The Role of MicroRNAs in Arrhythmogenic Cardiomyopathy: Biomarkers or Innocent Bystanders of Disease Progression?

 , and
, and
Abstract
:1. Introduction
1.1. AC Pathogenesis
1.2. AC Diagnosis
1.3. AC Genetics
1.4. AC Biomarkers
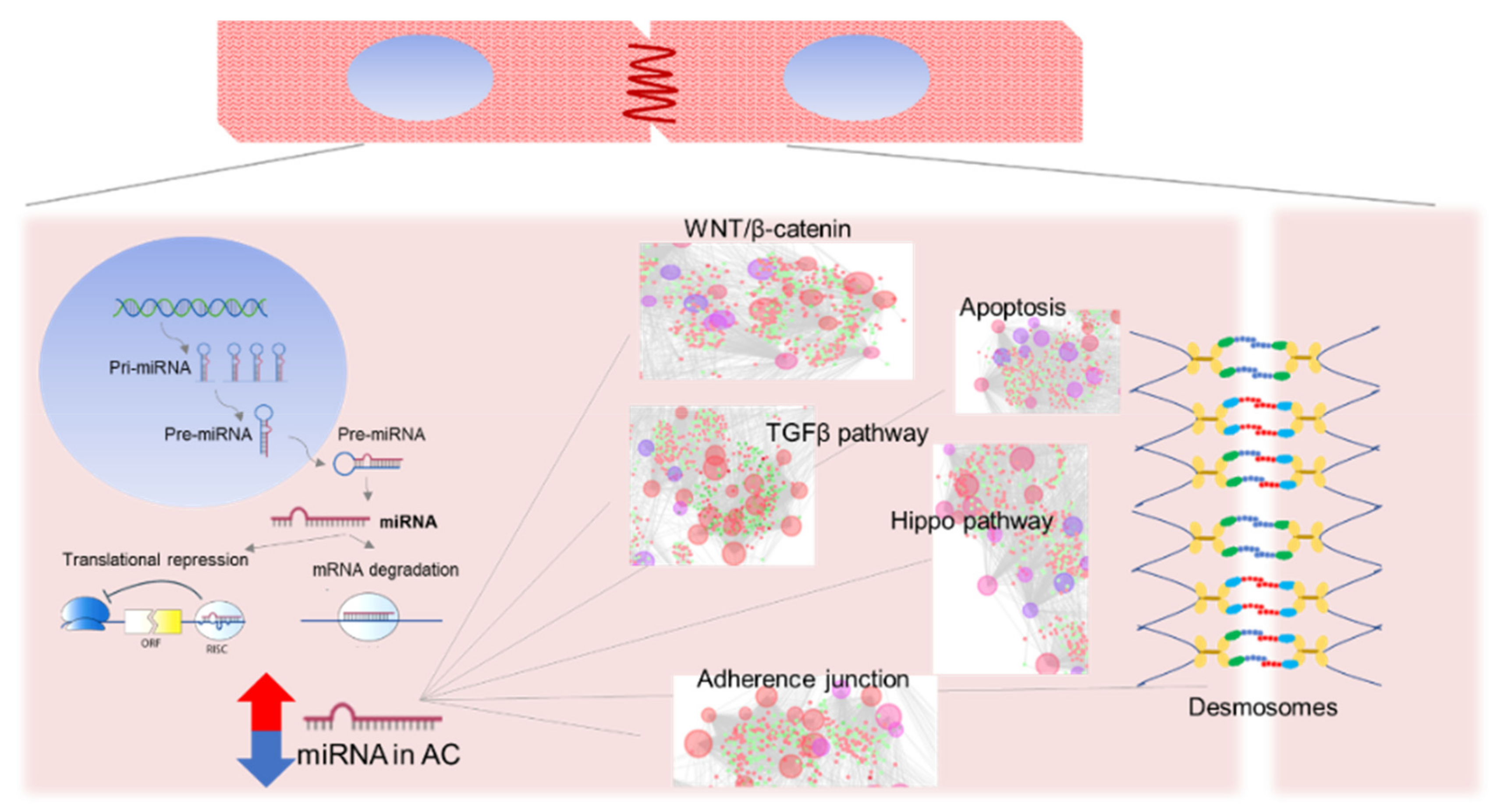
1.5. MicroRNAs
2. MicroRNAs in AC Human Samples
3. MiRNAs in Cell Culture and AC Animal Models
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Thiene, G.; Nava, A.; Rossi, L.; Pennelli, N. Sudden death in young competitive athletes: Clinicopathologic correlations in 22 cases. Am. J. Med. 1990, 89, 588–596. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Nava, A.; Bauce, B.; Basso, C.; Muriago, M.; Rampazzo, A.; Villanova, C.; Daliento, L.; Buja, G.; Corrado, D.; Danieli, G.A.; et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2000, 36, 2226–2233. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J. The 2006 american heart association classification of cardiomyopathies is the gold standard. Circ. Heart Fail 2008, 1, 72–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, R.; Dong, J.; Rodriguez, G.; Bell, A.; Leung, T.K.; Schwartz, R.J.; Willerson, J.T.; Brugada, R.; Marian, A.J. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2009, 104, 1076–1084. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Basso, C.; Pilichou, K.; Bauce, B.; Corrado, D.; Thiene, G. Diagnostic criteria, genetics, and molecular basis of arrhythmogenic cardiomyopathy. Heart Fail Clin. 2018, 14, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task force of the working group myocardial and pericardial disease of the european society of cardiology and of the scientific council on cardiomyopathies of the international society and federation of cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [PubMed] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The padua criteria. Int. J. Cardiol. 2020, in press. [Google Scholar] [CrossRef]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Perazzolo Marra, M.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 33. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac abnormalities in familial palmoplantar keratosis. Br. Heart J. 1986, 56, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [Green Version]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Pilichou, K.; Lazzarini, E.; Rigato, I.; Celeghin, R.; de Bortoli, M.; Perazzolo Marra, M.; Cason, M.; Jongbloed, J.; Calore, M.; Rizzo, S.; et al. Large genomic rearrangements of desmosomal genes in italian arrhythmogenic cardiomyopathy patients. Circ. Arrhythm. Electrophysiol. 2017, 10, e005324. [Google Scholar] [CrossRef] [PubMed]
