3.1. Chemical Methods
Melting points were determined in open capillaries on a Gallenkamp electrothermal apparatus and are uncorrected. Mass spectra were recorded on a HP MS 6890-5973 MSD spectrometer, electron impact 70 eV, equipped with a HP ChemStation or with an Agilent LC–MS 1100 Series LC–MSD Trap System VL spectrometer, electrospray ionization (ESI).
1H-NMR spectra were recorded using the suitable deuterated solvent on a Varian Mercury 300 NMR Spectrometer or with an Agilent VNMRS500. Chemical shifts (δ) are expressed as parts per million (ppm) and the coupling constants (J) in Hertz (Hz). Microanalyses of solid compounds were carried out with a Eurovector Euro EA 3000 model analyzer. The analytical results are within ±0.4% of theoretical values. Column chromatography was performed using Geduran silica gel 60 A° (63–200 µm) as a stationary phase. Optical rotations were determined with a Perkin-Elmer 341 polarimeter at room temperature (20 °C). Concentrations are expressed as g/100mL. Chemicals were purchased from Aldrich Chemicals (Milan, Italy) and were used without any further purification. Spectral analysis data (GC-MS, HRMS and NMR) can be found in the
Supplementary Information.
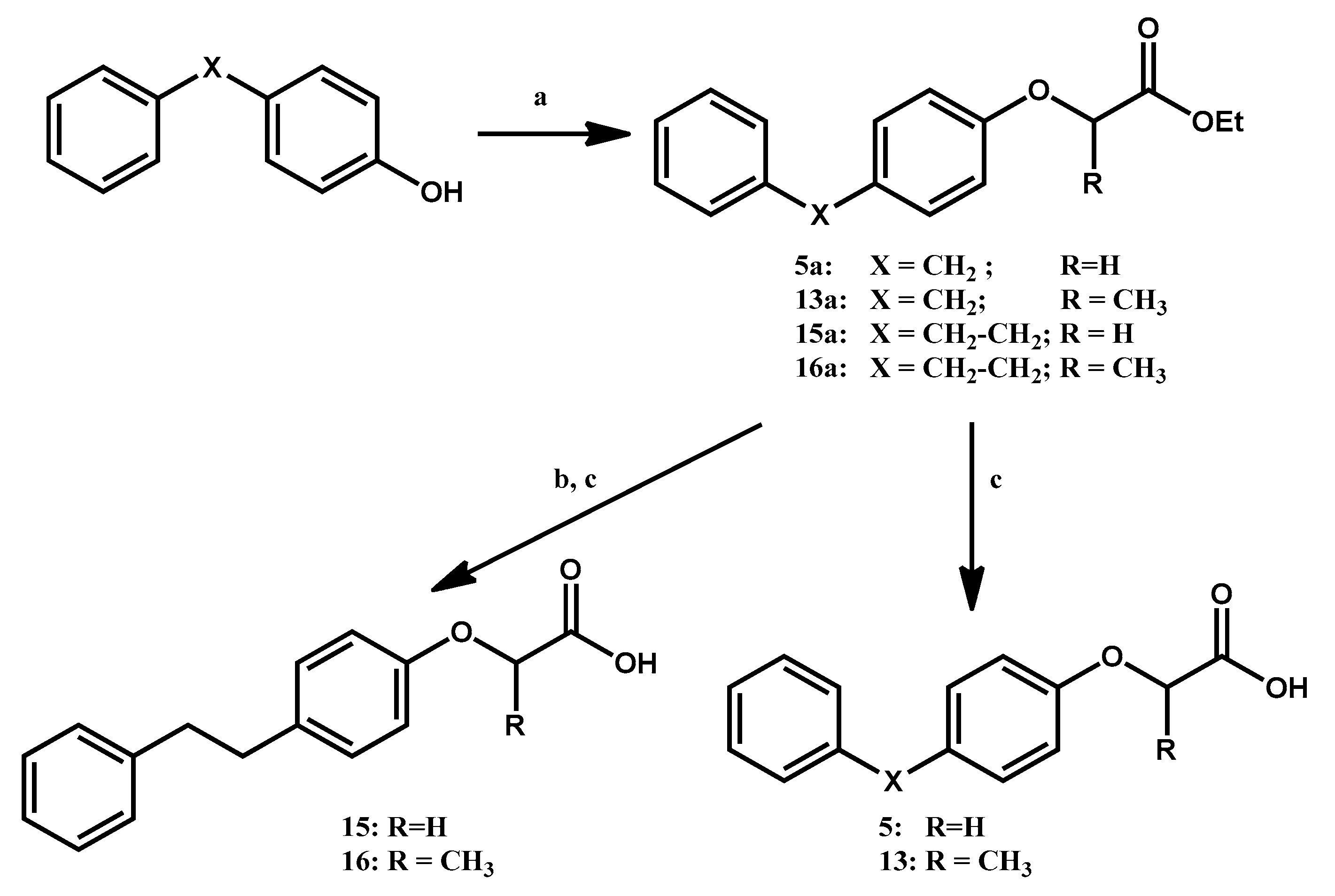
3.1.1. General Procedure for the Preparation of Ethyl Aryloxyacetates 5a, (R,S)-15a and Ethyl 2-Aryloxy-propanoates (R,S)-13a, (R,S)-16a
At T = 0 °C, using anhydrous DMF as solvent, NaH is mixed with the appropriate phenol (either 4-benzylphenol or 4-hydroxy-trans-stilbene) in a 5:1 stoichiometric ratio. After 30 min, the resulting suspension is brought to room temperature. A DMF solution of ethyl 2-bromoacetate or ethyl 2-bromopropanoate is subsequently added dropwise in a 2:1 stoichiometric ratio with the phenol. The mixture is stirred at 65 °C for 2–6 h, then the solvent is evaporated under vacuum. The residue is dissolved in EtOAc, washed once with NH4Cl, once with NaOH 0.5N, and once with NaClss. The organic phase is dried over Na2SO4, filtered over cotton and the solvent is evaporated under vacuum. The crude product is then purified via column chromatography with eluent Hex/EtOAc 95:5.
