The Molecular Floodgates of Stress-Induced Senescence Reveal Translation, Signalling and Protein Activity Central to the Post-Mortem Proteome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
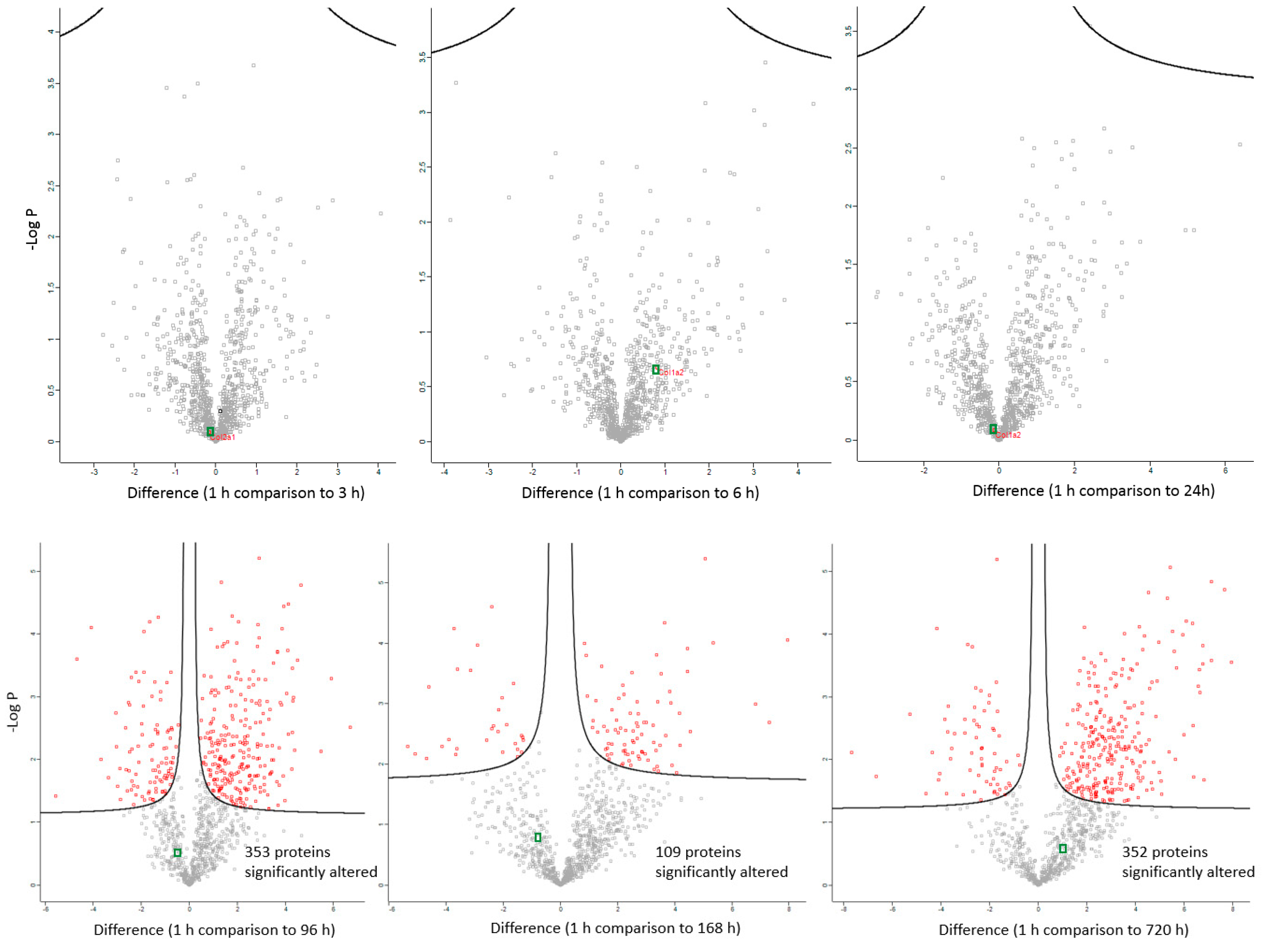
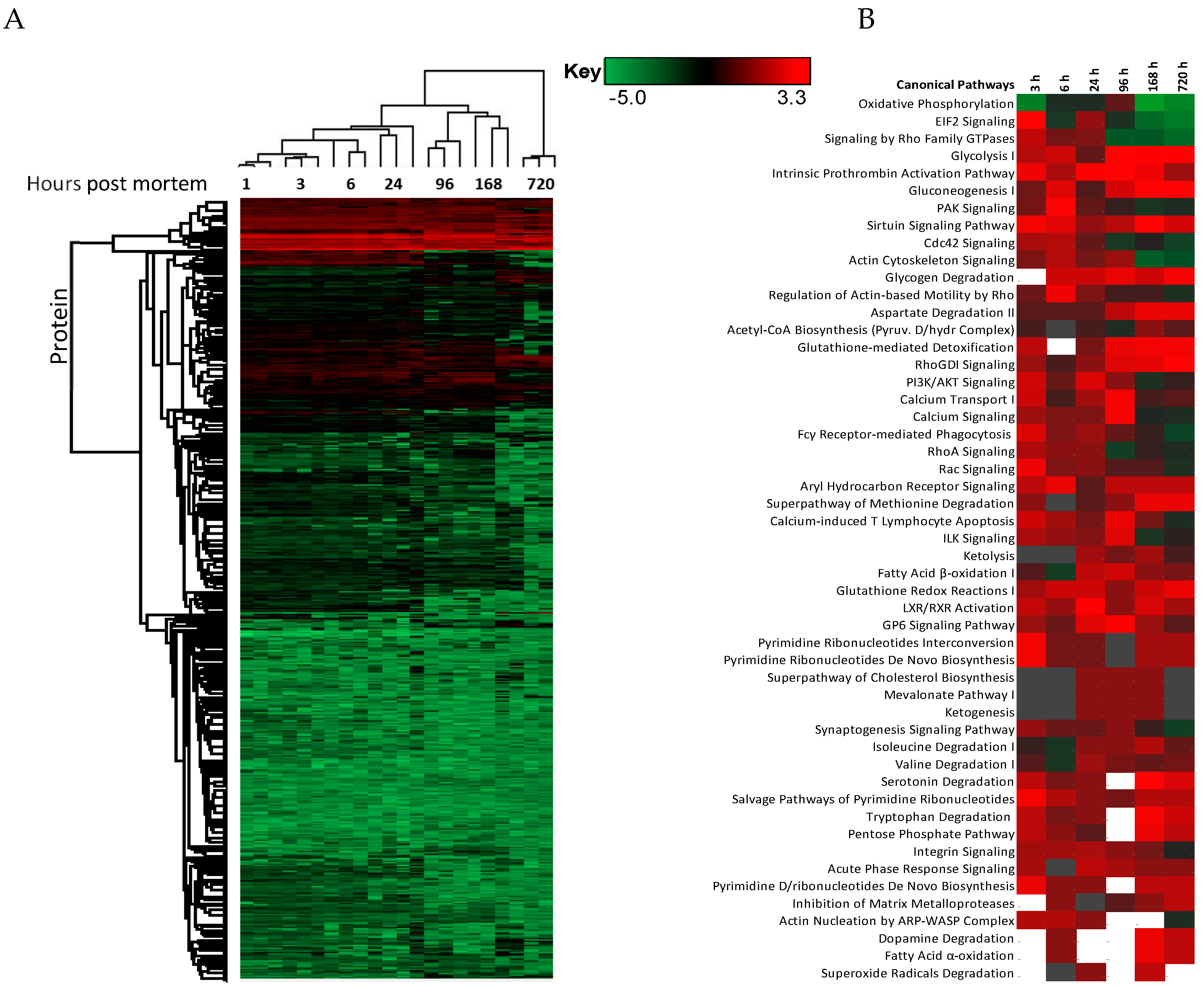
2. Results
3. Discussion
3.1. Relationship to Ageing and Senescence