- Celeghin, R.; Thiene, G.; Bauce, B.; Basso, C.; Pilichou, K. Genetics in cardiovascular diseases. Ital. J. Med. 2019, 13, 137–151. [Google Scholar] [CrossRef] [Green Version]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef] [Green Version]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (arvd2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the tmem43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Van Tintelen, J.P.; van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the des gene. Heart Rhythm. 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban r14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the lamin a/c gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bortoli, M.; Calore, C.; Lorenzon, A.; Calore, M.; Poloni, G.; Mazzotti, E.; Rigato, I.; Marra, M.P.; Melacini, P.; Iliceto, S.; et al. Co-inheritance of mutations associated with arrhythmogenic cardiomyopathy and hypertrophic cardiomyopathy. Eur. J. Human Genet. EJHG 2017, 25, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.; Hoorntje, E.T.; Te Riele, A.; Tichnell, C.; van der Heijden, J.F.; Tandri, H.; van den Berg, M.P.; Jongbloed, J.D.H.; Wilde, A.A.M.; Hauer, R.N.W.; et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (arvc). J. Cardiovasc. Electrophysiol. 2018, 29, 1004–1009. [Google Scholar] [CrossRef]
- Sakamoto, N.; Natori, S.; Hosoguchi, S.; Minoshima, A.; Noro, T.; Akasaka, K.; Sato, N.; Ohno, S.; Ikeda, Y.; Ishibashi-Ueda, H.; et al. Left-dominant arrhythmogenic cardiomyopathy with heterozygous mutations in dsp and mybpc3. Circ. Cardiovasc. Imaging 2019, 12, e008913. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Ortiz-Genga, M.; Gomez-Milanes, I.; Lopez-Cuenca, D.; Ruiz-Espejo, F.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Monserrat, L.; Gimeno, J.R. A mutation in the z-line cypher/zasp protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2015, 88, 172–176. [Google Scholar] [CrossRef]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of scn5a mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; de Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphat-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of cadherin 2 (cdh2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef] [Green Version]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates cdh2-encoded n-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef] [PubMed]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-exome sequencing identifies pathogenic variants in tjp1 gene associated with arrhythmogenic cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating flnc mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; McKenna, W.J. The utility of magnetic resonance imaging in the evaluation of arrhythmogenic right ventricular cardiomyopathy. Curr. Opin. Cardiol. 2008, 23, 38–45. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Leoni, L.; Tokajuk, B.; Turrini, P.; Bauce, B.; Migliore, F.; Pavei, A.; Tarantini, G.; Napodano, M.; et al. Three-dimensional electroanatomical voltage mapping and histologic evaluation of myocardial substrate in right ventricular outflow tract tachycardia. J. Am. Coll. Cardiol. 2008, 51, 731–739. [Google Scholar] [CrossRef]
- Perazzolo Marra, M.; Rizzo, S.; Bauce, B.; de Lazzari, M.; Pilichou, K.; Corrado, D.; Thiene, G.; Iliceto, S.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz 2015, 40, 600–606. [Google Scholar] [CrossRef]