Ethyl 4-benzyl-phenoxyacetate 5a
61% yield.
(R,S)-Ethyl 2-(4-benzylphenoxy)-propanoate 13a
83% yield.
Ethyl trans-(4-styryl-phenoxy)-acetate 15a
73% yield.
(R,S)-Ethyl 2-trans-(4-styryl-phenoxy)-propanoate 16a
71% yield.
3.1.2. General Procedure for the Preparation of Ethyl (4-Phenethyl-phenoxy)-acetate 15b and (R,S)-Ethyl 2-(4-Phenethyl-phenoxy)-propanoate 16b
A suspension of Wilkinson’s catalyst (0.15 mmol) in absolute EtOH is added to a solution of the appropriate ester 15a or 16a (3 mmol) in THF. The resulting mixture is then placed in an autoclave at 15atm of H2 pressure and stirred for 48 h. Afterwards, the catalyst is removed via filtration over celite and the solvent is evaporated under vacuum. The crude product is then purified via column chromatography over silica gel (eluent Hex/EtOAc 95:5), affording the purified product.
Ethyl (4-phenethyl-phenoxy)-acetate 15b
79% yield.
(R,S)-Ethyl 2-(4-phenethyl-phenoxy)-acetate 16b
58% yield.
3.1.3. General Procedure for the Preparation of 4-Aryl-phenoxyacetic Acids 5 and 15 and (R,S)-2-(4-Arylphenoxy)-propanoic Acids 13 and 16
The appropriate ester is dissolved in 30 mL THF and mixed with a 10 mL solution of NaOH 6N. The solution is stirred at room temperature for 6–12 h. The solvent is then evaporated under vacuum and the residue is brought to acid pH with HCl 6N. The resulting solution is extracted three times with diethyl ether, and the organic phases are joined, washed once with NaClss, dried over Na2SO4, filtered over cotton, and the solvent is evaporated under vacuum. The desired product is obtained via crystallization from CHCl3/Hex.
4-benzyl-phenoxyacetic acid 5
56% yield. m.p. 117–118 °C.
