Mitochondrial Function and Protein Turnover in the Diaphragm are Altered in LLC Tumor Model of Cancer Cachexia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
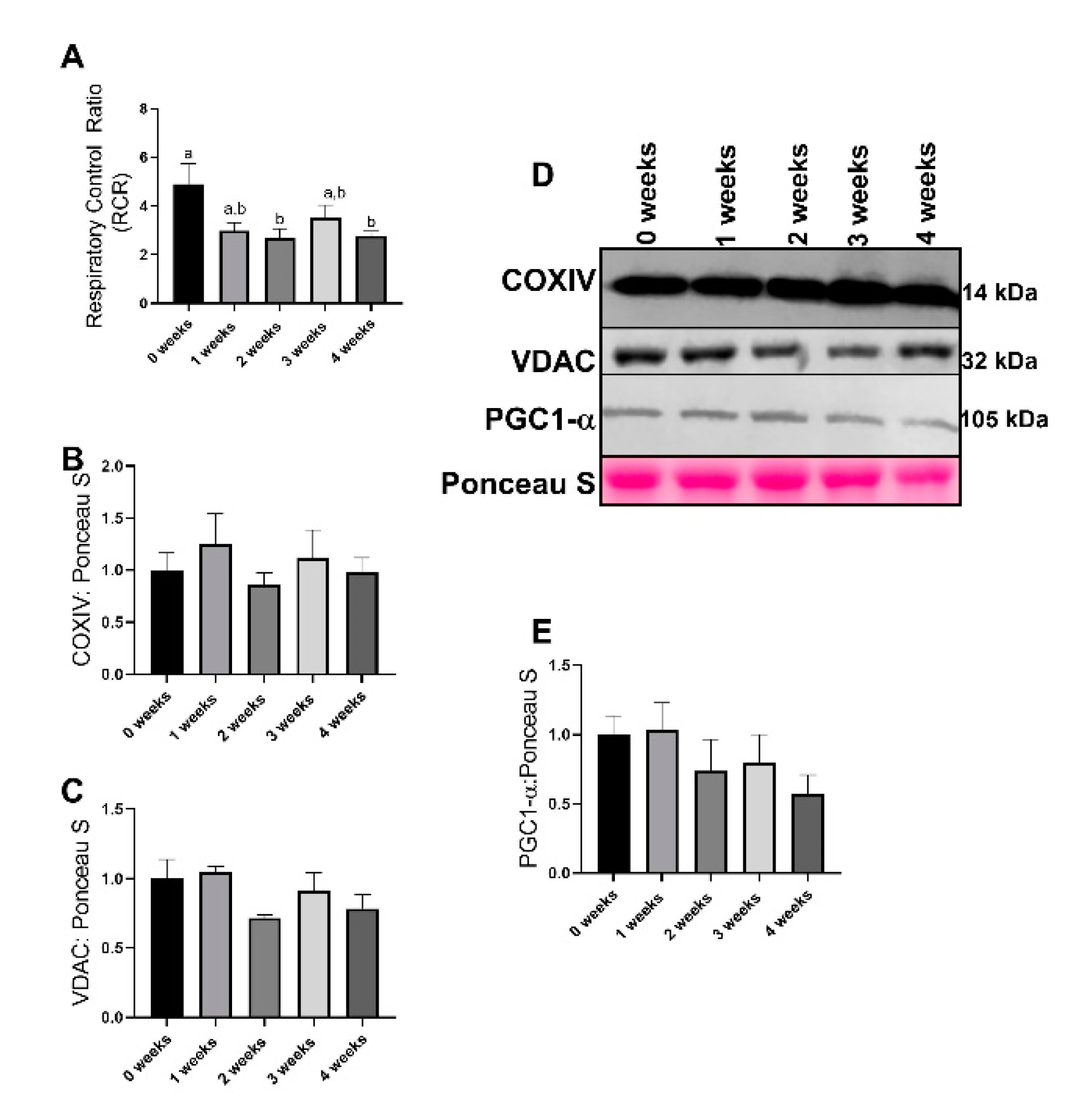
2.1. Lewis Lung Carcinoma (LLC) Resulted in Alterations to Mitochondrial Function without Changes to Mitochondrial Content
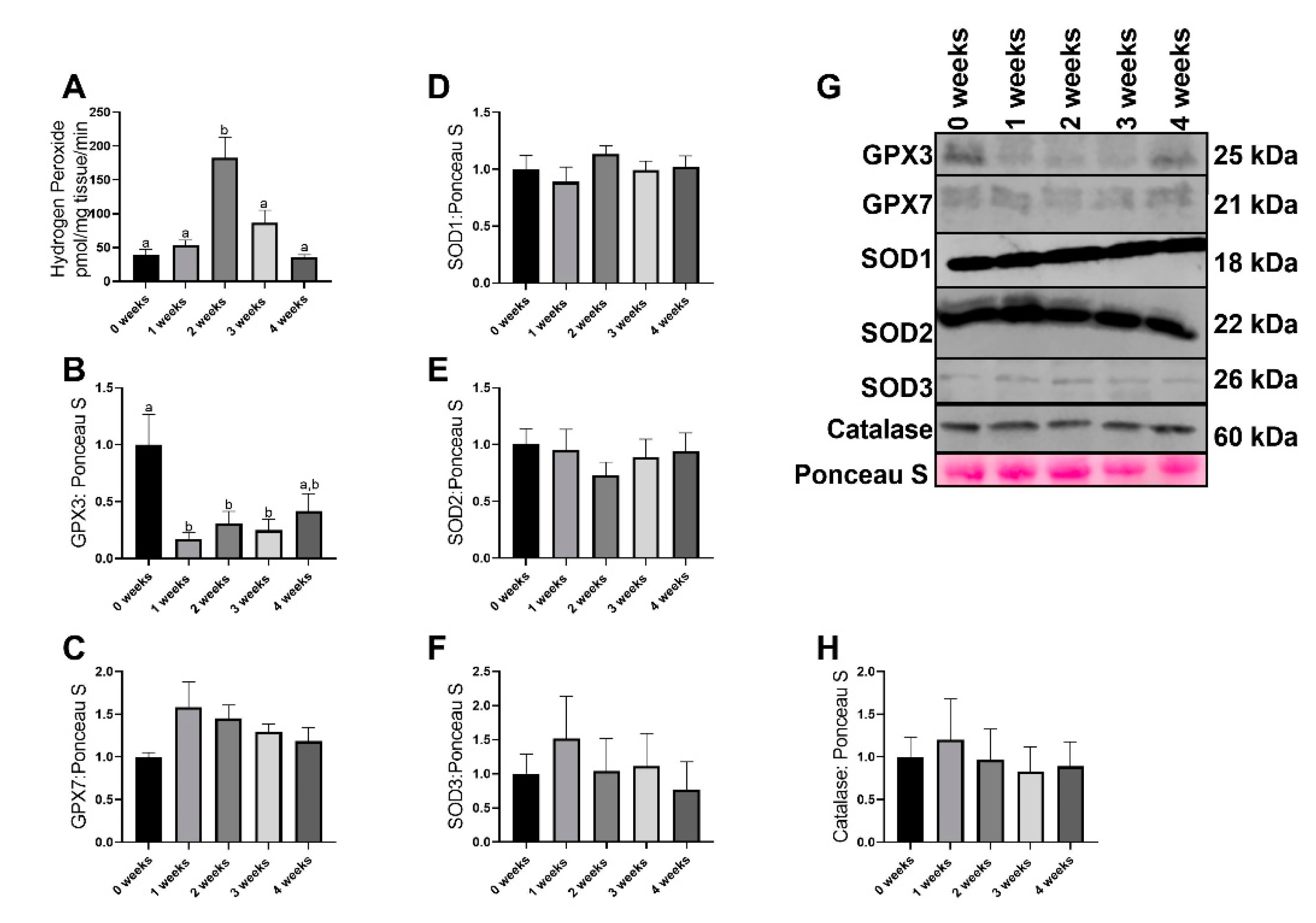
2.2. LLC Resulted in Greater Reactive Oxgyen Species Generation without any Changes in most Antioxidant Related Proteins
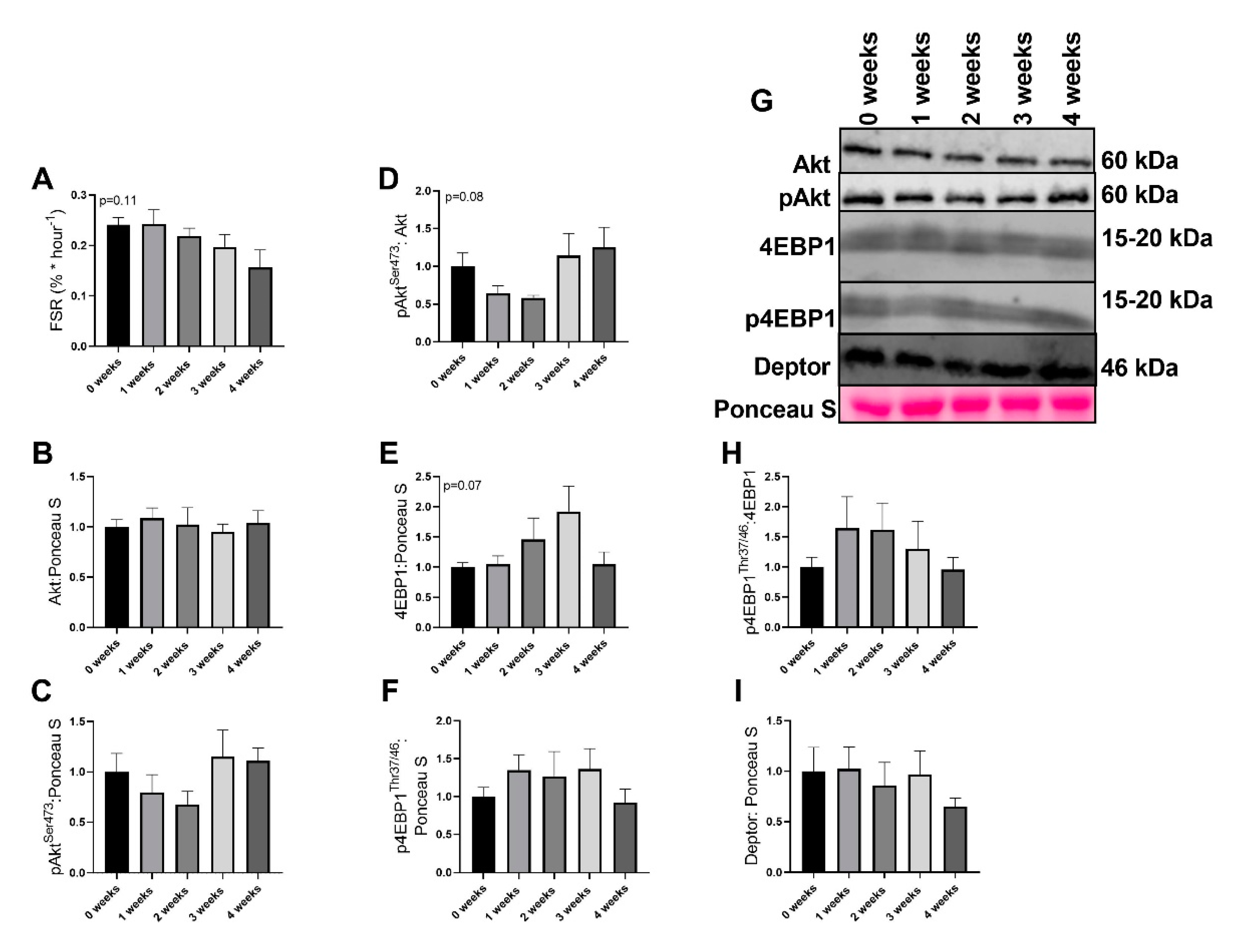
2.3. LLC Did not Have Significant Effects on Protein Synthesis Signaling
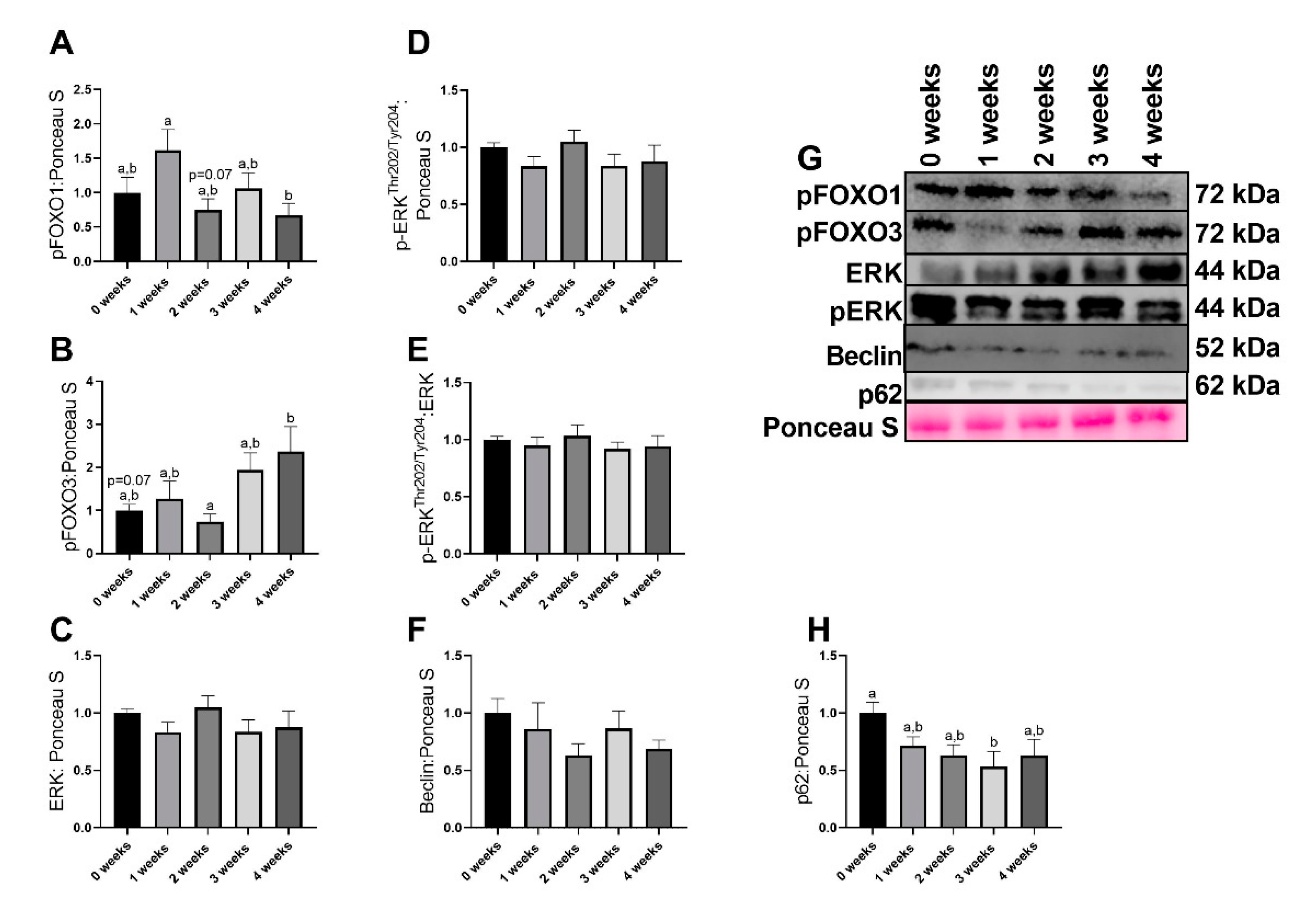
2.4. LLC Appeared to Increase FOXO3-Dependent Signaling
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Study Design
4.3. Determination of Muscle Protein Synthesis
4.4. Mitochondrial Function
4.5. Reactive Oxygen Species (ROS) Emission
4.6. Western Blotting
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mokdad, A.H.; Dwyer-Lindgren, L.; Fitzmaurice, C.; Stubbs, R.W.; Bertozzi-Villa, A.; Morozoff, C.; Charara, R.; Allen, C.; Naghavi, M.; Murray, C.J. Trends and Patterns of Disparities in Cancer Mortality Among US Counties, 1980–2014. JAMA 2017, 317, 388–406. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Colorectal Cancer Mortality Rates in Adults Aged 20 to 54 Years in the United States, 1970–2014. JAMA 2017, 318, 572–574. [Google Scholar] [CrossRef]
- Evans, W.J. Skeletal muscle loss: Cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 2010, 91, 1123s–1127s. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Vanhoutte, G.; van de Wiel, M.; Wouters, K.; Sels, M.; Bartolomeeussen, L.; De Keersmaecker, S.; Verschueren, C.; De Vroey, V.; De Wilde, A.; Smits, E.; et al. Cachexia in cancer: What is in the definition? BMJ Open Gastroenterol. 2016, 3, e000097. [Google Scholar] [CrossRef] [PubMed]
