Cellular Signaling and Anti-Apoptotic Effects of Prolactin-Releasing Peptide and Its Analog on SH-SY5Y Cells

Abstract
:1. Introduction
2. Results
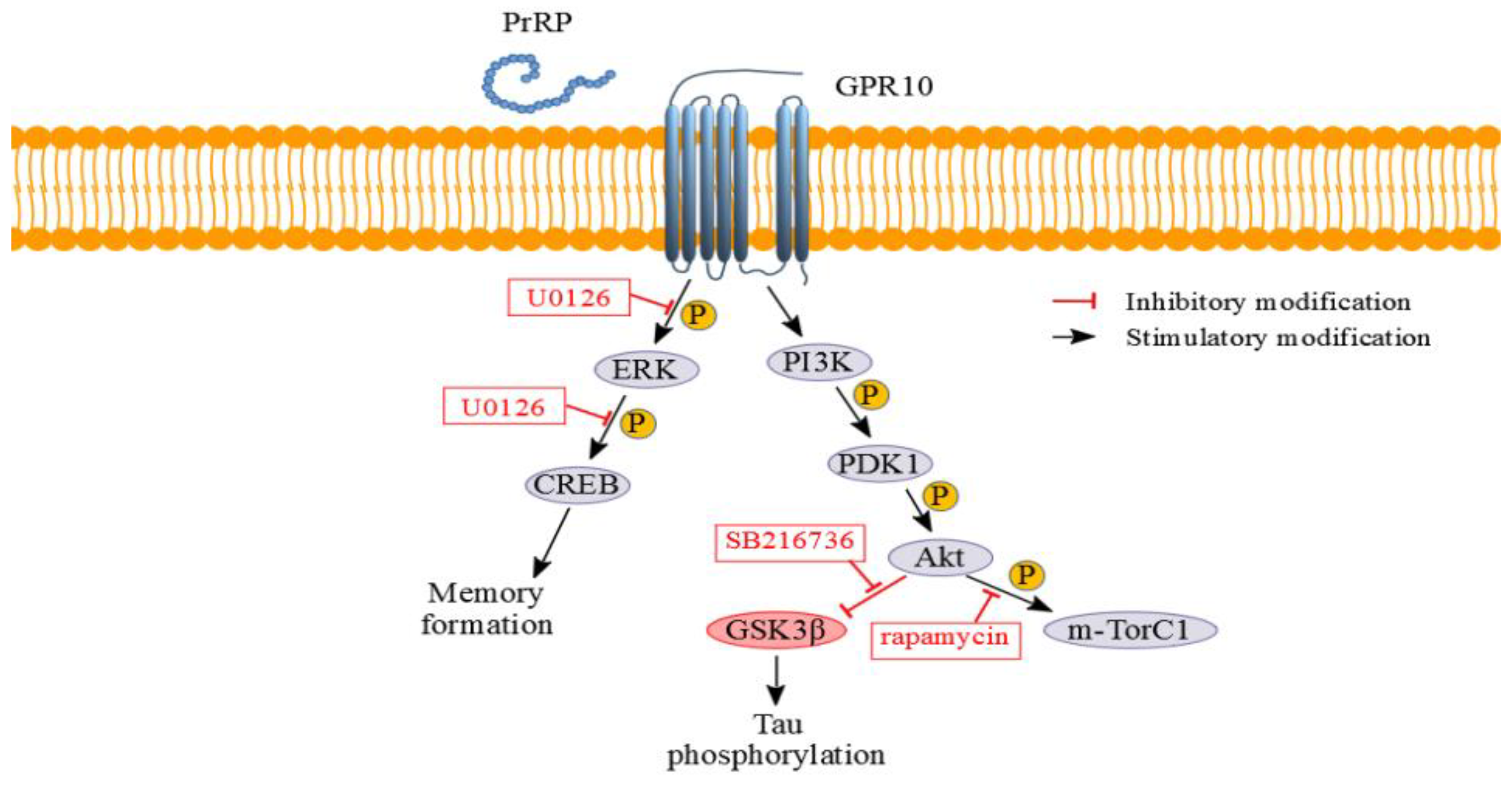
2.1. PrRP31 and Palm11-PrRP31 Activated the PI3K/Akt Signaling Pathway in SH-SY5Y Cells
2.2. PrRP31 and Palm11-PrRP31 Activated the ERK-CREB Signaling Pathway in SH-SY5Y Cells
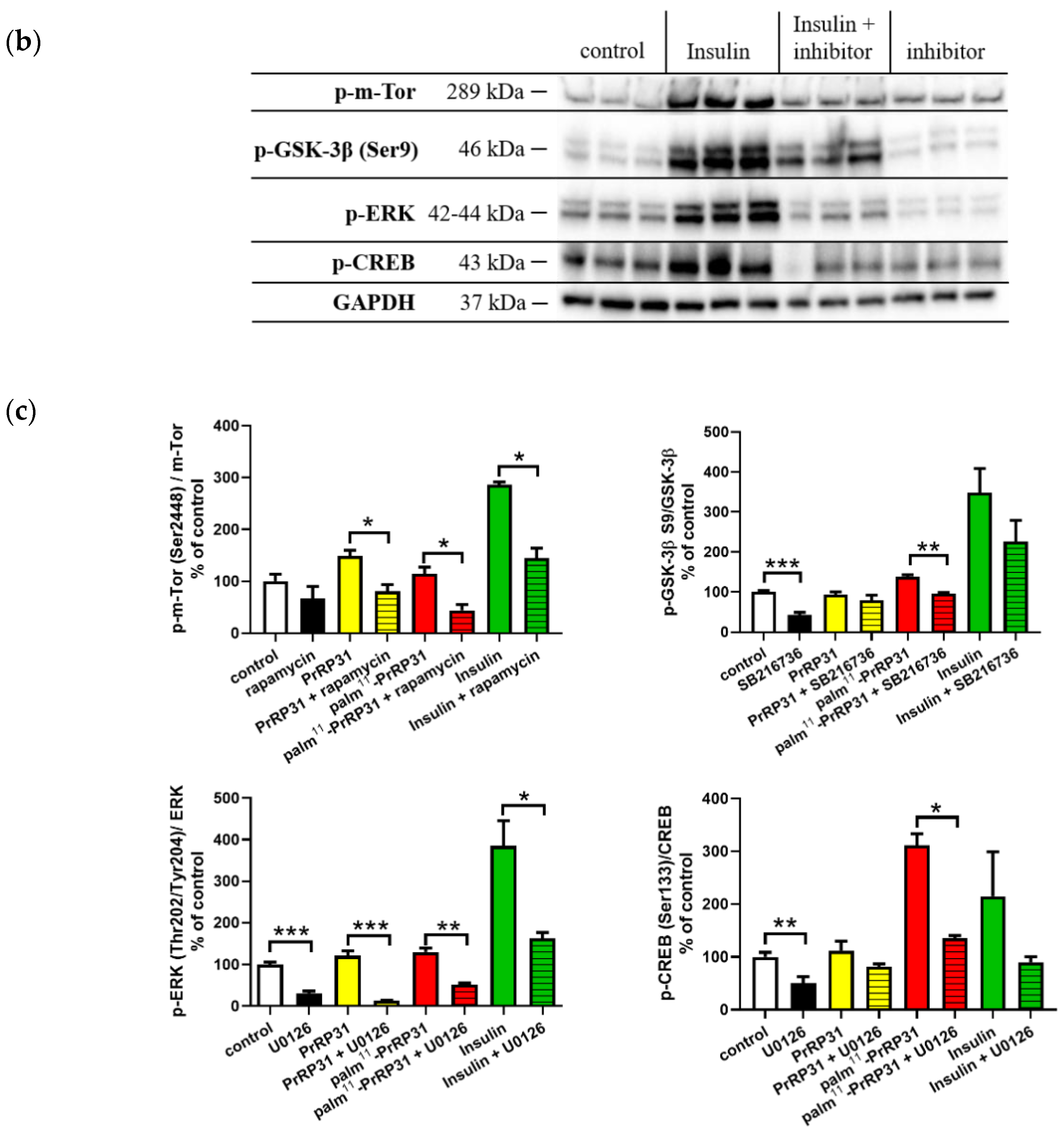
2.3. Inhibitors of Signaling Pathways Proved That Activation of Signaling Pathways Is Mediated Specifically via PrRP31 and Its Palmitoylated Analog
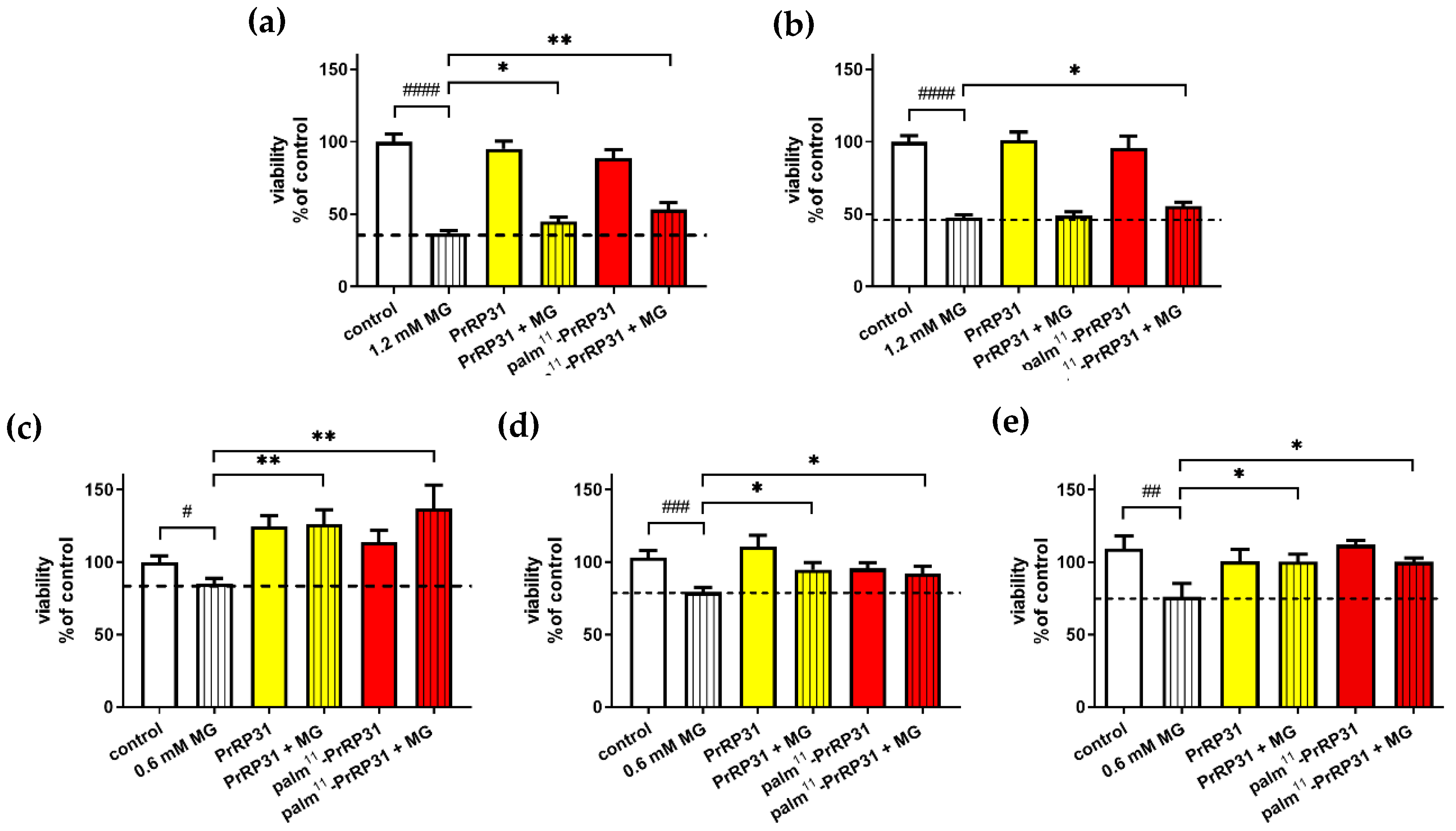
2.4. Methylglyoxal-Induced Cytotoxicity
2.4.1. PrRP31 and Its Palmitoylated Analog Protected SH-SY5Y Cells from the Cytotoxic Effects of MG
2.4.2. PrRP and Its Palmitoylated Analog Increased the Viability of SH-SY5Y Cells after 16 h of Exposure to MG
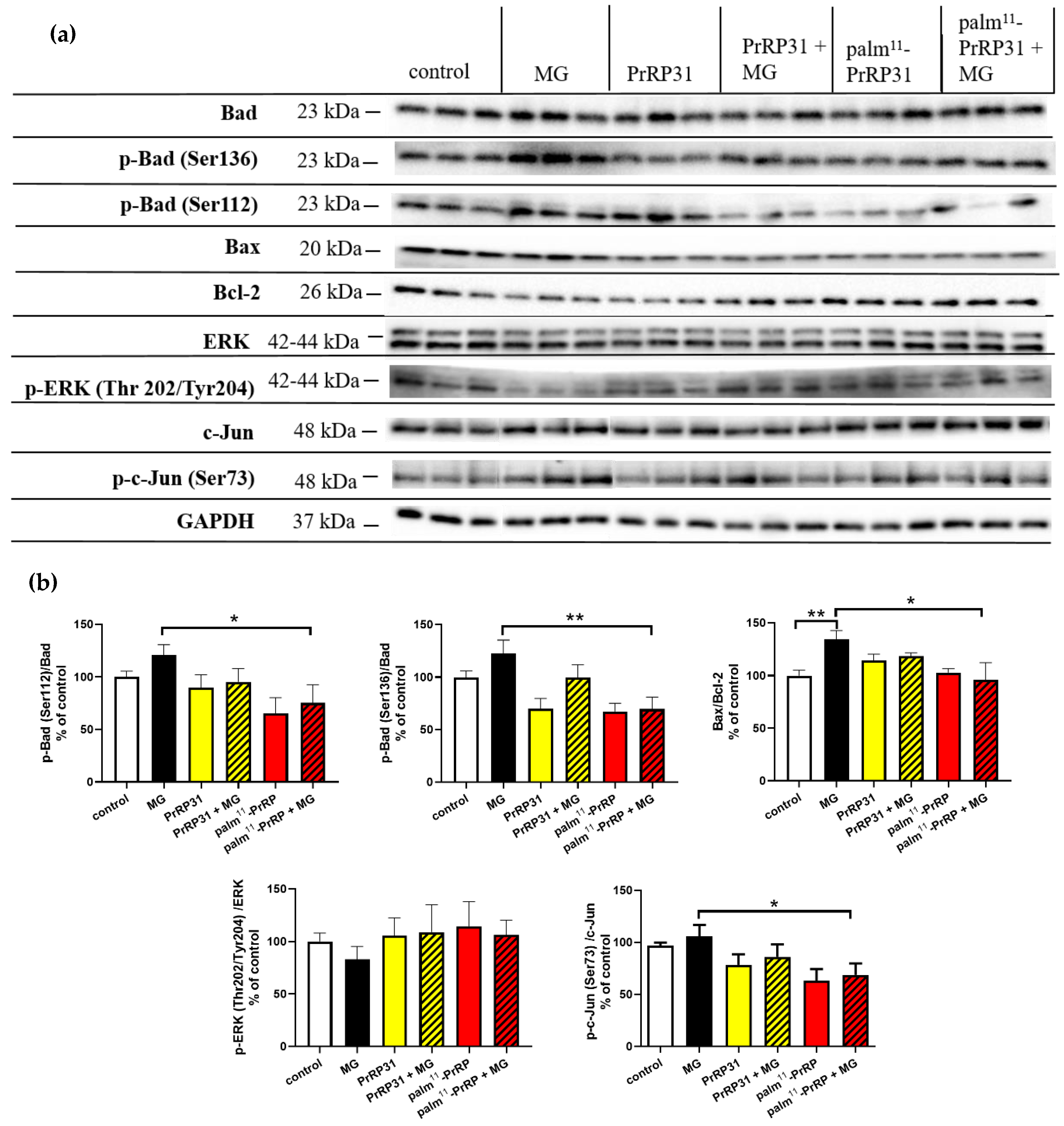
2.5. PrRP31 and Palm11-PrRP31 Induced Anti-Apoptotic Signaling in SH-SY5Y Cells
2.6. PrRP31 and Palm11-PrRP31 Attenuated Tau Hyperphosphorylation at Different Epitopes in SH-SY5Y Cells
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Chemicals
4.3. Cell Lines
4.4. Differentiation of SH-SY5Y Cells
4.5. Cell Viability Measurement
4.6. Western Blotting
4.7. Analysis of Data and Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bjursell, M.; Lenneras, M.; Goransson, M.; Elmgren, A.; Bohlooly, Y.M. GPR10 deficiency in mice results in altered energy expenditure and obesity. Biochem. Biophys. Res. Commun. 2007, 363, 633–638. [Google Scholar] [CrossRef]
- Pražienková, V.; Popelová, A.; Kuneš, J.; Maletínská, L. Prolactin-Releasing Peptide: Physiological and Pharmacological Properties. Int. J. Mol. Sci. 2019, 20, 5297. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, C.B.; Celsi, F.; Brennand, J.; Luckman, S.M. Alternative role for prolactin-releasing peptide in the regulation of food intake. Nat. Neurosci. 2000, 3, 645–646. [Google Scholar] [CrossRef]