(R,S)-2-(4-benzyl)-phenoxypropanoic acid 13
71% yield. m.p. 102–104 °C
4-phenethyl-phenoxyacetic acid 15
59% yield. m.p. 130–131 °C
(R,S)-2-(4-phenethyl)-phenoxyacetic acid 16
62% yield. m.p. 106–107 °C
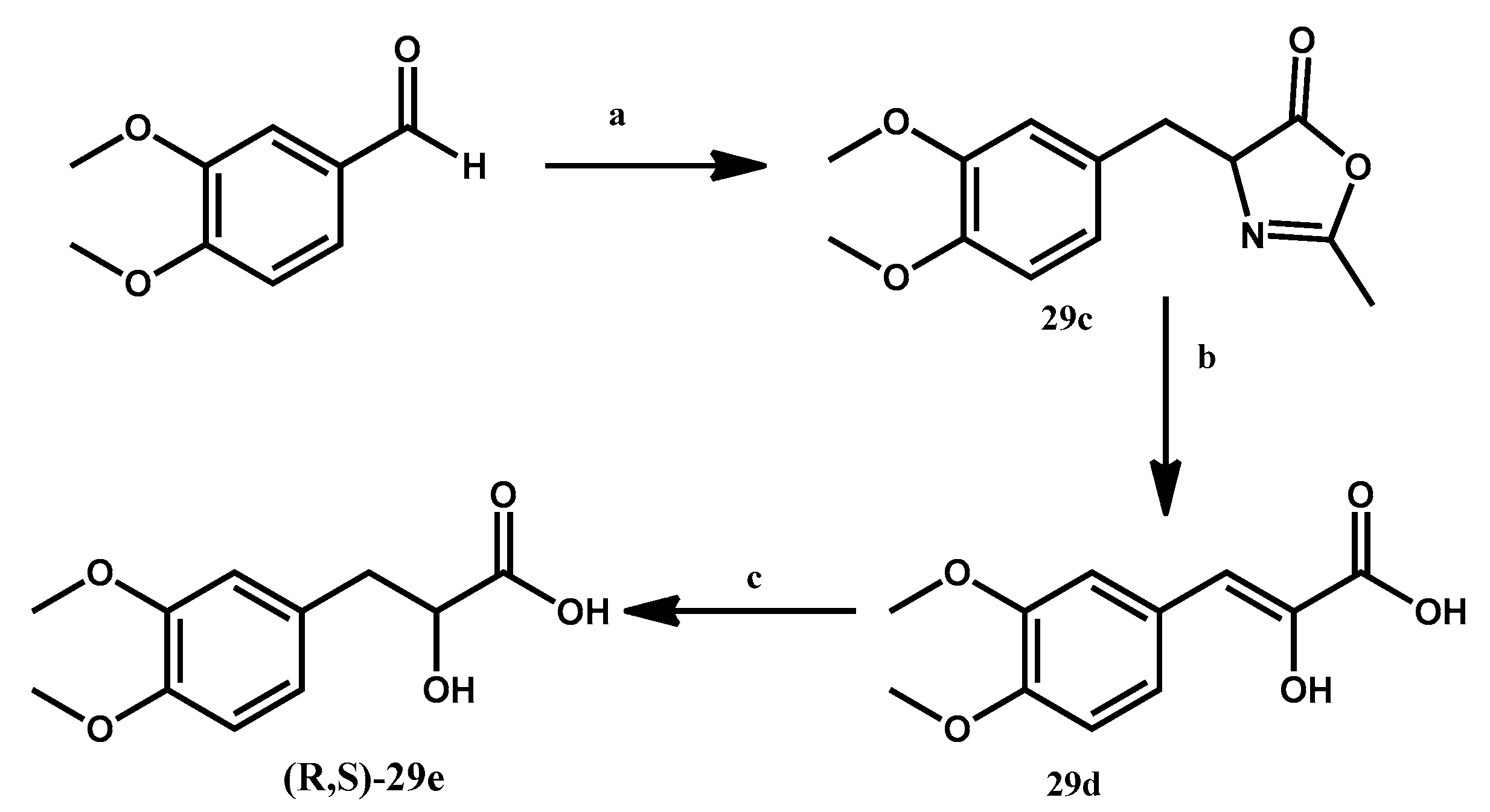
3.1.4. Preparation of 4-[(3,4-Dimethoxyphenyl)methyl-idene]-2-methyl-4,5-dihydro-1,3-oxazol-5-one 29c
N-Acetylglycine (15 mmol), sodium acetate (15 mmol) and acetic anhydride (60 mmol) were added to 3,4-dimethoxybenzaldehyde (15 mmol). The reaction is stirred for 18 h at 110 °C. Upon cooling, the formation of a yellow solid precipitate is observed. This solid is filtered, then washed with 10 mL of an EtOH/H2O 1:1 solution, affording the desired compound as a yellow solid. 25% yield.
