D.; Romano, M.; Tuccillo, C.; Palomba, L.; Medaglia, C.; De Seta, M.; Esposito, N.; et al. Interaction between MyD88, TIRAP and IL1RL1 against Helicobacter pylori infection. Sci. Rep. 2020, 10, 15831. [Google Scholar] [CrossRef]
- Papaianni, M.; Cuomo, P.; Fulgione, A.; Albanese, D.; Gallo, M.; Paris, D.; Motta, A.; Iannelli, D.; Capparelli, R. Bacteriophages Promote Metabolic Changes in Bacteria Biofilm. Microorganisms 2020, 8, 480. [Google Scholar] [CrossRef] [Green Version]
- Sansone, C.; Nuzzo, G.; Galasso, C.; Casotti, R.; Fontana, A.; Romano, G.; Ianora, A. The Marine Dinoflagellate Alexandrium andersoni Induces Cell Death in Lung and Colorectal Tumor Cell Lines. Mar. Biotechnol. 2018, 20, 343–352. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Name | Gene ID | Gene Name | Fold Regulation 1 h | Fold Regulation 2 h |
|---|---|---|---|---|
| mTOR signaling pathway | RPTOR | Regulatory associated protein of mTOR complex 1 | −1.42 | 286.04 |
| MLST8 | mTOR associated protein, LST8 homolog (S. cerevisiae) | −1.42 | 398.95 | |
| AKT1 | V-akt murine thymoma viral oncogene homolog 1 | −1.42 | 50.13 | |
| AKT2 | V-akt murine thymoma viral oncogene homolog 2 | −1.42 | 504.97 | |
| INSR | Insulin receptor | −1.42 | 257.79 | |
| IRS1 | Insulin receptor substrate 1 | −1.42 | 278.22 | |
| PLD1 | Phospholipase D1, phosphatidylcholine-specific | −6.31 | 130.70 | |
| RPS6KA2 | Ribosomal protein S6 kinase, 90 kDa, polypeptide 2 | −1.24 | 3.37 | |
| PDPK1 | 3-phosphoinositide dependent protein kinase-1 | −1.53 | 28.25 | |
| PIK3CB | Phosphoinositide-3-kinase, catalytic, beta polypeptide | −1.42 | 16.34 | |
| PIK3CD | Phosphoinositide-3-kinase, catalytic, delta polypeptide | 3.37 | 184.83 | |
| PIK3CG | Phosphoinositide-3-kinase, catalytic, gamma polypeptide | −1.42 | 215.28 | |
| CHUK | Conserved helix-loop-helix ubiquitous kinase | −4.08 | 181.03 | |
| EIF4E | Eukaryotic translation initiation factor 4E | −1.42 | 922.92 | |
| HIF1A | Hypoxia inducible factor 1, alpha subunit | 192.93 | 955.47 | |
| Inflammatory pathway | CXCL8 | Interleukin 8 | −3.29 | 2.96 |
| IL-6 | Interleukin 6 | 14.45 | 114.56 | |
| TLR2 | Toll-like receptor 2 | 58 | 72.18 | |
| TLR9 | Toll-like receptor 9 | 3.29 | 134.55 | |
| TNF | Tumor necrosis factor | 12.9 | 154.26 | |
| Oxidative stress pathway | ATOX1 | ATX1 antioxidant protein 1 homolog (yeast) | 3.57 | 37.69 |
| GPX2 | Glutathione peroxidase 2 (gastrointestinal) | 3.57 | 37.69 | |
| GPX4 | Glutathione peroxidase 4 (gastrointestinal) | 3.57 | 37.69 | |
| GSS | Glutathione synthetase | 3.57 | 9.54 | |
| NOX5 | NADPH oxidase. EF-hand calcium binding domain 5 | 3.57 | 7.54 | |
| SOD1 | Superoxide dismutase 1. soluble | −28.68 | −9.67 | |
| SOD2 | Superoxide dismutase 2. mitochondrial | 3.96 | 4.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuomo, P.; Papaianni, M.; Sansone, C.; Iannelli, A.; Iannelli, D.; Medaglia, C.; Paris, D.; Motta, A.; Capparelli, R. An In Vitro Model to Investigate the Role of Helicobacter pylori in Type 2 Diabetes, Obesity, Alzheimer’s Disease and Cardiometabolic Disease. Int. J. Mol. Sci. 2020, 21, 8369. https://doi.org/10.3390/ijms21218369
Cuomo P, Papaianni M, Sansone C, Iannelli A, Iannelli D, Medaglia C, Paris D, Motta A, Capparelli R. An In Vitro Model to Investigate the Role of Helicobacter pylori in Type 2 Diabetes, Obesity, Alzheimer’s Disease and Cardiometabolic Disease. International Journal of Molecular Sciences. 2020; 21(21):8369. https://doi.org/10.3390/ijms21218369
Chicago/Turabian StyleCuomo, Paola, Marina Papaianni, Clementina Sansone, Antonio Iannelli, Domenico Iannelli, Chiara Medaglia, Debora Paris, Andrea Motta, and Rosanna Capparelli. 2020. "An In Vitro Model to Investigate the Role of Helicobacter pylori in Type 2 Diabetes, Obesity, Alzheimer’s Disease and Cardiometabolic Disease" International Journal of Molecular Sciences 21, no. 21: 8369. https://doi.org/10.3390/ijms21218369