- Leitner, L.M.; Wilson, R.J.; Yan, Z.; Godecke, A. Reactive Oxygen Species/Nitric Oxide Mediated Inter-Organ Communication in Skeletal Muscle Wasting Diseases. Antioxid. Redox Signal 2017, 26, 700–717. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Quan, X.Q.; Yu, S. An Epidemiological Survey of Cachexia in Advanced Cancer Patients and Analysis on Its Diagnostic and Treatment Status. Nutr. Cancer 2015, 67, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Senesse, P.; Gioulbasanis, I.; Antoun, S.; Bozzetti, F.; Deans, C.; Strasser, F.; Thoresen, L.; Jagoe, R.T.; Chasen, M.; et al. Diagnostic criteria for the classification of cancer-associated weight loss. J. Clin. Oncol. 2015, 33, 90–99. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Rosa-Caldwell, M.E.; Fix, D.K.; Washington, T.A.; Greene, N.P. Muscle Alterations in the Development and Progression of Cancer-Induced Muscle Atrophy: A Review. J. Appl. Physiol. 2020, 128, 25–41. [Google Scholar] [CrossRef]
- Hermans, G.; Van Mechelen, H.; Clerckx, B.; Vanhullebusch, T.; Mesotten, D.; Wilmer, A.; Casaer, M.P.; Meersseman, P.; Debaveye, Y.; Van Cromphaut, S.; et al. Acute outcomes and 1-year mortality of intensive care unit-acquired weakness. A cohort study and propensity-matched analysis. Am. J. Respir. Crit. Care Med. 2014, 190, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Li, L.F. Ventilator-induced Diaphragm Dysfunction in Critical Illness. Exp. Biol. Med. 2018, 243, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Goligher, E.C.; Dres, M.; Eddy Fan, E.; Gordon, D.; Rubenfeld, G.D.; Scales, D.C.; Herridge, M.S.; Vorona, S.; Sklar, M.C.; Rittayamai, N.; et al. Mechanical Ventilation-induced Diaphragm Atrophy Strongly Impacts Clinical Outcomes. Am. J. Respir. Crit. Care Med. 2018, 197, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Degracia, A.; Busquets, S.; Argilés, J.M.; López-Soriano, F.J.; Esther