3.1.5. Preparation of 3-(3,4-Dimethoxyphenyl)-2-hydroxyacrylic Acid 29d
Breifly, 25 mL of HCl 3N are added to compound 15c (3.75 mmol). The reaction is stirred for 6 h at 100 °C. Upon cooling, a red solid is precipitated. The desired compound is crystallized in MeOH/H2O. 31% yield.
3.1.6. Preparation of (R,S)-3-(3,4-Dimethoxyphenyl)-2-hydroxypropanoic Acid 29e
At T = 0 °C, a 25% solution of MeOH in H2O is added to 15d (1.16 mmol), NaOH is slowly added until pH = 10, then NaBH4 (1.74 mmol) is added. The reaction is stirred for 17 h at room temperature. The resulting solution is acidified with HCl 1N, then saturated with NaCl, and extracted with EtOAc (5 × 10 mL). The organic phases are joined, dried over anhydrous Na2SO4 and filtered over cotton, after which the solvent is evaporated in vacuo, affording a light brown oil. This crude product is purified via column chromatography with eluent CHCl3/MeOH 10:1, affording a yellow oil. 89% yield.
3.1.7. General Procedure for the Preparation of Allyl 3-Phenyl-2-hydroxypropanoates (R)-28a, (S)-28a, (R,S)29a
The appropriate 3-phenyl-2-hydroxypropanoic acid is dissolved in anhydrous toluene and mixed with allyl alcohol and paratoluensulfonic acid in a stoichiometric ratio of 1:1.2:0.2. The resulting mixture is stirred at 100 °C for 6 h. The solvent is removed in vacuo, the crude product is dissolved in EtOAc and washed twice with NaHCO3ss and one with NaClss. The organic phase is dried over Na2SO4, filtered over cotton and the solvent is evaporated in vacuo.
(R)- and (S)-Allyl 3-phenyl-2-hydroxypropanoate 28a
56–89% yield.
Allyl 3-(3,4-dimethoxyphenyl)-2-hydroxypropanoate 29a
46% yield.
3.1.8. General Procedure for the Preparation of 1-(Allyloxy)-1-oxo-3-phenylpropan-2-yl-2-phenylacrylates
The appropriate allyl 3-phenyl-2-hydroxypropanoate (1 mmol), the appropriate cinnamic acid (1.1 mmol), EDCI (1.3 mmol), and DMAP (0.1 mmol) are dissolved in CH2Cl2 and stirred for 24 h at RT. The resulting mixture is washed once with NH4Clss, diluted with more CH2Cl2 and washed once with NaClss. The organic phase is dried over anhydrous Na2SO4, filtered, and the solvent is evaporated under vacuum, affording a yellow crude oil which is subsequently purified via column chromatography on silica gel (eluent Hex:EtOAc 9:1), yielding the desired product as a yellow oil.
(R)- and (S)-1-(allyloxy)-1-oxo-3-phenylpropan-2-yl-2-phenylacrylate (R)-28b
39–52% yield.
1-(allyloxy)-1-oxo-3-(3,4-dimethoxyphenyl)-propan-2-yl 2-phenylacrylate 29b
56% yield.
(R)-1-(allyloxy)-1-oxo-3-phenyl-propan-2-yl-2-(3,4-dimethoxyphenyl)acrylate (R)-30b
37% yield.