- Matsuo, K.; Nishikimi, T.; Yutani, C.; Kurita, T.; Shimizu, W.; Taguchi, A.; Suyama, K.; Aihara, N.; Kamakura, S.; Kangawa, K.; et al. Diagnostic value of plasma levels of brain natriuretic peptide in arrhythmogenic right ventricular dysplasia. Circulation 1998, 98, 2433–2440. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Lu, M.; Hou, C.; Chen, X.; Wang, J.; Yin, G.; Chu, J.; Zhang, S.; Prasad, S.K.; Pu, J.; et al. Relation between n-terminal pro-brain natriuretic peptide and cardiac remodeling and function assessed by cardiovascular magnetic resonance imaging in patients with arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2015, 115, 341–347. [Google Scholar] [CrossRef]
- Martins, D.; Ovaert, C.; Khraiche, D.; Boddaert, N.; Bonnet, D.; Raimondi, F. Myocardial inflammation detected by cardiac mri in arrhythmogenic right ventricular cardiomyopathy: A paediatric case series. Int. J. Cardiol. 2018, 271, 81–86. [Google Scholar] [CrossRef]
- Wei, Y.J.; Huang, Y.X.; Shen, Y.; Cui, C.J.; Zhang, X.L.; Zhang, H.; Hu, S.S. Proteomic analysis reveals significant elevation of heat shock protein 70 in patients with chronic heart failure due to arrhythmogenic right ventricular cardiomyopathy. Mol. Cell Biochem. 2009, 332, 103–111. [Google Scholar] [CrossRef]
- Stadiotti, I.; Pompilio, G.; Maione, A.S.; Pilato, C.A.; D’Alessandra, Y.; Sommariva, E. Arrhythmogenic cardiomyopathy: What blood can reveal? Heart Rhythm 2019, 16, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.T.; Cogswell, R.; James, C.A.; Kang, G.; Pullinger, C.R.; Malloy, M.J.; Kane, J.P.; Wojciak, J.; Calkins, H.; Scheinman, M.M.; et al. Plasma bin1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012, 9, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A. Bin1: A new biomarker to track arvc? Heart Rhythm 2012, 9, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Broch, K.; Leren, I.S.; Saberniak, J.; Ueland, T.; Edvardsen, T.; Gullestad, L.; Haugaa, K.H. Soluble st2 is associated with disease severity in arrhythmogenic right ventricular cardiomyopathy. Biomarkers 2017, 22, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Oz, F.; Onur, I.; Elitok, A.; Ademoglu, E.; Altun, I.; Bilge, A.K.; Adalet, K. Galectin-3 correlates with arrhythmogenic right ventricular cardiomyopathy and predicts the risk of ventricular -arrhythmias in patients with implantable defibrillators. Acta Cardiol. 2017, 72, 453–459. [Google Scholar] [CrossRef]
- Ambros, V. Micrornas: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lai, E.C. Micro rnas are complementary to 3’ utr sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Latronico, M.V.; Condorelli, G. Micrornas and cardiac pathology. Nat. Rev. Cardiol. 