- Hinuma, S.; Habata, Y.; Fujii, R.; Kawamata, Y.; Hosoya, M.; Fukusumi, S.; Kitada, C.; Masuo, Y.; Asano, T.; Matsumoto, H.; et al. A prolactin-releasing peptide in the brain. Nature 1998, 393, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Kuneš, J.; Pražienková, V.; Popelová, A.; Mikulášková, B.; Zemenová, J.; Maletínská, L. Prolactin-releasing peptide: A new tool for obesity treatment. J. Endocrinol. 2016, 230, R51–R58. [Google Scholar] [CrossRef]
- Maletinska, L.; Nagelova, V.; Ticha, A.; Zemenova, J.; Pirnik, Z.; Holubova, M.; Spolcova, A.; Mikulaskova, B.; Blechova, M.; Sykora, D.; et al. Novel lipidized analogs of prolactin-releasing peptide have prolonged half-lives and exert anti-obesity effects after peripheral administration. Int. J. Obes. 2015, 39, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Holubova, M.; Zemenova, J.; Mikulaskova, B.; Panajotova, V.; Stohr, J.; Haluzik, M.; Kunes, J.; Zelezna, B.; Maletinska, L. Palmitoylated PrRP analog decreases body weight in DIO rats but not in ZDF rats. J. Endocrinol. 2016, 229, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Mikulaskova, B.; Holubova, M.; Prazienkova, V.; Zemenova, J.; Hruba, L.; Haluzik, M.; Zelezna, B.; Kunes, J.; Maletinska, L. Lipidized prolactin-releasing peptide improved glucose tolerance in metabolic syndrome: Koletsky and spontaneously hypertensive rat study. Nutr. Diabetes 2018, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spolcova, A.; Mikulaskova, B.; Holubova, M.; Nagelova, V.; Pirnik, Z.; Zemenova, J.; Haluzik, M.; Zelezna, B.; Galas, M.C.; Maletinska, L. Anorexigenic lipopeptides ameliorate central insulin signaling and attenuate tau phosphorylation in hippocampi of mice with monosodium glutamate-induced obesity. J. Alzheimers Dis. 2015, 45, 823–835. [Google Scholar] [CrossRef]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Begard, S.; Brion, J.P.; Hamdane, M.; Buee, L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Popelova, A.; Prazienkova, V.; Neprasova, B.; Kasperova, B.J.; Hruba, L.; Holubova, M.; Zemenova, J.; Blum, D.; Zelezna, B.; Galas, M.C.; et al. Novel Lipidized Analog of Prolactin-Releasing Peptide Improves Memory Impairment and Attenuates Hyperphosphorylation of Tau Protein in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2018, 62, 1725–1736. [Google Scholar] [CrossRef]