3.1.9. General Procedure for the Preparation of 1-(Allyloxy)-1-oxo-3-phenylpropan-2-yl 2-Arylacrylic Acids
Allyl esters (14b–16b), morpholine and Pd(PPH3)4 are dissolved in anhydrous THF in a 1:20:0.1 stoichiometric ratio. Stirring occurred for 2 h at RT. The solvent is removed in vacuo and the residue is dissolved in CH2Cl2 and extracted with NaHCO3ss, the aqueous phase is then brought to acidic pH with HCl 2N and is finally extracted with CH2Cl2. The organic phase is washed with NaClss, dried with Na2SO4, filtered, and the solvent is removed in vacuo, affording a yellow oil which is further purified via column chromatography over silica gel.
(R)- and (S)-1-(allyloxy)-1-oxo-3-phenylpropan-2-yl-2-phenylacrylic acid 28b
Column chromatography was performed using an eluent composed of Hex/EtOAc/Acetic acid (6.6:3.3:0.1) affording a transparent oil which was further purified forming the sodium salt of the desired product. 36–66% yield. (R)-14b [α]D20 = +57.2 (c = 2, MeOH). (S)-14b [α]D20 = −58.6 (c = 0.958, MeOH).
1-(allyloxy)-1-oxo-3-(3,4-dimethoxyphenyl)-propan-2-yl-2-phenylacrylate 29b
The crude oil is purified via the preparation of a cyclohexylamine salt without column chromatography. 84% yield.
(R)-1-(allyloxy)-1-oxo-3-phenyl-propan-2-yl 2-(3,4-dimethoxyphenyl)acrylate (R)-30b
Column chromatography was conducted a CH2Cl2/MeOH 9:1 mixture as eluent, affording a transparent oil which was further purified forming the cyclohexylamine salt of the desired product.
20% yield. [α]D20 = +15.2 (c = 0.25; MeOH).
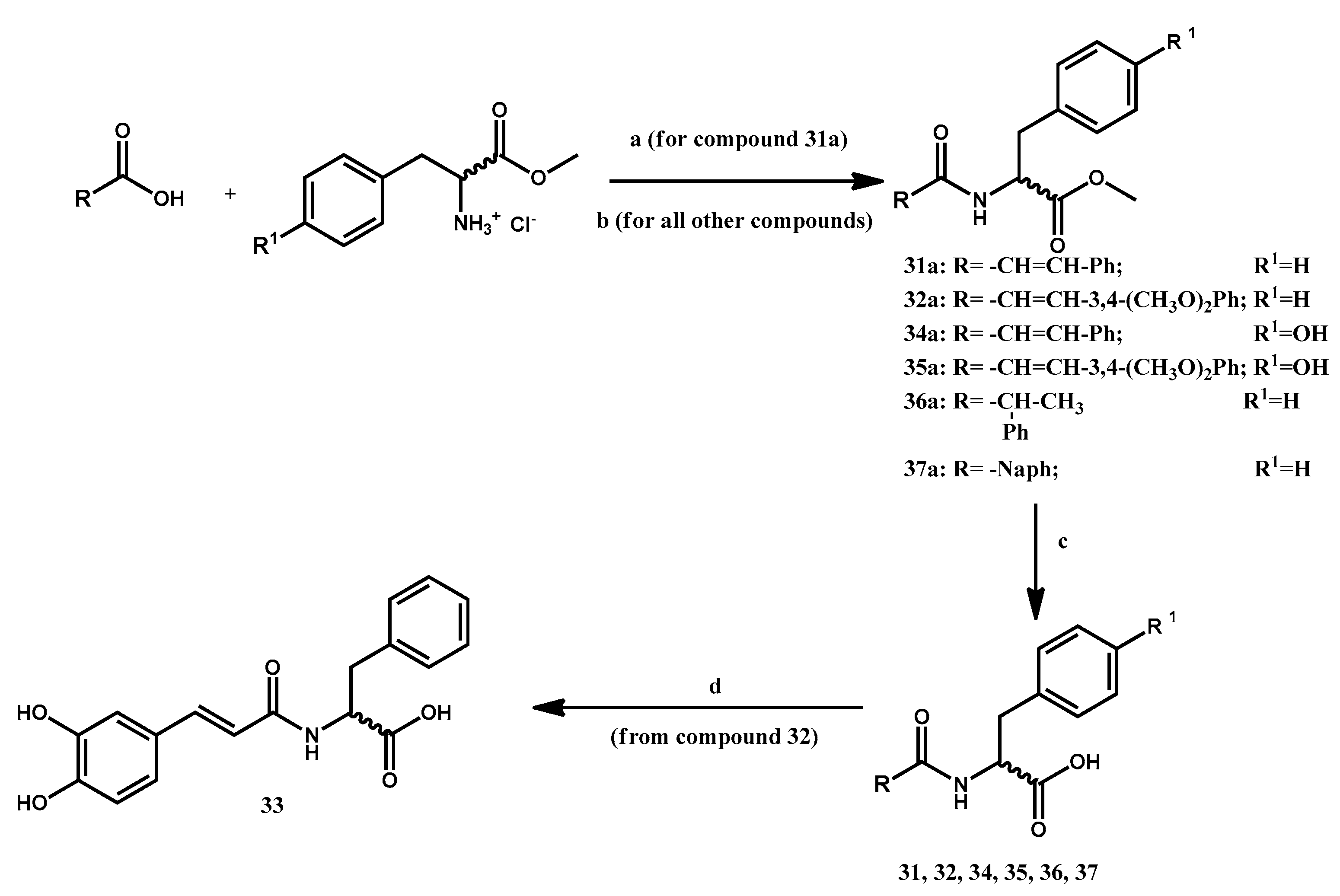
3.1.10. Preparation of Methyl 2-Cinnamamido-3-phenylpropanoate 31a