Barreiro, E. Formoterol Attenuates Increased Oxidative Stress and Myosin Protein Loss in Respiratory and Limb Muscles of Cancer Cachectic Rats. PeerJ 2017, 5, e4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacon-Cabrera, A.; Mateu-Jimenez, M.; Langohr, K.; Fermoselle, C.; García-Arumí, E.; Andreu, A.L.; Yelamos, J.; Barreiro, E. Role of PARP Activity in Lung Cancer-Induced Cachexia: Effects on Muscle Oxidative Stress, Proteolysis, Anabolic Markers, and Phenotype. J. Cell. Physiol. 2017, 232, 3744–3761. [Google Scholar] [CrossRef]
- Judge, A.R.; Wu, C.-L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-wide Identification of FoxO-dependent Gene Networks in Skeletal Muscle During C26 Cancer Cachexia. BMC Cancer 2014, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Fermoselle, C.; García-Arumí, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; Joffé, E.D.B.D.K.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial Dysfunction and Therapeutic Approaches in Respiratory and Limb Muscles of Cancer Cachectic Mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and Ventilatory Dysfunction During Cancer Cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Degracia, A.; Blanco, D.; Vilà-Ubach, M.; De Biurrun, G.; De Solórzano, C.O.; Montuenga, L.M.; Barreiro, E. Phenotypic and Metabolic Features of Mouse Diaphragm and Gastrocnemius Muscles in Chronic Lung Carcinogenesis: Influence of Underlying Emphysema. J. Transl. Med. 2016, 14, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.T.; Chee, A.; Trieu, J.; Naim, T.; Lynch, G.S. Importance of Functional and Metabolic Impairments in the Characterization of the C-26 Murine Model of Cancer Cachexia. Dis. Model. Mech. 2012, 5, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig-Vilanova, E.; Rodriguez, D.A.; Lloreta, J.; Ausin, P.; Pascual-Guardia, S.; Broquetas, J.; Roca, J.; Gea, J.; Barreiro, E. Oxidative