The Cytogenomic “Theory of Everything”: Chromohelkosis May Underlie Chromosomal Instability and Mosaicism in Disease and Aging

 ,
,
Abstract
:1. Introduction
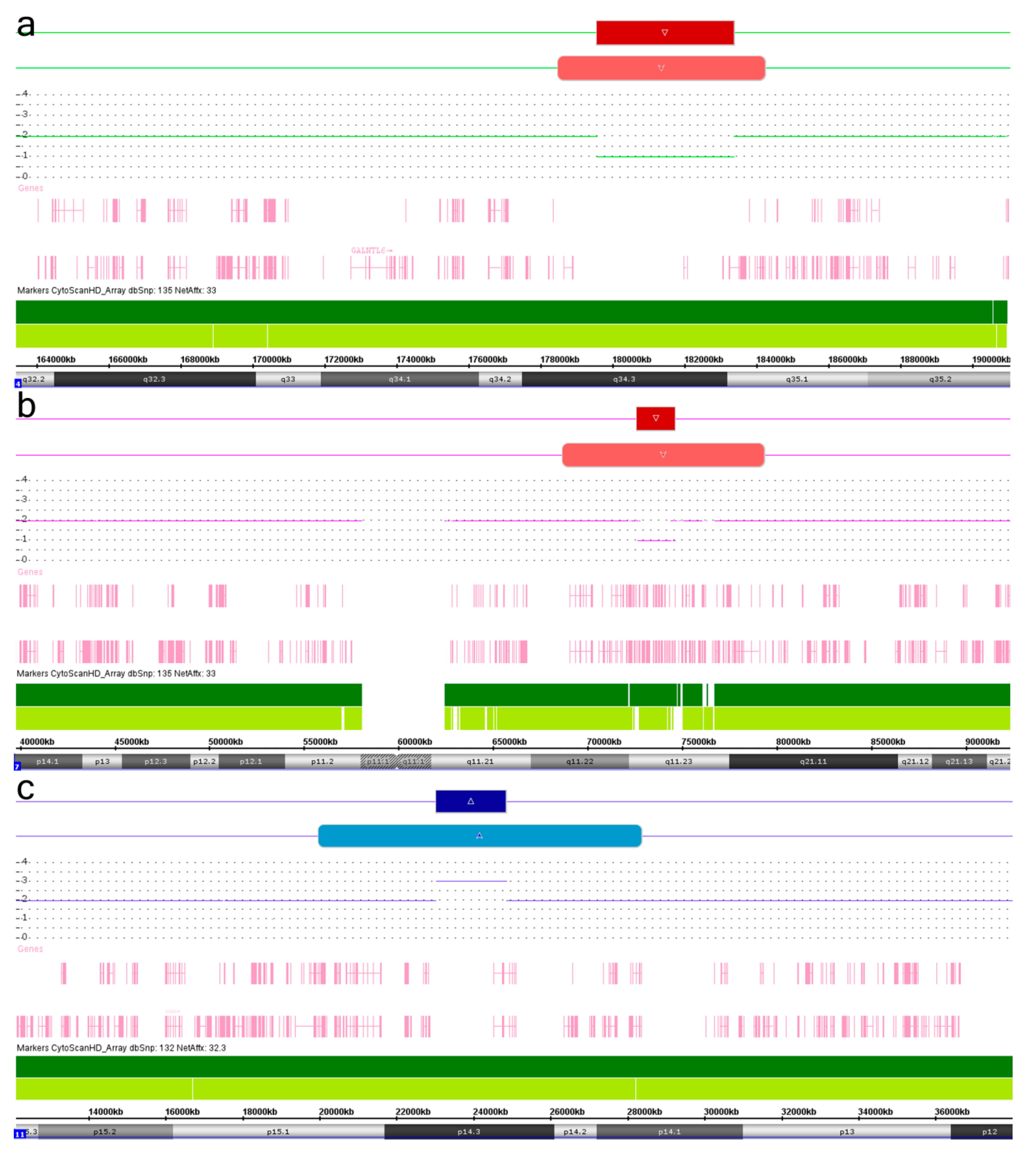
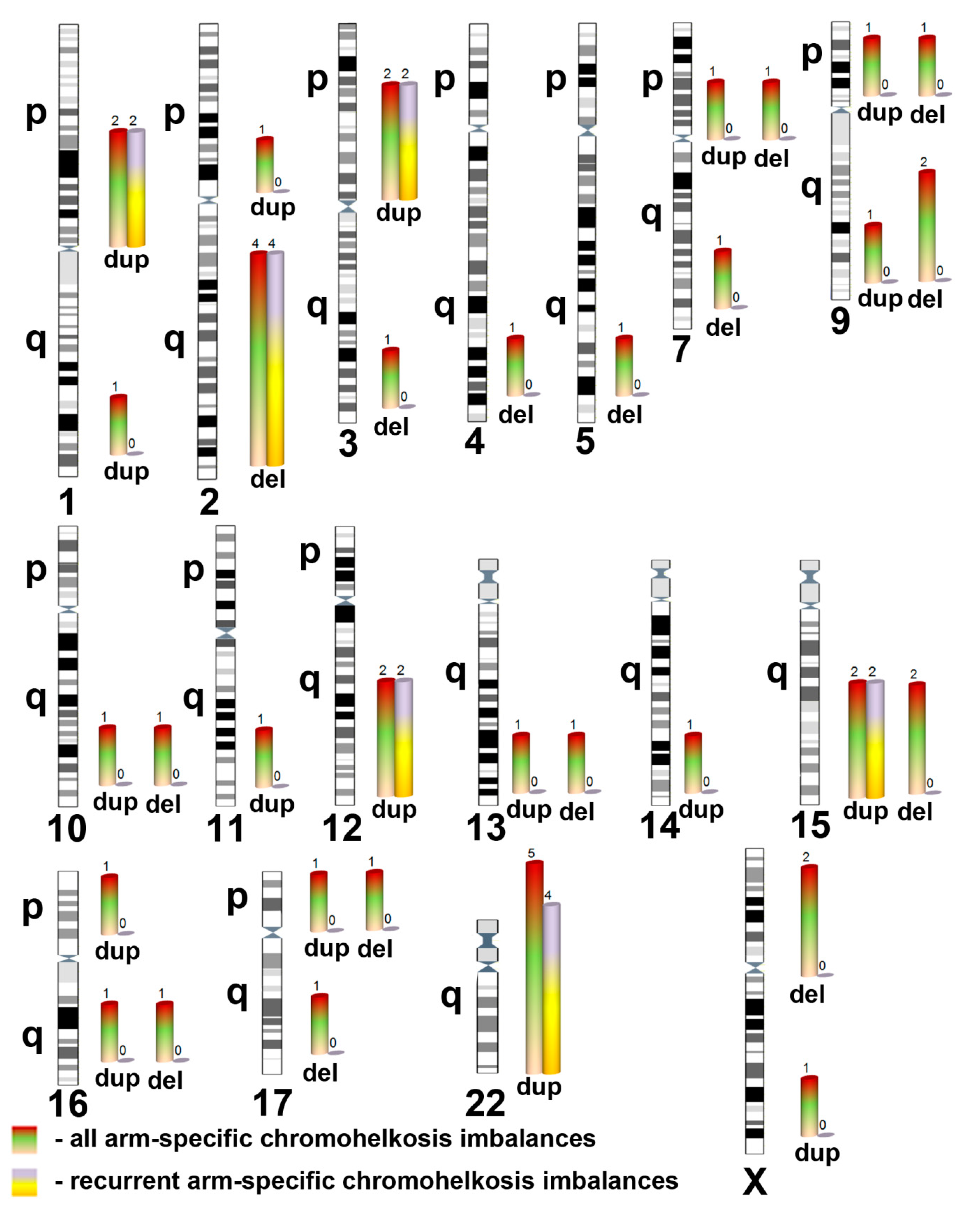
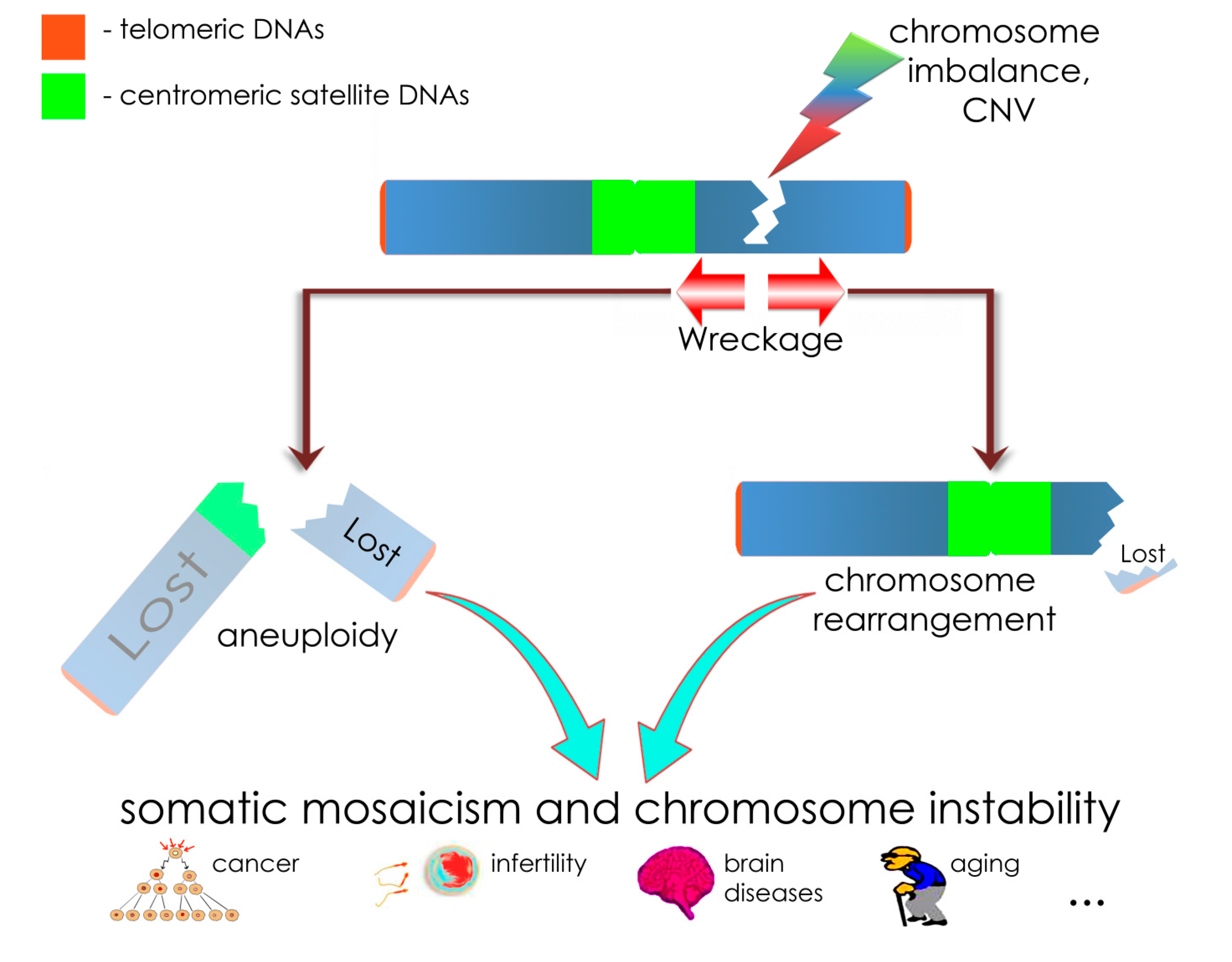
2. Results and Discussion
3. Materials and Methods
3.1. Patients and Samples
3.2. SNP-Array
3.3. Bioinformatic Analysis
Pathway Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ChAS | Chromosome Analysis Suite |
| CIN | Chromosomal instability |
| CNV | Copy number variations |
| Del | deletion (tables & figures) |
| dup | duplication (tables & figures) |
| GIN | Genome instability |
| IPP | Index of pathway prioritization |
| SCM | Somatic chromosomal mosaicism |
| SNP | Single nucleotide polymorphism |
References
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, H.H.Q.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Kutsev, S.I. Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism. Genes 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, M.L.; Zhang, C.-Z.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet. 2015, 49, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Pellestor, F. Chromoanagenesis: Cataclysms behind complex chromosomal rearrangements. Mol. Cytogenet. 2019, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iourov, I.; Vorsanova, S.G.; Yurov, Y.B. Somatic Genome Variations in Health and Disease. Curr. Genom. 2010, 11, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Somatic Cell Genomics of Brain Disorders: A New Opportunity to Clarify Genetic-Environmental Interactions. Cytogenet. Genome Res. 2013, 139, 181–188. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural variant calling: The long and the short of it. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Fukami, M.; Shima, H.; Suzuki, E.; Ogata, T.; Matsubara, K.; Kamimaki, T. Catastrophic cellular events leading to complex chromosomal rearrangements in the germline. Clin. Genet. 2017, 91, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Zhang, C.; Li, D.; Carvalho, C.M.; Ji, H.; Xiao, J.; Wu, Y.; Zhou, W.; Wang, H.; et al. Efficient CNV breakpoint analysis reveals unexpected structural complexity and correlation of dosage-sensitive genes with clinical severity in genomic disorders. Hum. Mol. Genet. 2017, 26, 1927–1941. [Google Scholar] [CrossRef]