Cinnamic acid (2.07 mmol) is dissolved in anhydrous THF at 0 °C, to which L-phenylalanine methyl ester hydrochloride (1.86 mmol), triethylamine (1.67 mmol) in CHCl3, HOBt x H2O (1.43 mmol) in THF and DCC (1.78 mmol) in CHCl3 are added. The solution is stirred for 1 h at 0 °C, then at RT overnight. Afterwards, a precipitate is observed and filtered away; the solvent is evaporated away under vacuum and the residue is dissolved in EtOAc, washed once with distilled water, then once with a 10% aqueous solution of citric acid, once with NaHCO3ss, three times with NaOH 0.1N, and finally once with NaClss. The organic phase is dried over Na2SO4, filtered and the solvent is removed in vacuo, affording the desired product as a pale yellow solid. 27% yield.
3.1.11. General Procedure for the Preparation of Methyl 2-(3-Arylacrylamido)-3-arylpropanoates 32a, 34a, 35a
Appropriately substituted cinnamic acids (1 mmol) are dissolved in DMF; the system is cooled to 0 °C and then HOBt x H2O (1 mmol) and EDCI (1 mmol) are added. In a separate vessel, the appropriate L-aminoacid methyl ester hydrochloride (1 mmol) was mixed with N-methylmorpholine (1 mmol) in CH2Cl2 for 15 min, then this mixture is added to the main reaction vessel. Stirring occurred at RT overnight. The crude reaction mixture is diluted with NaHCO3 5%, extracted five times with CH2Cl2, and the organic phase is washed once with NaClss, dried over anhydrous Na2SO4, filtered, and the solvent is evaporated in vacuo, affording a solid.
Methyl 2-(3-(3,4-dimethoxyphenyl)acrylamido)-(S)-3-phenylpropanoate 32a
The crude solid was purified via column chromatography over silica gel (eluent EtOAc/Hex 6:4), affording a white solid. 54% yield.
Methyl 2-cinnamamido-(S)-3-(4-hydroxyphenyl)propanoate 34a:
The crude product did not require further purification and appeared as a white solid. 99% yield.
Methyl 2- (3-(3,4-dimethoxyphenyl)acrylamido)-(S)-3-(4-hydroxyphenyl)propanoate 35a
The crude solid is purified via column chromatography over silica gel using a EtOAc/Hex 6:4 mixture as eluent. The purified product appears as a white solid.