Stress, Redox Signaling Pathways, and Autophagy in Cachectic Muscles of Male Patients With Advanced COPD and Lung Cancer. Free. Radic. Biol. Med. 2015, 79, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T.; Chee, A.; Gleeson, B.G.; Naim, T.; Swiderski, K.; Koopman, R.; Lynch, G.S. Antibody-directed Myostatin Inhibition Enhances Muscle Mass and Function in Tumor-Bearing Mice. Am. J. Physiol. Integr. Comp. Physiol. 2011, 301, R716–R726. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, L.; Costelli, P.; Bonetti, G.; Baccino, F.M. Cancer cachexia, malnutrition, and tissue protein turnover in experimental animals. Arch. Biochem. Biophys. 1993, 306, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.A.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Ryan, Z.C.; Craig, T.A.; Wang, X.; Delmotte, P.; Salisbury, J.L.; Lanza, I.R.; Sieck, G.C.; Kumar, R. 1alpha,25-dihydroxyvitamin D3 mitigates cancer cell mediated mitochondrial dysfunction in human skeletal muscle cells. Biochem. Biophys. Res. Commun. 2018, 496, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Barret, M.; Antoun, S.; Dalban, C.; Malka, D.; Mansourbakht, T.; Zaanan, A.; Latko, E.; Taieb, J. Sarcopenia is linked to treatment toxicity in patients with metastatic colorectal cancer. Nutr. Cancer 2014, 66, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Huillard, O.; Mir, O.; Peyromaure, M.; Tlemsani, C.; Giroux, J.; Boudou-Rouquette, P.; Ropert, S.; Delongchamps, N.B.; Zerbib, M.; Goldwasser, F. Sarcopenia and body mass index predict sunitinib-induced early dose-limiting toxicities in renal cancer patients. Br. J. Cancer 2013, 108, 1034–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.L.; Lee, D.E.; Rosa-Caldwell, M.E.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Huseman, K.; Sataranatarajan, K.; Van Remmen, H.; Washington, T.A.; et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Nicholls, D. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Hooijman, P.E.; Beishuizen, A.; Witt, C.C.; de Waard, M.C.; Girbes, A.R.J.; Spoelstra-de Man, A.M.E.; Niessen, H.W.M.; Manders, E.; van Hees, H.W.H.; van den Brom, C.E.; et al. Diaphragm Muscle Fiber Weakness and Ubiquitin–Proteasome Activation in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2015, 191, 1126–1138. [Google Scholar] [CrossRef]
- Halle, J.L.; Pena, G.S.; Paez, H.G.; Castro, A.J.; Rossiter, H.B.; Visavadiya, N.P.; Whitehurst, M.A.; Khamoui, A.V. Tissue-specific dysregulation of mitochondrial respiratory capacity and coupling control in colon-26 tumor-induced cachexia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R68–R82. [Google Scholar] [CrossRef] [PubMed]
- Nosacka, R.L.; Delitto, A.E.; Delitto, D.; Patel, R.; Judge, A.R.; Trevino, J.G.; Judge, A.R. Distinct cachexia profiles in response to human pancreatic tumours in mouse limb and respiratory muscle. J. Cachexia Sarcopenia Muscle 2020, 11, 820–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Lu, L.; Wu, S.; Zuo, L. Effects of Ionizing Irradiation on Mouse Diaphragmatic Skeletal Muscle. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig-Vilanova, E.; Aguilo, R.; Rodríguez-Fuster, A.; Martínez-Llorens, J.; Gea, J.; Barreiro, E. Epigenetic mechanisms in respiratory muscle dysfunction of patients with chronic obstructive pulmonary disease. PLoS ONE 2014, 9, e111514. [Google Scholar] [CrossRef]
- Kritsiligkou, P.; Chatzi, A.; Charalampous, G.; Mironov, A.; Grant, C.M.; Tokatlidis, K. Unconventional Targeting of a Thiol Peroxidase to the Mitochondrial Intermembrane Space Facilitates Oxidative Protein Folding. Cell Rep. 2017, 18, 2729–2741. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Chacon-Cabrera, A.; Fermoselle, C.; Urtreger, A.J.; Mateu-Jimenez, M.; Diament, M.J.; de Kier Joffe, E.D.; Sandri, M.; Barreiro, E. Pharmacological strategies in lung cancer-induced cachexia: Effects on muscle proteolysis, autophagy, structure, and weakness. J. Cell Physiol. 2014, 229, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Kwon, I.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y.; Yeom, D.C. Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis. J. Exerc. Nutr. Biochem. 2015, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Fields, D.P.; Roberts, B.M.; Simon, A.K.; Judge, A.R.; Fuller, D.D.; Mitchell, G.S. Cancer cachexia impairs neural respiratory drive in hypoxia but not hypercapnia. J. Cachexia Sarcopenia Muscle 2018, 10, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.E.; Brown, J.L.; Rosa-Caldwell, M.E.; Perry, R.A.; Brown, L.A.; Haynie, W.S.; Washington, T.A.; Wiggs, M.P.; Rajaram, N.; Greene, N.P. Cancer-induced cardiac atrophy adversely affects myocardial redox state and mitochondrial oxidative characteristics. JCSM Rapid Commun. 2020. [Google Scholar] [CrossRef]
- Rosa-Caldwell, M.E.; Brown, J.L.; Lee, D.E.; Wiggs, M.P.; Perry, R.A., Jr.; Haynie, W.S.; Caldwell, A.R.; Washington, T.A.; Lo, W.J.; Greene, N.P. Hepatic alterations during the development and progression of cancer cachexia. Appl. Physiol. Nutr. Metab. 2020, 45, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.I.; Greene, N.P.; Dobson, J.P.; Wiggs, M.P.; Gasier, H.G.; Macias, B.R.; Shimkus, K.L.; Fluckey, J.D. Insulin resistance syndrome blunts the mitochondrial anabolic response following resistance exercise. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E466–E474. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [Green Version]