- Balachandran, P.; Beck, C.R. Structural variant identification and characterization. Chromosom. Res. 2020, 28, 31–47. [Google Scholar] [CrossRef]
- Hu, Q.; Maurais, E.G.; Ly, P. Cellular and genomic approaches for exploring structural chromosomal rearrangements. Chromosom. Res. 2020, 28, 19–30. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, 0712. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Cleveland, D.W.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Korostelev, S.A.; Zelenova, M.A.; Yurov, Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol. Cytogenet. 2015, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Zelenova, M.A.; Vasin, K.S.; Kurinnaia, O.S.; Korostelev, S.A.; Yurov, Y.B. Epigenomic variations manifesting as a loss of heterozygosity affecting imprinted genes represent a molecular mechanism of autism spectrum disorders and intellectual disability in children. Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova 2019, 119, 91–97. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Henry, H.H. New data collection priority: Focusing on genome-based bioinformation. Res. Result. Biomed. 2020, 6, 5–8. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. The variome concept: Focus on CNVariome. Mol. Cytogenet. 2019, 12, 52–56. [Google Scholar] [CrossRef]
- Zelenova, M.A.; Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Laundering CNV data for candidate process prioritization in brain disorders. Mol. Cytogenet. 2019, 12, 54–56. [Google Scholar] [CrossRef] [Green Version]
- Padeken, J.; Zeller, P.; Gasser, S.M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015, 31, 12–19. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef] [Green Version]