3.1.12. Preparation of Methyl (R,S)-2-phenylpropanoylamido-(S)-3-phenylpropanoate 36a
Briefly, 2-phenylpropanoic acid (2.32 mmol) is dissolved in DMF. EDCI (2.32 mmol) and HOBt x H2O (2.32 mmol) are added at 0 °C, while L-phenylalanine methyl ester hydrochloride (2.32 mmol) and N-methylmorpholine (2.32) are mixed in a separate vessel in CH2Cl2 for 10 min. The two mixtures are joined and stirred overnight at RT. The resulting solution is diluted with NaHCO3 5%, extracted five times with CH2Cl2 and the organic phase is washed once with NaClss, dried over anhydrous Na2SO4, filtered and the solvent is evaporated away in vacuo. The residue, appearing as a yellow crude solid, is dissolved in EtOAc and washed with HCl 2N. Once the solvent is removed, the desired product is obtained as a white solid. 79% yield.
3.1.13. Preparation of Methyl 2-(2-Naphthamido)-(S)-3-phenylpropanoate 37a
2-naphthoic acid (1.16 mmol) is dissolved in DMF; the system is cooled to 0 °C, after which EDCI (1.16 mmol) and HOBt x H2O (1.16 mmol) are added. In a separate vessel, L-phenylalanine methyl ester hydrochloride (1.16 mmol) is mixed with N-methylmorpholine (1.16 mmol) in CH2Cl2 for 10 min. This mixture is then added to the naphtoic acid solution and the reaction is stirred overnight at RT. The resulting solution is diluted with NaHCO3 5%, extracted 4 times with CH2Cl2 and the organic phase is washed once with NaClss, dried over anhydrous Na2SO4, filtered and the solvent is removed under vacuum. The residue is dissolved in EtOAc and washed with HCl 2N; the desired product appears as a pale yellow solid. 80% yield.
3.1.14. General Procedure for the Preparation of 2-Substituted-3-arylpropanoic Acids 31, 32, 34–37
The appropriate 2-substituted methyl 3-arylpropanoate (1 mmol) is dissolved in a THF/H2O 1:1 solution; the system is cooled to 0 °C and LiOH x H2O (5 mmol) is added. The mixture is stirred for 5 h at RT, then it is acidified with HCl 1N and extracted with EtOAc 4 times. The organic phase is separated, dried over anhydrous Na2SO4, filtered, and the solvent is removed under vacuum giving the crude product.
2-cinnamamido-3-phenylpropanoic acid 31:
The crude product appears as a transparent oil which is treated with Et2O/Hex 1:1 giving a white solid that is further purified via crystallization with Hex + EtOAc(gtt), affording the desired product as white crystals. 24% yield. mp: 194.2–198.2 °C
(S)-2-(3-(3,4-dimethoxyphenyl)acrylamido)-3-phenylpropanoic acid 32:
The crude product appears as an oil mixed with a white solid and is treated with Et2O/Hex 1:1 affording a white solid. 78.7% yield.
(S)-2-cinnamamido-3-(4-hydroxyphenyl)propanoic acid 34:
The crude product appears as a pale yellow oil which is treated with Et2O/Hex 1:1 affording a pale yellow solid. 76% yield.
(S)-2-(3-(3,4-dimethoxyphenyl)acrylamido)-3-(4-hydroxyphenyl)propanoic acid 35:
The crude product appears as a colorless oil and is treated with CHCl3/Hex 1:1 affording the desired product as a white powder. 96.6% yield. mp: 105.0–108.5 °C. [α]D20 = −29.66 (c = 1.00, MeOH).
(R,S)-2-phenylpropanoylamido-(S)-3-phenylpropanoic acid 36:
The crude product, a yellow solid, is crystallized in Et2O + Hex (gtt), affording the desired product as white crystals. 34% yield. mp: 66.5–69.3 °C
(S)-2-(2-naphthamido)-3-phenylpropanoic acid 37:
The crude product appears as a white solid that is further purified via crystallization at 5 °C in CHCl3 + Hex (gtt), affording white crystals. 70% yield. mp: 156.5–157.3 °C. [α]D20 = −84.61 (c = 1.001, MeOH).