2009, 6, 419–429. [Google Scholar] [CrossRef]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating micrornas: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: Microrna biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free micrornas in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, S.; Dong, T.; Yang, J.; Xie, Y.; Wu, Y.; Kang, K.; Hu, S.; Gou, D.; Wei, Y. Profiling of differentially expressed micrornas in arrhythmogenic right ventricular cardiomyopathy. Sci. Rep. 2016, 6, 28101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommariva, E.; D’Alessandra, Y.; Farina, F.M.; Casella, M.; Cattaneo, F.; Catto, V.; Chiesa, M.; Stadiotti, I.; Brambilla, S.; Dello Russo, A.; et al. Mir-320a as a potential novel circulating biomarker of arrhythmogenic cardiomyopathy. Sci. Rep. 2017, 7, 4802. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hsiao, Y.W.; Chang, S.L.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chiang, S.J.; Hu, Y.F.; Tuan, T.C.; Chao, T.F.; et al. Circulating micrornas in arrhythmogenic right ventricular cardiomyopathy with ventricular arrhythmia. Europace 2018, 20, f37–f45. [Google Scholar] [CrossRef] [PubMed]
- Bueno Marinas, M.; Celeghin, R.; Cason, M.; Bariani, R.; Frigo, A.C.; Jager, J.; Syrris, P.; Elliott, P.M.; Bauce, B.; Thiene, G.; et al. A microrna expression profile as non-invasive biomarker in a large arrhythmogenic cardiomyopathy cohort. Int. J. Mol. Sci. 2020, 21, 1536. [Google Scholar] [CrossRef] [Green Version]
- Gurha, P.; Chen, X.; Lombardi, R.; Willerson, J.T.; Marian, A.J. Knockdown of plakophilin 2 downregulates mir-184 through cpg hypermethylation and suppression of the e2f1 pathway and leads to enhanced adipogenesis in vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S.R.; Calway, T.; Harmon, C.; Farrell, P.; Kim, G.H. Microrna-130a regulation of desmocollin 2 in a novel model of arrhythmogenic cardiomyopathy. Microrna 2017, 6, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabatelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of wnt signaling and mirna dysregulation. Cardiovasc. Res. 2019, 8, 737. [Google Scholar] [CrossRef]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Rainer, J.; Meraviglia, V.; Blankenburg, H.; Piubelli, C.; Pramstaller, P.P.; Paolin, A.; Cogliati, E.; Pompilio, G.; Sommariva, E.; Domingues, F.S.; et al. The arrhythmogenic cardiomyopathy-specific coding and non-coding transcriptome in human cardiac stromal cells. BMC Genom. 2018, 19, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzzi, L.; Borin, D.; Gurha, P.; Lombardi, R.; Martinelli, V.; Weiss, M.; Andolfi, L.; Lazzarino, M.; Mestroni, L.; Marian, A.J.; et al. Knock down of plakophillin 2 dysregulates adhesion pathway through upregulation of mir200b and alters the mechanical properties in cardiac cells. Cells 2019, 8, 1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; da Graca Cabreira-Hansen, M.; Bell, A.; Fromm, R.R.; Willerson, J.T.; Marian, A.J. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ. Res. 2011, 109, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Khudiakov, A.A.; Smolina, N.A.; Perepelina, K.I.; Malashicheva, A.B.; Kostareva, A.A. Extracellular micrornas and mitochondrial DNA as potential biomarkers of arrhythmogenic cardiomyopathy. Biochem. Biokhimiia 2019, 84, 272–282. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Kassiri, Z.; Virag, J.A.I.; de Castro Bras, L.E.; Scherrer-Crosbie, M. Guidelines for measuring cardiac physiology in mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H733–H752. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| I. Global or Regional Dysfunction and Structural Alterations * | |