- Vorsanova, S.G.; Yurov, Y.B.; Soloviev, I.; Iourov, I. Molecular Cytogenetic Diagnosis and Somatic Genome Variations. Curr. Genom. 2010, 11, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Potter, H.P. Beyond Trisomy 21: Phenotypic Variability in People with Down Syndrome Explained by Further Chromosome Mis-segregation and Mosaic Aneuploidy. J. Down Syndr. Chromosom. Abnorm. 2016, 2, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Qiu, L.; Mrasek, K.; Zhang, J.; Liehr, T.; Quintana, L.G.; Li, Z. Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11974–11999. [Google Scholar] [CrossRef] [Green Version]
- Puliti, A.; Rizzato, C.; Conti, V.; Bedini, A.; Gimelli, G.; Barale, R.; Sbrana, I. Low-copy repeats on chromosome 22q11.2 show replication timing switches, DNA flexibility peaks and stress inducible asynchrony, sharing instability features with fragile sites. Mutat. Res. Mol. Mech. Mutagen. 2010, 686, 74–83. [Google Scholar] [CrossRef]
- Yadav, V.; Sun, S.; Coelho, M.A.; Heitman, J. Centromere scission drives chromosome shuffling and reproductive isolation. Proc. Natl. Acad. Sci. USA 2020, 117, 7917–7928. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Zelenova, M.A.; Korostelev, S.A.; Yurov, Y.B. Genomic Copy Number Variation Affecting Genes Involved in the Cell Cycle Pathway: Implications for Somatic Mosaicism. Int. J. Genom. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Wevrick, R.; Fisher, R.B.; Ferguson-Smith, M.A.; Lin, C.C. Human centromeric DNAs. Hum. Genet. 1997, 100, 291–304. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.M.; Sullivan, B.A. Alpha satellite DNA biology: Finding function in the recesses of the genome. Chromosom. Res. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Bolzán, A.D. Using telomeric chromosomal aberrations to evaluate clastogen-induced genomic instability in mammalian cells. Chromosom. Res. 2020, 1–18. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Chromosome Instability in the Neurodegenerating Brain. Front. Genet. 2019, 10, 892. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and Confined Chromosomal Mosaicism in the Developing Human Brain. PLoS ONE 2007, 2, 558. [Google Scholar] [CrossRef]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef]
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I. Ontogenetic Variation of the Human Genome. Curr. Genom. 2010, 11, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiela, M.J. Mosaicism, aging and cancer. Curr. Opin. Oncol. 2019, 31, 108–113. [Google Scholar] [CrossRef]