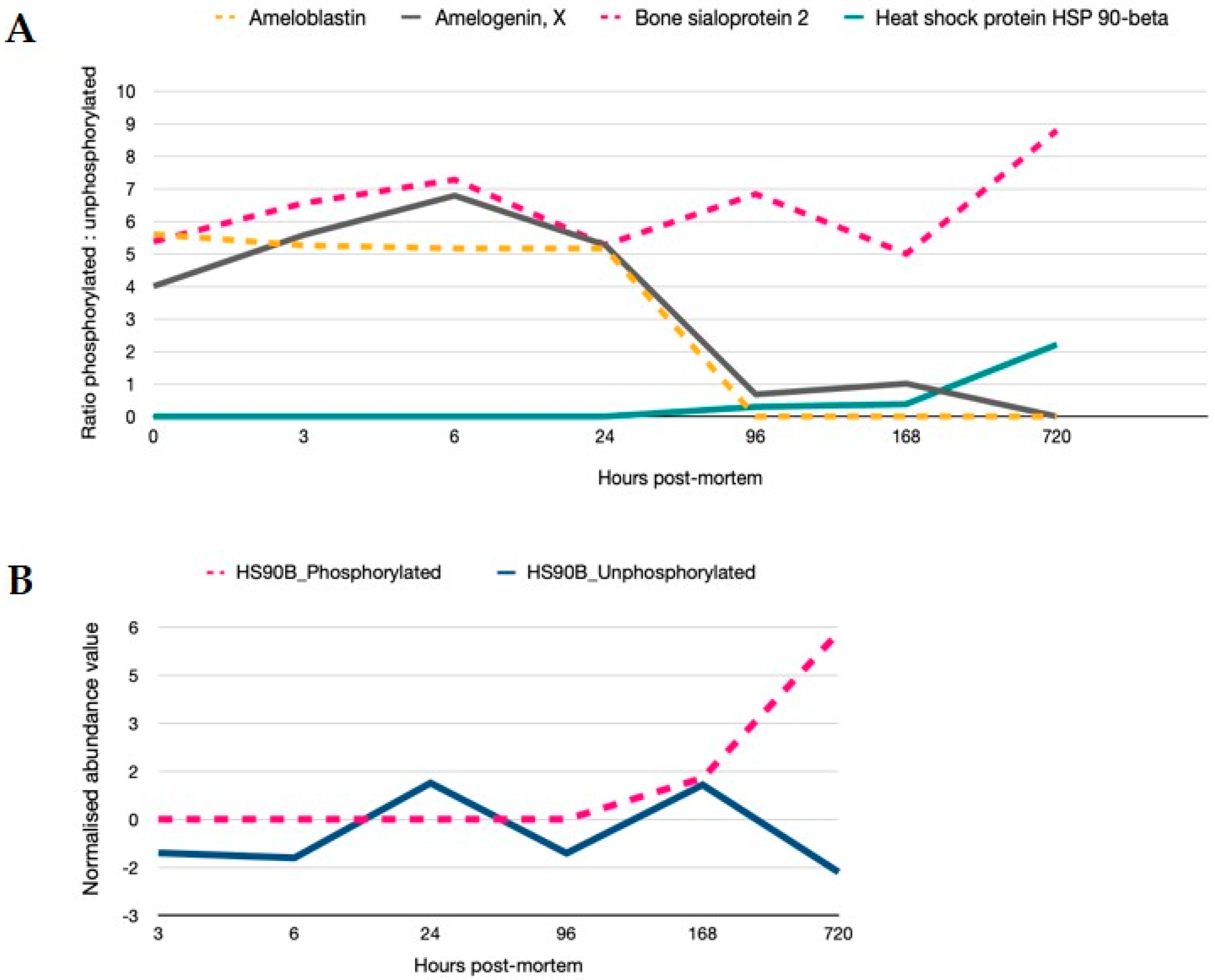
3.2. The Stress Response
3.3. Signalling Cascades in PM Time
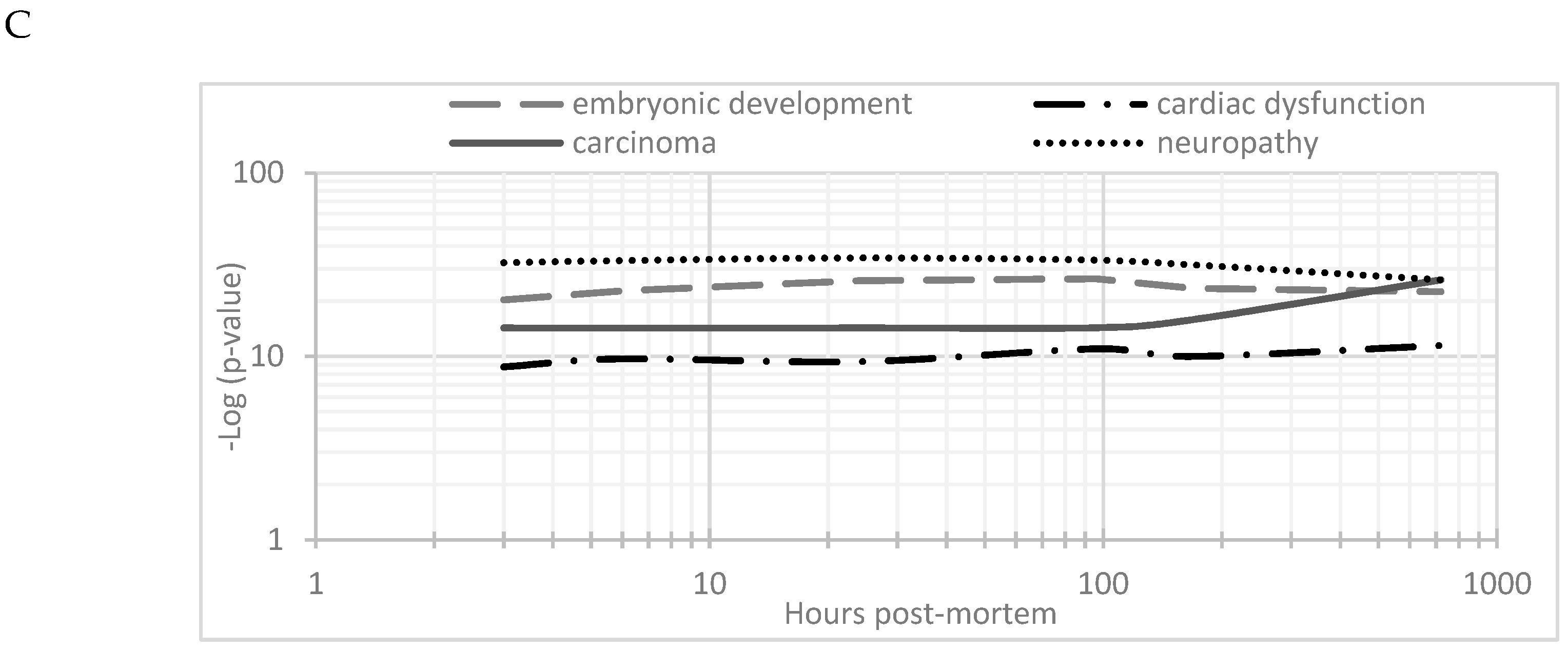
3.4. Disease Activation Pathways and Functional Enrichment
3.5. Commonalities between the “Death” Proteome and the Stress-Induced Senescence Phenotype
4. Materials and Methods
4.1. Experimental Design and Statistical Rationale
4.2. Sample Preparation
4.3. Mass Spectrometry of Samples
4.4. Protein Identification Relative Quantitation
4.5. Immunoblotting
4.6. PGM Colourimetric Activity Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PM | Post-Mortem |
| SIPS | Stress-induced premature senescence |
| RBP | ribosomal binding protein |
References
- Begg, S.; Vos, T.; Barker, B.; Stevenson, C.; Stanley, L.; Lopez, A.D. The Burden of Disease and Injury in Australia 2003; Australian Institute of Health and Welfare: Surry Hills, Australia, 2007; Available online: http://hdl.handle.net/10536/DRO/DU:30046702 (accessed on 9 July 2020).