- Holubová, M.; Hrubá, L.; Popelová, A.; Bencze, M.; Pražienková, V.; Gengler, S.; Kratochvílová, H.; Haluzík, M.; Železná, B.; Kuneš, J.; et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of β-amyloid pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef]
- Kimura, A.; Ohmichi, M.; Tasaka, K.; Kanda, Y.; Ikegami, H.; Hayakawa, J.; Hisamoto, K.; Morishige, K.; Hinuma, S.; Kurachi, H.; et al. Prolactin-releasing peptide activation of the prolactin promoter is differentially mediated by extracellular signal-regulated protein kinase and c-Jun N-terminal protein kinase. J. Biol. Chem. 2000, 275, 3667–3674. [Google Scholar] [CrossRef] [Green Version]
- Maletinska, L.; Ticha, A.; Nagelova, V.; Spolcova, A.; Blechova, M.; Elbert, T.; Zelezna, B. Neuropeptide FF analog RF9 is not an antagonist of NPFF receptor and decreases food intake in mice after its central and peripheral administration. Brain Res. 2013, 1498, 33–40. [Google Scholar] [CrossRef]
- Hayakawa, J.; Ohmichi, M.; Tasaka, K.; Kanda, Y.; Adachi, K.; Nishio, Y.; Hisamoto, K.; Mabuchi, S.; Hinuma, S.; Murata, Y. Regulation of the PRL promoter by Akt through cAMP response element binding protein. Endocrinology 2002, 143, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9 (Suppl 3), 309–317. [Google Scholar] [CrossRef]
- Wickelgren, I. Tracking insulin to the mind. Science 1998, 280, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [Green Version]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Bosch-Morató, M.; Valls-Comamala, V.; Muñoz, F.J. Methylglyoxal reduces mitochondrial potential and activates Bax and caspase-3 in neurons: Implications for Alzheimer’s disease. Neurosci. Lett. 2014, 580, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; Xie, J.Z.; Jiang, X.; Lv, B.L.; Cheng, X.S.; Du, L.L.; Zhang, J.Y.; Wang, J.Z.; Zhou, X.W. Methylglyoxal induces tau hyperphosphorylation via promoting AGEs formation. Neuromolecular Med. 2012, 14, 338–348. [Google Scholar] [CrossRef]