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2018, 144, 8–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal Variation in Mammalian Neuronal Cells: Known Facts and Attractive Hypotheses. Int. Rev. Cytol. 2006, 249, 143–191. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Single Cell Genomics of the Brain: Focus on Neuronal Diversity and Neuropsychiatric Diseases. Curr. Genom. 2012, 13, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Liehr, T.; Al-Rikabi, A. Mosaicism: Reason for Normal Phenotypes in Carriers of Small Supernumerary Marker Chromosomes with Known Adverse Outcome. A Systematic Review. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef]
- Potter, H.; Chial, H.J.; Caneus, J.; Elos, M.; Elder, N.; Borysov, S.; Granic, A. Chromosome Instability and Mosaic Aneuploidy in Neurodegenerative and Neurodevelopmental Disorders. Front. Genet. 2019, 10, 1092. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, J.; Osei-Owusu, I.A.; Avigdor, B.E.; Tupler, R.; Pevsner, J. Mosaicism in Human Health and Disease. Annu. Rev. Genet. 2020, 54, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol. Cytogenet. 2020, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hochstenbach, R.; Buizer-Voskamp, J.; Vorstman, J.; Ophoff, R. Genome Arrays for the Detection of Copy Number Variations in Idiopathic Mental Retardation, Idiopathic Generalized Epilepsy and Neuropsychiatric Disorders: Lessons for Diagnostic Workflow and Research. Cytogenet. Genome Res. 2011, 135, 174–202. [Google Scholar] [CrossRef]
- Schang, A.-L.; Saberan-Djoneidi, D.; Mezger, V. The impact of epigenomic next-generation sequencing approaches on our understanding of neuropsychiatric disorders. Clin. Genet. 2017, 93, 467–480. [Google Scholar] [CrossRef]
- Hawking, S.; Mlodinow, L. The (elusive) theory of everything. Sci. Am. 2010, 303, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Wang, H.; Li, H.; Song, X.; Wang, T.; Xiang, J.; Li, H. Genetic analysis of products of conception using a HLPA/SNP-array strategy. Mol. Cytogenet. 2019, 12, 1–7. [Google Scholar] [CrossRef]