- Greene, N.P.; Lee, D.E.; Brown, J.L.; Rosa, M.E.; Brown, L.A.; Perry, R.A.J.; Henry, J.N.; Washington, T.A. Mitochondrial quality control, driven by PGC-1α, is dysregulated by Western Diet-induced obesity and partially restored by moderate physical activity in mice. Physiol. Rep. 2015, 3, e12470. [Google Scholar] [CrossRef]
- Rosa-Caldwell, M.E.; Lee, D.E.; Brown, J.L.; Brown, L.A.; Perry, J.R.A.; Greene, E.S.; Carvallo Chaigneau, F.R.; Washington, T.A.; Greene, N.P. Moderate Physical Activity Promotes Basal Hepatic Autophagy in Diet-Induced Obese Mice. Appl. Physiol. Nutr. Metab. 2017, 42, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Caldwell, M.E.; Brown, J.L.; Lee, D.E.; Blackwell, T.A.; Turner, K.W.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Washington, T.A.; Greene, N.P. Autophagy activation, not peroxisome proliferator-activated receptor gamma coactivator 1alpha, may mediate exercise-induced improvements in glucose handling during diet-induced obesity. Exp. Physiol. 2017, 102, 1194–1207. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosa-Caldwell, M.E.; Benson, C.A.; Lee, D.E.; Brown, J.L.; Washington, T.A.; Greene, N.P.; Wiggs, M.P. Mitochondrial Function and Protein Turnover in the Diaphragm are Altered in LLC Tumor Model of Cancer Cachexia. Int. J. Mol. Sci. 2020, 21, 7841. https://doi.org/10.3390/ijms21217841
Rosa-Caldwell ME, Benson CA, Lee DE, Brown JL, Washington TA, Greene NP, Wiggs MP. Mitochondrial Function and Protein Turnover in the Diaphragm are Altered in LLC Tumor Model of Cancer Cachexia. International Journal of Molecular Sciences. 2020; 21(21):7841. https://doi.org/10.3390/ijms21217841
Chicago/Turabian StyleRosa-Caldwell, Megan E., Conner A. Benson, David E. Lee, Jacob L. Brown, Tyrone A. Washington, Nicholas P. Greene, and Michael P. Wiggs. 2020. "Mitochondrial Function and Protein Turnover in the Diaphragm are Altered in LLC Tumor Model of Cancer Cachexia" International Journal of Molecular Sciences 21, no. 21: 7841. https://doi.org/10.3390/ijms21217841