- Chapman, C.D.; Schioth, H.B.; Grillo, C.A.; Benedict, C. Intranasal insulin in Alzheimer’s disease: Food for thought. Neuropharmacology 2018, 136, 196–201. [Google Scholar] [CrossRef]
- Maixnerová, J.; Špolcová, A.; Pýchová, M.; Blechová, M.; Elbert, T.; Řezáčová, M.; Železná, B.; Maletínská, L. Characterization of prolactin-releasing peptide: Binding, signaling and hormone secretion in rodent pituitary cell lines endogenously expressing its receptor. Peptides 2011, 32, 811–817. [Google Scholar] [CrossRef]
- Holubova, M.; Hruba, L.; Neprasova, B.; Majercikova, Z.; Lacinova, Z.; Kunes, J.; Maletinska, L.; Zelezna, B. Prolactin-releasing peptide improved leptin hypothalamic signaling in obese mice. J. Mol. Endocrinol. 2018, 60, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korinkova, L.; Holubova, M.; Neprasova, B.; Hruba, L.; Prazienkova, V.; Bencze, M.; Haluzik, M.; Kunes, J.; Maletinska, L.; Zelezna, B. Synergistic effect of leptin and lipidized PrRP on metabolic pathways in ob/ob mice. J. Mol. Endocrinol. 2020, 64, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Varghese, B.V.; Koohestani, F.; McWilliams, M.; Colvin, A.; Gunewardena, S.; Kinsey, W.H.; Nowak, R.A.; Nothnick, W.B.; Chennathukuzhi, V.M. Loss of the repressor REST in uterine fibroids promotes aberrant G protein-coupled receptor 10 expression and activates mammalian target of rapamycin pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 2187–2192. [Google Scholar] [CrossRef] [Green Version]