- Xue, H.; Huang, H.L.; Wang, Y.; An, G.; Zhang, M.; Xu, L.; Lin, Y. Molecular cytogenetic identification of small supernumerary marker chromosomes using chromosome microarray analysis. Mol. Cytogenet. 2019, 12, 13. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. In silico molecular cytogenetics: A bioinformatic approach to prioritization of candidate genes and copy number variations for basic and clinical genome research. Mol. Cytogenet. 2014, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Chromosomal Localization | Deletion (del)/Duplication (dup) | Genomic Localization 1 | Size (Mb), Regular | Size (Mb), Mosaic | Mosaicism Rate (%) 2 | ||
|---|---|---|---|---|---|---|---|
| Regular | Mosaic | Regular | Mosaic | ||||
| 1p36.32 | 1p36.33p36.32 | Dup | 2,793,846–3,123,524 | 849,466–3,586,707 | 0.329 | 2.737 | 20 |
| 1p36.33 | 1p36.33p36.32 | Dup | 1,134,091–1,207,463 | 849,466–5,278,786 | 0.073 | 4.429 | 25 |
| 1q21.1q21.2 | 1p12q21.2 | Dup | 146,003,044–147,398,560 | 118,506,747–149,965,913 | 1.395 | 31.459 | 20 |
| 2p25.3 | 2p25.3 | Dup | 1,611,691–1,861,548 | 12,770–3,007,511 | 0.249 | 2.994 | 25 |
| 2q22.2 q22.3 | 2q22.1q23.3 | Del | 143,410,303–145,299,945 | 140,410,739–150,635,360 | 1.889 | 10.224 | 50 |
| 2q23.1q23.3 | 2q22.2q24.1 | Del | 148,851,963–151,316,465 | 143,753,727–155,408,790 | 2.464 | 11.655 | 40 |
| 2q23.1q23.3 | 2q22.2q24.1 | Del | 149,073,384–151,886,100 | 144,007,224–156,393,001 | 2.812 | 12.385 | 45 |
| 2q24.1 | 2q23.3q24.2 | Del | 155,684,576–157,919,431 | 151,497,654–162,200,234 | 2.234 | 10.702 | 35 |
| 3p26.1 | 3p26.3p26.1 | Dup | 4,311,166–7,256,278 | 2,788,170–8,587,443 | 2.945 | 5.799 | 40 |
| 3p26.3p26.2 | 3p26.3p26.1 | Dup | 1,839,722–3,372,758 | 61,891–4,693,249 | 1.533 | 4.631 | 60 |
| 3q26.2q26.31 | 3q26.1q26.31 | Del | 170,316,791–171,650,195 | 165,957,466–175,300,706 | 1.333 | 9.343 | 30 |
| 4q34.3q35.1 | 4q34.3q35.1 | Del | 179,568,373–183,377,810 | 178,503,425–184,251,370 | 3.809 | 5.747 | 55 |
| 5q35.2q35.3 | 5q35.1q35.3 | Del | 175,029,372–177,324,736 | 171,538,904–180,719,789 | 3.395 | 9.180 | 30 |
| 7p22.1p15.2 | 7p22.1p15.2 | Dup | 4,783,314–26,275,210 | 4,790,968–26,522,153 | 21.491 | 21.731 | 80 |