- Hunter, M.C.; Pozhitkov, A.E.; Noble, P.A. Accurate predictions of postmortem interval using linear regression analyses of gene meter expression data. Forensic Sci. Int. 2017, 275, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, P.A.; Pozhitkov, A.E. Cryptic sequence features in the active postmortem transcriptome. BMC Genom. 2018, 19, 675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, P.G.; Muñoz-Aguirre, M.; Reverter, F.; Sá Godinho, C.P.; Sousa, A.; Amadoz, A.; Sodaei, R.; Hidalgo, M.R.; Pervouchine, D.; Carbonell-Caballero, J.; et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 2018, 9, 490. [Google Scholar] [CrossRef] [PubMed]
- Hadj-Moussa, H.; Watts, A.J.; Storey, K.B. Genes of the un-dead: Hibernation and death display different gene profiles. Fed. Eur. Biochem. Soc. Lett. 2019, 593, 527–532. [Google Scholar] [CrossRef]
- Zimmermann, A.; Tadic, J.; Kainz, K.; Hofer, S.J.; Bauer, M.A.; Carmona-Gutierrez, D.; Madeo, F. Transcriptional and epigenetic control of regulated cell death in yeast. Int. Rev. Cell Mol. Biol. 2020, 352, 55–82. [Google Scholar]
- Stampone, E.C.I.; Zullo, A.; Bencivenga, D.; Mancini, F.P.; Della Ragione, F.; Borriello, A. Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. Int. J. Mol. Sci. 2018, 19, 1055. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Mizumura, K.; Choi, A.M. The impact of autophagy on cell death modalities. Int. J. Cell Biol. 2014, 2014, 502676. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Silva, G.M.; Marcotte, E.M. Protein expression regulation under oxidative stress. Mol. Cell. Proteom. 2011, 10, M111.009217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozhitkov, A.E.; Neme, R.; Domazet-Lošo, T.; Leroux, B.G.; Soni, S.; Tautz, D.; Noble, P.A. Thanatotranscriptome: Genes actively expressed after organismal death. BioRxiv 2016, 058305. [Google Scholar] [CrossRef] [Green Version]
- Pozhitkov, A.E.; Neme, R.; Domazet-Lošo, T.; Leroux, B.G.; Soni, S.; Tautz, D.; Noble, P.A. Tracing the dynamics of gene transcripts after organismal death. Open Biol. 2017, 7, 160267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 255–270. [Google Scholar] [CrossRef] [Green Version]
- Powley, I.R.; Kondrashov, A.; Young, L.A.; Dobbyn, H.C.; Hill, K.; Cannell, I.G.; Stoneley, M.; Kong, Y.W.; Cotes, J.A.; Smith, G.C.; et al. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev. 2009, 23, 1207–1220. [Google Scholar] [CrossRef] [Green Version]
- Harvey, R.; Dezi, V.; Pizzinga, M.; Willis, A.E. Post-transcriptional control of gene expression following stress: The role of RNA-binding proteins. Biochem. Soc. Trans. 2017, 45, 1007–1014. [Google Scholar] [CrossRef]
- Cao, S.Q.; Zheng, H.; Sun, B.C.; Wang, Z.L.; Liu, T.; Guo, D.H.; Shen, Z.Y. Long non-coding RNA highly up-regulated in liver cancer promotes exosome secretion. World J. Gastroenterol. 2019, 25, 5283–5299. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Willimott, S.; Wagner, S.D. Post-transcriptional and post-translational regulation of Bcl2. Biochem. Soc. Trans. 2010, 38, 1571–1575. [Google Scholar] [CrossRef] [Green Version]
- Cagnetta, R.; Wong, H.H.; Frese, C.K.; Mallucci, G.R.; Krijgsveld, J.; Holt, C.E. Noncanonical Modulation of the eIF2 Pathway Controls an Increase in Local Translation during Neural Wiring. Mol. Cell 2019, 73, 474–489. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.K.; Bressler, K.; Patel, H.; Balasingam, N.; Thakor, N. Role of Eukaryotic Initiation Factors during Cellular Stress and Cancer Progression. J. Nucleic Acids 2016, 2016, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, D.A.N.; Kimball, S.R.; Cavener, D.R.; Jefferson, L.S. eIF2alpha kinases GCN2 and PERK modulate transcription and translation of distinct sets of mRNAs in mouse liver. Physiol. Genom. 2009, 38, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Noor, E.; Eden, E.; Milo, R.; Alon, U. Central Metabolism- A minimal walk between precursors. Mol. Cell 2010, 39, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Glycolysis Is an Energy-Conversion Pathway in Many Organisms; W H Freeman: New York, NY, USA, 2002. [Google Scholar]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acid Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Dobberstein, R.C.; Collins, M.J.; Craig, O.E.; Taylor, G.; Penkman, K.E.H.; Ritz-Timme, S. Archeological Collagen: Why worry about collagen diagenesis? Hist. Archaeol. 2009, 1, 31–42. [Google Scholar]