- Sauter, A.; Goldstein, M.; Engel, J.; Ueta, K. Effect of insulin on central catecholamines. Brain Res. 1983, 260, 330–333. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Tamatani, M.; Matsuzaki, H.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001, 276, 5256–5264. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of transcription factor FKHRL1 is mediated by phosphatidylinositol 3-kinase/Akt kinase and role of this pathway in insulin-like growth factor-1-induced survival of cultured hippocampal neurons. Mol. Pharmacol. 2002, 62, 225–233. [Google Scholar] [CrossRef]
- Beattie, M.S.; Li, Q.; Bresnahan, J.C. Cell death and plasticity after experimental spinal cord injury. Prog. Brain Res. 2000, 128, 9–21. [Google Scholar]
- Man, H.Y.; Lin, J.W.; Ju, W.H.; Ahmadian, G.; Liu, L.; Becker, L.E.; Sheng, M.; Wang, Y.T. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 2000, 25, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.A.; Birnbaum, M.J. A role for the serine/threonine kinase, Akt, in insulin-stimulated glucose uptake. Biochem. Soc. Trans. 1997, 25, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Huang, C.C.; Hsu, K.S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011, 61, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys. Acta 2008, 1784, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, J.; Sheng, M. The growing role of mTOR in neuronal development and plasticity. Mol. Neurobiol. 2006, 34, 205–219. [Google Scholar] [CrossRef]
- Franco, R.; Martinez-Pinilla, E.; Navarro, G.; Zamarbide, M. Potential of GPCRs to modulate MAPK and mTOR pathways in Alzheimer’s disease. Prog. Neurobiol. 2017, 149–150, 21–38. [Google Scholar] [CrossRef]
- Nicolia, V.; Fuso, A.; Cavallaro, R.A.; Di Luzio, A.; Scarpa, S. B vitamin deficiency promotes tau phosphorylation through regulation of GSK3beta and PP2A. J. Alzheimers Dis. 2010, 19, 895–907. [Google Scholar] [CrossRef]
- Baum, L.; Hansen, L.; Masliah, E.; Saitoh, T. Glycogen synthase kinase 3 alteration in alzheimer disease is related to neurofibrillary tangle formation. Mol. Chem. Neuropathol. 1996, 29, 253–261. [Google Scholar] [CrossRef]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [Green Version]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [Green Version]