|---|---|
| Major | Minor |
| By 2D echo | By 2D echo |
| Regional RV akinesia, dyskinesia, or aneurysm and 1 of the following (end diastole): | Regional RV akinesia or dyskinesia and 1 of the following (end diastole): |
|
|
| By CMR | By CMR |
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following: | Regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following: |
|
|
| By RV angiography | |
| Regional RV akinesia, dyskinesia, or aneurysm | |
| II. Tissue Characterization of Wall | |
| Major | Minor |
| Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on EMB | Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on EMB |
| III. Repolarization Abnormalities | |
| Major | Minor |
|
|
| IV. Depolarization/Conduction Abnormalities | |
| Major | Minor |
|
|
| V. Arrhythmias | |
| Major | Minor |
|
|
| VI. Family History | |
| Major | Minor |
|
|
| Locus | Disease gene | Gene | Mode of Transmission | Reference | Comment |
|---|---|---|---|---|---|
| Desmosomal genes | |||||
| 17q21.2 | Plakoglobin | JUP | AD/AR | Mckoy et al. [23] | AR form: cardiocutaneous syndrome |
| 6p24.3 | Desmoplakin | DSP | AD/AR | Rampazzo et al. [20] | AR form: cardiocutaneous syndrome |
| 12p11.21 | Plakophilin-2 | PKP2 | AD/AR | Gerull et al. [24] | |
| 18q12.1 | Desmoglein-2 | DSG2 | AD/AR | Pilichou et al. [25] | |
| 18q12.1 | Desmocollin-2 | DSC2 | AD/AR | Syrris et al. [26] | |
| Non-desmosomal genes | |||||
| 1q43 | Cardiac Ryanodine Receptor 2 | RYR2 | AD | Tiso et al. [27] | CPVT |
| 14q24.3 | Transforming growth factor-beta-3 | TGFB3 | AD | Beffagna et al. [28] | Debated |
| 3p25.1 | Transmembrane Protein 43 | TMEM43 | AD | Merner et al. [29] | Limited to the founder p.Ser358Leu variant leading to fully disease penetrance. |
| 2q35 | Desmin | DES | AD | Van Tintelen et al. [30] | Overlap syndrome (DCM and HCM phenotype, early conduction disease) |
| 6q22.31 | Phospholamban | PLN | AD | Van der Zwaag et al. [31] | Limited to the founder p.Arg14del variant. |
| 2q31.2 | Titin | TTN | AD | Taylor et al. [32] | Overlap syndrome (early conduction disease, AF) |
| 1q22 | Lamin A/C | LMNA | AD | Quarta et al. [33] | Overlap syndrome (DCM, early conduction disease) |
| 14q11.2 | Myosin heavy chain 7 | MYH7 | De Bortoli et al. [34] | Debated overlap HCM | |
| Murray et al. [35] | |||||
| 3p21.31 | Myosin light Chain 3 | MYL3 | Murray et al. [35] | Debated overlap HCM | |
| 11p11.2 | Myosin binding protein C3 | MYBPC3 | Sakamoto et al. [36] | Debated overlap HCM | |
| Murray et al. [35] | |||||
| 10q23.2 | LIM domain binding 3 | LDB3 | Lopez-Ayala et al. [37] | Debated | |
| 3p22.2 | Sodium voltage-gated channel alpha subunit 5 | SCN5A | Te Riele et al. [38] | Debated overlap BrS | |
| 10q21.3 | Alpha-T-catenin | CTNNA3 | AD | Van Hengel et al. [39] | |