| 7p22.2p21.3 | 7p22.3p21.3 | Del | 3,235,409–7,970,015 | 43,360–8,320,635 | 4.734 | 8.277 | 20 |
| 7q11.23 | 7q11.22q21.11 | Del | 72,612,042–74,610,673 | 68,665,592–79,305,748 | 1.998 | 10.640 | 40 |
| 9p24.3 | 9p24.3p24.2 | Dup | 203,861–823,845 | 203,861–2,593,900 | 0.619 | 2.390 | 30 |
| 9p24.3 | 9p24.3 | Del | 203,861–410,357 | 203,861–1,074,830 | 0.206 | 0.870 | 25 |
| 9q22.31q22.33 | 9q22.31q22.33 | Del | 96,109,697–99,973,789 | 95,891,880–100,145,863 | 3.864 | 4.253 | 70 |
| 9q34.3 | 9q34.13q34.3 | Dup | 139,053,501–139,435,356 | 134,317,328–141,020,389 | 0.381 | 6.703 | 30 |
| 9q34.3 | 9q34.13q34.3 | Del | 139,784,913–141,020,389 | 135,282,452–141,020,389 | 1.235 | 5.737 | 40 |
| 10q21.1 | 10q11.23q21.1 | Dup | 53,156,807–57,931,080 | 52,693,425–58,936,553 | 4.774 | 6.243 | 75 |
| 10q23.1q23.2 | 10q23.1q23.2 | Del | 86,412,180–88,502,670 | 85,638,142–89,465,109 | 2.090 | 3.826 | 50 |
| 11p14.3 | 11p14.3 | Dup | 23,032,300–24,850,872 | 19,983,179–28,380,051 | 1.819 | 8.396 | 25 |
| 12q24.33 | 12q24.33 | Dup | 129,804,153–130,492,863 | 129,577,575–133,777,902 | 0.688 | 4.200 | 20 |
| 12q24.33 | 12q24.33 | Dup | 129,803,493–130,485,474 | 130,035,491–133,777,902 | 0.681 | 3.742 | 20 |
| 13q12.11 | 13q11q12.11 | Dup | 21,683,950–22,155,929 | 19,436,287–22,422,460 | 0.471 | 2.986 | 20 |
| 13q34 | 13q34 | Del | 114,085,478–115,107,733 | 110,963,086–115,107,733 | 1.022 | 4.144 | 40 |
| 14q32.2 | 14q32.13q32.2 | Dup | 99,153,952–101,024,454 | 95,563,168–100,095,249 | 1.870 | 4.532 | 20 |
| 15q11.2 | 15q11.2 | Dup | 22,770,421–23,082,328 | 22,770,421–25,083,880 | 0.311 | 2.313 | 20 |
| 15q11.2 | 15q11.2 | Dup | 22,770,421–23,288,350 | 22,770,421–25,318,376 | 0.517 | 2.547 | 25 |
| 15q11.2q13.1 | 15q11.2q13.1 | Del | 22,770,421–29,021,034 | 22,770,421–28,373,187 | 5.732 | 5.602 | 85 |
| 15q13.2q13.3 | 15q13.1q14 | Del | 30,913,573–32,914,239 | 28,394,840–36,544,674 | 2.518 | 8.149 | 25 |
| 16p11.2q11.2 | 16p11.2q12.1 | Dup | 32,038,693–46,463,769 | 34,448,198–51,124,520 | 14.425 | 16.676 | 20 |
| 16q23.1 | 16q22.3q23.3 | Dup | 77,496,014–78,916,839 | 73,357,720–82,335,001 | 1.420 | 8.977 | 30 |