- Basisty, N.; Kale, A.; Patel, S.; Campisi, J.; Schilling, B. The power of proteomics to monitor senescence-associated secretory phenotypes and beyond: Toward clinical applications. Expert Rev. Proteom. 2020, 17, 297–308. [Google Scholar] [CrossRef]
- Bhadra, M.; Howell, P.; Dutta, S.; Heintz, C.; Mair, W.B.A. Alternative splicing in aging and longevity. Hum. Genet. 2020, 139, 357–369. [Google Scholar] [CrossRef]
- Ubaida-Mohien, C.L.A.; Gonzalez-Freire, M.; Tharakan, R.; Shardell, M.; Moaddel, R.; Semba, R.D.; Chia, C.W.; Gorospe, M.; Sen, R.; Ferrucci, L. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. eLife 2019, 8, e49874. [Google Scholar] [CrossRef]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaquin, N.M.A.; Rodier, F. Keeping the senescence secretome under control: Molecular reins on the senescence-associated secretory phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef]
- Kitchlu, A.; Dixon, S.; Dirk, J.S.; Chanchlani, R.; Vasilevska-Ristovska, J.; Borges, K.; Dipchand, A.I.; Ng, V.L.; Hebert, D.; Solomon, M.; et al. Elevated Risk of Cancer After Solid Organ Transplant in Childhood: A Population-based Cohort Study. Transplantation 2019, 103, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Webster, A.C.; Wong, G. Cancer in the transplant recipient. Cold Spring Harb. Perspect. Med. 2013, 3, a015677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombline, G.G.J.; Macoretta, N.; Zacher, M.; Emmrich, S.; Zhao, Y.; Seluanov, A.; Gorbunova, V. Proteomics of Long-Lived Mammals. Proteomics 2020, 20, e1800416. [Google Scholar] [CrossRef]
- de Magalhaes, J.P.; Toussaint, O. GenAge: A genomic and proteomic network map of human ageing. Fed. Eur. Biochem. Soc. Lett. 2004, 571, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, F.M.; Ahmad, Y.; Gierliński, M.; Charrière, F.; Lamont, D.; Scott, M.; Barton, G.; Lamond, A.I. A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell. Proteom. 2012, 11, M111.011429. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, J.A.; Endo, K.; Fujita, Y.; Hayashi, K.; Takinoue, M.; Saito, H.; Inoue, T. Feedback control of protein expression in mammalian cells by tunable synthetic translational inhibition. ACS Synth. Biol. 2012, 1, 83–88. [Google Scholar] [CrossRef]
- Lijima, T.; Lijima, Y.; Witte, H.; Scheiffele, P. Neuronal cell type–specific alternative splicing is regulated by the KH domain protein SLM1. J. Cell Biol. 2014, 204, 331. [Google Scholar]
- Li, Y.; Liang, R.; Sun, M.; Li, Z.; Sheng, H.; Wang, J.; Xu, P.; Liu, S.; Yang, W.; Lu, B.; et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020, 478, 82–92. [Google Scholar] [CrossRef]
- Ma, D.W.J.; Zhao, Y.; Lee, W.N.; Xiao, J.; Go, V.L.; Wang, Q.; Recker, R.R.; Xiao, G.G. Inhibition of glycogen phosphorylation induces changes in cellular proteome and signaling pathways in MIA pancreatic cancer cells. Pancreas 2012, 41, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Favaro, E.B.K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Günther, U.L.; Buffa, F.M.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-SOD, CAT, GPX: Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2017, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Lemeshko, V.V. VDAC electronics: 3. VDAC-Creatine kinase-dependent generation of the outer membrane potential in respiring mitochondria. Biochim. Biophys. Acta 2016, 1858, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.L.; Li, Y.Q.; Gu, F.; Liu, F.F.; Li, W.D.; Zhang, X.M.; Fu, L. Overexpression of ubiquitous mitochondrial creatine kinase (uMtCK) accelerates tumor growth by inhibiting apoptosis of breast cancer cells and is associated with a poor prognosis in breast cancer patients. Biochem. Biophys. Res. Commun. 