3.1.15. Preparation of (S)-2-(3-(3,4-Dihydroxyphenyl)acrylamido-3-phenylpropanoic Acid 33
Briefly, 0.14 mmol of compound 32 are dissolved in CH2Cl2 and a 1 mL of a 1M solution of BBr3 is then added dropwise at room temperature. The reaction is stirred overnight at room temperature, after which it is quenched by adding H2O at −15 °C. The resulting mixture is extracted three times with EtOAc, dried over Na2SO4, filtered, and the solvent is evaporated under vacuum. The crude product is purified via column chromatography (eluent EtOAc:MeOH 7:3). 46% yield. m.p. 198–199 °C. [α]D20 = +6.465 (c = 1.00, MeOH).
3.2. Biological Methods
Reference compounds, cell culture mediums and other reagents, along with plates and other physical supports for cell culture and enzyme assays were purchased from Merck Sigma (Milan, Italy) and Invitrogen (Basel, Switzerland). The FAAH enzyme, the substrate AMC-AA and the reference compound JZL-195 for the FAAH inhibition assay were obtained from Cayman Chemical (Ann Arbor, MI, USA).
3.2.1. Plasmids
The expression vectors carrying the chimeric receptors composed of the yeast GAL4-DNA binding domain fused to human PPARα, PPARγ or PPARδ, and their respective reporter plasmid (pGAL5TKpGL3), presenting the GAL4 response element in five repeats upstream of a minimal thymidine kinase promoter, itself upstream to the luciferase gene, were previously described [
35]. These plasmids were kindly donated by Dr. Krister Bamberg (AstraZeneca, Mölndal, Sweden).
3.2.2. Cell Culture and Transfection
HepG2 cell line (human hepatoblastoma, Interlab Cell Line Collection, Genoa, Italy) was cultured in Minimum Essential Medium (MEM) containing 10% fetal bovine serum, penicillin (100 U/mL) and streptomycin sulfate (100 µg/mL) at 37 °C in a humidified incubator in 5% CO2 atmosphere. Transactivation assays were conducted seeding 105 cells/well in a 24-well plate. The cells were transfected after 24 h with the calcium phosphate method CAPHOS®, following the manufacturer’s guidelines. Plasmids encoding the fusion proteins GAL4-PPARa-LBD, GAL4-PPARg-LBD, GAL4-PPARd-LBD or GAL4-FXR-LBD (30 ng), pGAL5TKpGL3 (100 ng), and pCMVbgal (250 ng) were used. Treatment of the cells with the investigated compounds was carried out 4 h after transfection, in triplicate, and the cells were incubated in the treated medium for additional 20 h. Cells were then lysed and luciferase activity in the extract was determined via a VICTOR3 V Multilabel Plate Reader, PerkinElmer).
3.2.3. FAAH Inhibition Assay
Briefly, 96-well black flat-bottom microtiter NBS plates (COSTAR flat black) were utilized to perform the assays (in triplicate). The experiments in a total volume of 200 µL, first incubating different concentrations of each potential inhibitor in an appropriate fluorometric assay buffer (tris-HCl 125 mM, Na2EDTA · 2H2O 1 mM, pH = 9.0) with the enzyme (FAAH Human recombinant, Cayman Chemical, Ann Arbor, MI, USA) for 15 min at room temperature, keeping the plate in orbital shaking.
The substrate (7-amino-4-methyl-2H-1-benzopyran-2-one-5Z,8Z,11Z,14Z-eicosatetraen-amide, AMC-AA, 1 µM final concentration) was then added, and the assay was incubated for 2–3 h at 37 °C in a TECAN infinite M1000Pro plate reader (Tecan, Männedorf, Switzerland) which measured the fluorescence from each well every 30 s (λex = 340 nm, λem = 450 nm), determining FAAH activity as relative fluorescence units (RFU). Control wells lacking the inhibitor and blank wells lacking both inhibitor and enzyme were used to calculate percent inhibition for each tested compound. IC50 values were calculated via GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA) and are reported as mean ± SEM of at least two independent measurements performed in triplicate.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