- Scott Bitner, R. Cyclic AMP response element-binding protein (CREB) phosphorylation: A mechanistic marker in the development of memory enhancing Alzheimer’s disease therapeutics. Biochem. Pharmacol. 2012, 83, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.J.; Qian, X.; O’Rourke, D.M. Sustained mitogen-activated protein kinase activation is induced by transforming erbB receptor complexes. DNA Cell Biol. 1999, 18, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Maletinska, L.; Popelova, A.; Zelezna, B.; Bencze, M.; Kunes, J. The impact of anorexigenic peptides in experimental models of Alzheimer’s disease pathology. J. Endocrinol. 2019, 240, R47–R72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of methylglyoxal in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [Green Version]
- Bellier, J.; Nokin, M.J.; Larde, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcene, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef]
- Sharma, M.K.; Jalewa, J.; Hölscher, C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J. Neurochem. 2014, 128, 459–471. [Google Scholar] [CrossRef]
- Popelová, A.; Kákonová, A.; Hrubá, L.; Kuneš, J.; Maletínská, L.; Železná, B. Potential neuroprotective and anti-apoptotic properties of a long-lasting stable analog of ghrelin: An in vitro study using SH-SY5Y cells. Physiol. Res. 2018, 67, 339–346. [Google Scholar] [CrossRef]
- Salakou, S.; Kardamakis, D.; Tsamandas, A.C.; Zolota, V.; Apostolakis, E.; Tzelepi, V.; Papathanasopoulos, P.; Bonikos, D.S.; Papapetropoulos, T.; Petsas, T.; et al. Increased Bax/Bcl-2 ratio up-regulates caspase-3 and increases apoptosis in the thymus of patients with myasthenia gravis. In Vivo 2007, 21, 123–132. [Google Scholar]
- Stickles, X.B.; Marchion, D.C.; Bicaku, E.; Al Sawah, E.; Abbasi, F.; Xiong, Y.; Bou Zgheib, N.; Boac, B.M.; Orr, B.C.; Judson, P.L.; et al. BAD-mediated apoptotic pathway is associated with human cancer development. Int. J. Mol. Med. 2015, 35, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Vogt, P.K. Fortuitous convergences: The beginnings of JUN. Nat. Rev. Cancer 2002, 2, 465–469. [Google Scholar] [CrossRef]