| 16q24.3 | 16q24.2q24.3 | Del | 89,683,742–90,155,062 | 87,157,300–90,155,062 | 0.471 | 2.997 | 25 |
| 17p12 | 17p13.1p11.2 | Dup | 14,082,944–15,479,940 | 10,219,298–17,108,606 | 1.396 | 6.889 | 20 |
| 17p13.3 | 17p13.3p13.2 | Del | 525–1,323,904 | 525–4,375,742 | 1.323 | 4.375 | 40 |
| 17q25.3 | 17q25.3 | Del | 80,396,463–81,041,938 | 77,947,778–81,041,938 | 0.645 | 3.094 | 30 |
| 22q11.21 | 22q11.1q11.22 | Dup | 18,974,541–21,800,797 | 17,398,811–23,374,206 | 2.826 | 5.975 | 45 |
| 22q11.21 | 22q11.1q11.21 | Dup | 18,649,189–20,311,810 | 16,888,899–22,034,665 | 1.662 | 5.145 | 40 |
| 22q11.21 | 22q11.1q11.22 | Dup | 18,979,345–21,465,659 | 16,888,899–23,410,418 | 2.486 | 6.521 | 50 |
| 22q11.21 | 22q11.1q11.23 | Dup | 18,916,842–21,465,659 | 16,888,899–23,535,339 | 2.548 | 6.646 | 50 |
| 22q13.2q13.31 | 22q13.2q13.31 | Dup | 43,337,317–46,575,998 | 42,018,242–44,860,024 | 3.238 | 2.841 | 50 |
| Xp22.31 | Xp22.32p22.2 | Del | 6,784,550–7,495,395 | 4,931,788–9,634,138 | 0.710 | 4.702 | 25 |
| Xp21.1 | Xp21.1p11.4 | Del | 32,881,263–35,187,430 | 31,875,672–38,716,579 | 2.306 | 6.840 | 50 |
| Xq28 | Xq28 | Dup | 153,747,685–153,761,134 | 147,843,549–152,036,631 | 0.013 | 4.193 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Zelenova, M.A.; Kurinnaia, O.S.; Vasin, K.S.; Kutsev, S.I. The Cytogenomic “Theory of Everything”: Chromohelkosis May Underlie Chromosomal Instability and Mosaicism in Disease and Aging. Int. J. Mol. Sci. 2020, 21, 8328. https://doi.org/10.3390/ijms21218328
Iourov IY, Vorsanova SG, Yurov YB, Zelenova MA, Kurinnaia OS, Vasin KS, Kutsev SI. The Cytogenomic “Theory of Everything”: Chromohelkosis May Underlie Chromosomal Instability and Mosaicism in Disease and Aging. International Journal of Molecular Sciences. 2020; 21(21):8328. https://doi.org/10.3390/ijms21218328
Chicago/Turabian StyleIourov, Ivan Y., Svetlana G. Vorsanova, Yuri B. Yurov, Maria A. Zelenova, Oxana S. Kurinnaia, Kirill S. Vasin, and Sergei I. Kutsev. 2020. "The Cytogenomic “Theory of Everything”: Chromohelkosis May Underlie Chromosomal Instability and Mosaicism in Disease and Aging" International Journal of Molecular Sciences 21, no. 21: 8328. https://doi.org/10.3390/ijms21218328