| 18q12.1 | Cadherin-2 | CDH2 | AD | Mayosi et al. [40] | |
| Turkowski et al. [41] | |||||
| 15q13.1 | Tight junction protein 1 | TJP1 | AD | De Bortoli et al. [42] | |
| 7q32.1 | Filamin C | FLNC | AD | Ortiz-Genga et al. [43] | Overlap syndrome (DCM and adverse outcome) |
| miRNA | Specimen | Differential Diagnosis | AUC | Pathways | Reference |
|---|---|---|---|---|---|
| miR-21-5p | RV myocardial tissue | AC vs. ctrl | 0.944 | Wnt and Hippo pathways | [63] |
| miR-135b | RV myocardial tissue | AC vs. ctrl | 0.936 | ||
| miR-320a | Plasma | AC vs. ctrl | / | / | [64] |
| AC vs. IVT | 0.69 | ||||
| miR-144-3p | Plasma | AC definite vs. crtl AC definite vs. AC borderline/possible AC definite vs. IVT | / | / | |
| miR-145-5p | Plasma | / | [65] | ||
| miR-185-5p | Plasma | AC recurrent VA vs. no VA | 0.728 0.619 0.740 0.891 | ||
| miR-494 | Plasma | Activation of caspase 3, apoptosis related | |||
| miR-122-5p | RV myocardial tissue and blood | AC definite vs. ctrl | 0.995 | Adherence junction, Hippo pathway, TFGβ signaling pathway, AC pathway, Wnt/β-catenin pathway | |
| miR-142-3p | RV myocardial tissue and blood | AC definite vs. HCM | 0.804 | [66] | |
| miR-182-5p | RV myocardial tissue and blood | AC definite vs. DCM | 0.917 | ||
| miR-183-5p | RV myocardial tissue and blood | AC definite vs. AC gen + phen | 0.825 | ||
| miR-133a-3p | RV myocardial tissue and blood | AC definite vs. BrS | 0.981 | ||
| miR-133b | RV myocardial tissue and blood | AC definite vs. Myocarditis | 0.978 |
| miRNA | Specimen | Pathways | Reference |
|---|---|---|---|
| miR-184 | HL-1Pkp2-shRNA |
| [67] |
| Mouse Nkx2.5-Cre:Pkp2shRNA | |||
| Mouse Myh6:JupTr | |||
| miR-130 | Mouse αMHC-miR130a (overexpressing miR-130a) |
| [68] |
| miR-499-5p | Mouse Dsg2 Tg-hWT and Tg-hQ (transgenic WT and Dsg2 p.Q558X) |
| [69] |
| miR-217-5p | |||
| miR-708-5p | |||
| miR-29b-3p | CStCs from AC patients |
| [70] |
| miR-21 | Cardiomyocytes derived from iPSCs from AC patients |
| [71] |
| miR-29b-2 | |||
| miR-378a | |||
| miR-1 | |||
| miR-133a | |||
| miR-200b | HL-1 PKP2 deficiency |
| [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bueno Marinas, M.; Celeghin, R.; Cason, M.; Thiene, G.; Basso, C.; Pilichou, K. The Role of MicroRNAs in Arrhythmogenic Cardiomyopathy: Biomarkers or Innocent Bystanders of Disease Progression? Int. J. Mol. Sci. 2020, 21, 6434. https://doi.org/10.3390/ijms21176434
Bueno Marinas M, Celeghin R, Cason M, Thiene G, Basso C, Pilichou K. The Role of MicroRNAs in Arrhythmogenic Cardiomyopathy: Biomarkers or Innocent Bystanders of Disease Progression? International Journal of Molecular Sciences. 2020; 21(17):6434. https://doi.org/10.3390/ijms21176434
Chicago/Turabian StyleBueno Marinas, Maria, Rudy Celeghin, Marco Cason, Gaetano Thiene, Cristina Basso, and Kalliopi Pilichou. 2020. "The Role of MicroRNAs in Arrhythmogenic Cardiomyopathy: Biomarkers or Innocent Bystanders of Disease Progression?" International Journal of Molecular Sciences 21, no. 17: 6434. https://doi.org/10.3390/ijms21176434