- Du, J.; Suzuki, H.; Nagase, F.; Akhand, A.A.; Yokoyama, T.; Miyata, T.; Kurokawa, K.; Nakashima, I. Methylglyoxal induces apoptosis in Jurkat leukemia T cells by activating c-Jun N-terminal kinase. J. Cell Biochem. 2000, 77, 333–344. [Google Scholar] [CrossRef]
- Leppa, S.; Bohmann, D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene 1999, 18, 6158–6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Heimfarth, L.; Loureiro, S.O.; Pierozan, P.; de Lima, B.O.; Reis, K.P.; Torres, E.B.; Pessoa-Pureur, R. Methylglyoxal-induced cytotoxicity in neonatal rat brain: A role for oxidative stress and MAP kinases. Metab. Brain Dis. 2013, 28, 429–438. [Google Scholar] [CrossRef]
- Pražienková, V.; Schirmer, C.; Holubová, M.; Železná, B.; Kuneš, J.; Galas, M.C.; Maletínská, L. Lipidized Prolactin-Releasing Peptide Agonist Attenuates Hypothermia-Induced Tau Hyperphosphorylation in Neurons. J. Alzheimers Dis. 2019, 67, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Prazienkova, V.; Holubova, M.; Pelantova, H.; Buganova, M.; Pirnik, Z.; Mikulaskova, B.; Popelova, A.; Blechova, M.; Haluzik, M.; Zelezna, B.; et al. Impact of novel palmitoylated prolactin-releasing peptide analogs on metabolic changes in mice with diet-induced obesity. PLoS ONE 2017, 12, e0183449. [Google Scholar] [CrossRef] [PubMed]
- Maletínská, L.; Špolcová, A.; Maixnerová, J.; Blechová, M.; Železná, B. Biological properties of prolactin-releasing peptide analogs with a modified aromatic ring of a C-terminal phenylalanine amide. Peptides 2011, 32, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 53193. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analogs | Sequence |
|---|---|
| PrRP31 | SRTHRHSMEIR11TPDINPAWYASRGIRPVGRF-NH2 |
| Palm11-PrRP31 | SRTHRHSMEIK11 (γ-E (N-palm))TPDINPAWYASRGIRPVGRF-NH2 |
| Scrambled-palm11-PrRP31 | GHFTHSIRMI K11 (γ-E (N-palm))TPRNASVYARPCitDWWGICitPES |