2012, 427, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Dierick, J.F.; Kalume, D.E.; Wenders, F.; Salmon, M.; Dieu, M.; Raes, M.; Roepstorff, P.; Toussaint, O. Identification of 30 protein species involved in replicative senescence and stress-induced premature senescence. Fed. Eur. Biochem. Soc. Lett. 2002, 531, 499–504. [Google Scholar] [CrossRef]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, X.; Zhuo, W.; Fu, Y.; Shi, H.; Liang, Y.; Tong, M.; Chang, G.; Luo, Y. The regulatory mechanism of Hsp90alpha secretion and its function in tumor malignancy. Proc. Natl. Acad. Sci. USA 2009, 106, 21288–21293. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-G.; Gilmore, R.; Leone, G.; Coffey, M.C.; Weber, B.; Lee, P.W.K. Hsp90 Phosphorylation Is Linked to Its Chaperoning Function. J. Biol. Chem. 2001, 276, 32822–32827. [Google Scholar] [CrossRef] [Green Version]
- Coburn, C.; Allman, E.; Mahanti, P.; Benedetto, A.; Cabreiro, F.; Pincus, Z.; Matthijssens, F.; Araiz, C.; Mandel, A.; Vlachos, M.; et al. Anthranilate fluorescence marks a calcium-propagated necrotic wave that promotes organismal death in C. elegans. PLoS Biol. 2013, 11, e1001613. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [PubMed] [Green Version]
- Burton, D.G.A.; Kizhanovsky, V. Physiological and pathological consequences of cellular senescence. Cell Mol. Life Sci. 2014, 71, 4373–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, T.; Debacq-Chainiaux, F.; Chrétien, A.; Bastina, C.; Dabée, A.-F.; Bertholet, V.; Remacle, J.; Toussaint, O. Comparison of replicative senescence and stress-induced premature senescence combining differential display and low-density DNA arrays. Fed. Eur. Biochem. Soc. Lett. 2005, 579, 3651–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghuram, G.V.; Mishra, P.K. Stress induced premature senescence: A new culprit in ovarian tumorigenesis? Indian J. Med. Res. 2014, 140 (Suppl. 1), S120–S129. [Google Scholar] [PubMed]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell. Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Curnoe, D.; Bustamante, S.; Mendoza, R.; Shoocongdej, R.; Adler, L.; Baker, A.; Chintakanon, K.; Boel, C.; Tacon, P.S.C. Analysis of the Preserved Amino Acid Bias in Peptide Profiles of Iron Age Teeth from a Tropical Environment Enable Sexing of Individuals Using Amelogenin MRM. Proteomics 2019, 19, 1800341. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.L.; Whitmore, G.A. Power and sample size for DNA microarray studies. Stat. Med. 2002, 21, 3543–3570. [Google Scholar] [CrossRef]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef]
- Lundgren, D.H.; Hwang, S.I.; Wu, L.; Han, D.K. Role of spectral counting in quantitative proteomics. Exp. Rev. Prot. 2010, 7, 39–53. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Carlson, A.; Sinitcyn, P.; Mann, M.; Cox, J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 2015, 15, 1453–1456. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasinger, V.C.; Curnoe, D.; Boel, C.; Machin, N.; Goh, H.M. The Molecular Floodgates of Stress-Induced Senescence Reveal Translation, Signalling and Protein Activity Central to the Post-Mortem Proteome. Int. J. Mol. Sci. 2020, 21, 6422. https://doi.org/10.3390/ijms21176422
Wasinger VC, Curnoe D, Boel C, Machin N, Goh HM. The Molecular Floodgates of Stress-Induced Senescence Reveal Translation, Signalling and Protein Activity Central to the Post-Mortem Proteome. International Journal of Molecular Sciences. 2020; 21(17):6422. https://doi.org/10.3390/ijms21176422
Chicago/Turabian StyleWasinger, Valerie C., Darren Curnoe, Ceridwen Boel, Naomi Machin, and Hsiao Mei Goh. 2020. "The Molecular Floodgates of Stress-Induced Senescence Reveal Translation, Signalling and Protein Activity Central to the Post-Mortem Proteome" International Journal of Molecular Sciences 21, no. 17: 6422. https://doi.org/10.3390/ijms21176422