Myelosuppression in Patients Treated with the Telomerase Inhibitor Imetelstat Is Not Mediated through Activation of Toll-Like Receptors

Abstract
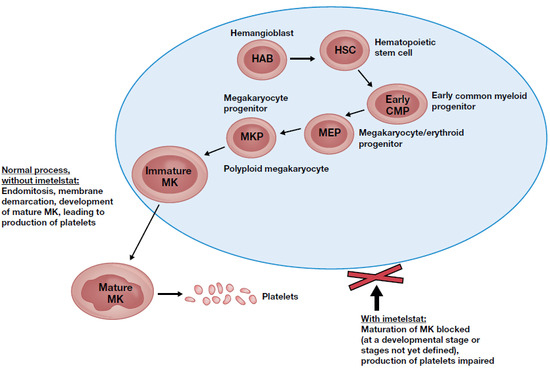
:
1. Background
2. Results and Discussion
3. Materials and Methods
Author Contributions
Funding
Conflicts of Interest
References
- Baerlocher, G.M.; Oppliger Leibundgut, E.; Ottmann, O.G.; Spitzer, G.; Odenike, O.; McDevitt, M.A.; Roth, A.; Daskalakis, M.; Burington, B.; Stuart, M.; et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N. Engl. J. Med. 2015, 373, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascarenhas, J.; Komrokji, R.S.; Cavo, M.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Imetelstat Is Effective Treatment for Patients with Intermediate-2 or High-Risk Myelofibrosis Who Have Relapsed on or Are Refractory to Janus Kinase Inhibitor Therapy: Results of a Phase 2 Randomized Study of Two Dose Levels. Blood 2018, 132, 685. [Google Scholar] [CrossRef]
- Tefferi, A.; Al-Kali, A.; Begna, K.H.; Patnaik, M.M.; Lasho, T.L.; Rizo, A.; Wan, Y.; Hanson, C.A. Imetelstat therapy in refractory anemia with ring sideroblasts with or without thrombocytosis. Blood Cancer J. 2016, 6, e405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U.; Fenaux, P.; Steensma, D.P.; Eygen, K.V.; Raza, A.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Treatment with imetelstat provides durable transfusion independence (TI) in heavily transfused non-del(5Q) lower risk MDS (LRMDS) relapsed/refractory (R/R) to erythropoiesis stimulating agents (ESA). Eha25 Virtual 2020, 11–21. [Google Scholar]
- Herbert, B.S.; Gellert, G.C.; Hochreiter, A.; Pongracz, K.; Wright, W.E.; Zielinska, D.; Chin, A.C.; Harley, C.B.; Shay, J.W.; Gryaznov, S.M. Lipid modification of GRN163, an N3’-->P5’ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 2005, 24, 5262–5268. [Google Scholar] [CrossRef] [Green Version]
- Shammas, M.A.; Koley, H.; Bertheau, R.C.; Neri, P.; Fulciniti, M.; Tassone, P.; Blotta, S.; Protopopov, A.; Mitsiades, C.; Batchu, R.B.; et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 2008, 22, 1410–1418. [Google Scholar] [CrossRef]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.A.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar]
- Bruedigam, C.; Bagger, F.O.; Heidel, F.H.; Paine Kuhn, C.; Guignes, S.; Song, A.; Austin, R.; Vu, T.; Lee, E.; Riyat, S.; et al. Telomerase inhibition effectively targets mouse and human AML stem cells and delays relapse following chemotherapy. Cell Stem Cell 2014, 15, 775–790. [Google Scholar] [CrossRef] [Green Version]
- Chao, W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1–H12. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Pohar, J.; Kuznik Krajnik, A.; Jerala, R.; Bencina, M. Minimal sequence requirements for oligodeoxyribonucleotides activating human TLR9. J. Immunol. 2015, 194, 3901–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxadé, M.; Lunazzi, G.; Minguillón, J.; Iborra, S.; Berga-Bolaños, R.; Del Val, M.; Aramburu, J.; López-Rodríguez, C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 2012, 209, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, R.G.; Young, R.A. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 2005, 3, 281–294. [Google Scholar] [CrossRef]
- Armanios, M.; Greider, C.W. Treating Myeloproliferation--On Target or Off? N. Engl. J. Med. 2015, 373, 965–966. [Google Scholar] [CrossRef]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, L.M.; Freedman, J.E. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb. Res. 2009, 125, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Flaujac, C.; Boukour, S.; Cramer-Bordé, E. Platelets and viruses: An ambivalent relationship. Cell. Mol. Life Sci. 2010, 67, 545–556. [Google Scholar] [CrossRef]
- Akilesh, H.M.; Buechler, M.B.; Duggan, J.M.; Hahn, W.O.; Matta, B.; Sun, X.; Gessay, G.; Whalen, E.; Mason, M.; Presnell, S.R.; et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 2019, 363, eaao5213. [Google Scholar] [CrossRef]
- Nakatake, M.; Sasaki, N.; Murakami-Murofushi, K.; Yamada, O. Transient posttranslational up-regulation of telomerase activity during megakaryocytic differentiation of K562 cells. Biochem. Biophys. Res. Commun. 2004, 314, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Mosoyan, G.; Kraus, T.; Ye, F.; Eng, K.; Crispino, J.D.; Hoffman, R.; Iancu-Rubin, C. Imetelstat, a telomerase inhibitor, differentially affects normal and malignant megakaryopoiesis. Leukemia 2017, 31, 2458–2467. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, G.M.; Haubitz, M.; Braschler, T.R.; Brunold, C.; Burington, B.; Oppliger Leibundgut, E.; Go, N. Imetelstat inhibits growth of megakaryocyte colony-forming units from patients with essential thrombocythemia. Blood Adv. 2019, 3, 3724–3728. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, C.S.; Petersen, B.; Qiu, J.; Ye, F.; Houldsworth, J.; Eng, K.; Huang, F.; Hoffman, R. Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells. Blood Adv. 2018, 2, 2378–2388. [Google Scholar] [CrossRef] [Green Version]
- Schuller, C.; Jankowski, K.; MacKenzie, K. Telomere length of cord blood-derived CD34+ progenitors predicts erythroid proliferative potential. Leukemia 2007, 21, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Weng, N. Interplay between telomere length and telomerase in human leukocyte differentiation and aging. J. Leukoc. Biol. 2001, 70, 861–867. [Google Scholar] [CrossRef]
- Norrback, K.F.; Roos, G. Telomeres and telomerase in normal and malignant haematopoietic cells. Eur. J. Cancer 1997, 33, 774–780. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Experimental Agents a | ||
|---|---|---|
| Imetelstat | 5′-R-TAGGGTTAGACAA-NH2-3′ | |
| Mismatch oligonucleotide (with 4 nucleotides mismatched to imetelstat) | 5′-R-TAGGTGTAAGCAA-NH2-3′ | |
| Sense oligonucleotide (with reverse sequence of imetelstat) | 5′-AACAGATTGGGAT-R-3′ | |
| Positive Control Agents | ||
| Positive Control Ligand | Receptor | Ligand Concentration |
| Heat-killed Listeria monocytogenes | hTLR2 | 108 cells/mL |
| Poly(I:C) HMW | hTLR3 | 1 μg/mL |
| Escherichia coli K12 lipopolysaccharide | hTLR4 | 100 ng/mL |
| Salmonella typhimurium flagellin | hTLR5 | 100 ng/mL |
| Imiquimod | hTLR7 | 1 μg/mL |
| CL075 | hTLR8 | 1 μg/mL |
| CpG ODN 2006 | hTLR9 | 100 ng/mL |
| Requirement | Imetelstat | Conclusion |
|---|---|---|
| At least two CpG islands separated by 6 to 10 nucleotides, e.g., 5′-TCGTTTTTTTCGTTTTTTTTTTTT-3′ | 13-mer oligonucleotide: 5′-TAGGGTTAGACAA-NH2-3′ | Criteria not met |
| First CpG island is preceded by a 5′ thymidine, e.g., 5′-TCGTTTTTTTCGTTTTTTTTTTTT-3′ | Criteria not met | |
| Poly-thymidine tail of ≥10 nucleotides at the 3′ end, e.g., 5′-TCGTTTTTTTCGTTTTTTTTTTTT-3′ | Criteria not met | |
| Overall length ≥21 nucleotides | Criteria not met |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baerlocher, G.M.; Rusbuldt, J.; Bussolari, J.; Huang, F. Myelosuppression in Patients Treated with the Telomerase Inhibitor Imetelstat Is Not Mediated through Activation of Toll-Like Receptors. Int. J. Mol. Sci. 2020, 21, 6550. https://doi.org/10.3390/ijms21186550
Baerlocher GM, Rusbuldt J, Bussolari J, Huang F. Myelosuppression in Patients Treated with the Telomerase Inhibitor Imetelstat Is Not Mediated through Activation of Toll-Like Receptors. International Journal of Molecular Sciences. 2020; 21(18):6550. https://doi.org/10.3390/ijms21186550
Chicago/Turabian StyleBaerlocher, Gabriela M., Joshua Rusbuldt, Jacqueline Bussolari, and Fei Huang. 2020. "Myelosuppression in Patients Treated with the Telomerase Inhibitor Imetelstat Is Not Mediated through Activation of Toll-Like Receptors" International Journal of Molecular Sciences 21, no. 18: 6550. https://doi.org/10.3390/ijms21186550