| Antibody | Blocking | Dilution for Western Blot | Provider |
|---|---|---|---|
| Akt rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-Akt (Ser473) rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-Akt (Thr308) rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| Bad rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-Bad (Ser112) rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-Bad (Ser136) rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| Bax rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| Bcl2 rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| CREB mouse Ab | 5% milk | 1:1000, 5% milk | Cell signaling Technology, Beverly MA, USA |
| p-CREB (Ser133) mouse Ab | 5% milk | 1:1000, 5% milk | Cell signaling Technology, Beverly MA, USA |
| ERK mouse Ab | 5% milk | 1:2000, 5% milk | Cell signaling Technology, Beverly MA, USA |
| p-ERK (Thr202/Tyr204) mouse Ab | 5% milk | 1:2000, 5% milk | Cell signaling Technology, Beverly MA, USA |
| GSK-3β rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-GSK-3β (Ser9) rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| GAPDH mouse Ab | 5% milk | 1:1000, 5% milk | Cell signaling Technology, Beverly MA, USA |
| c-Jun rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-c-Jun (Ser73) rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| m-Tor rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-m-Tor (Ser2448) rabbit Ab | 5% BSA | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| PDK1 rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| p-PDK1 (Ser241) rabbit AB | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| PI3K p85 rabbit Ab | 5% milk | 1:1000, 5% BSA | Cell signaling Technology, Beverly MA, USA |
| Tau 1 mouse Ab | 5% milk | 1:10000, 5% milk | Merck Millipore, Burlington MA, USA |
| Tau 5 mouse Ab | 5% milk | 1:5000, 5% milk | Thermo Fisher Scientific Inc., Waltham, MA, USA |
| p-Tau (Ser396) rabbit Ab | 5% BSA | 1:10000, 5% BSA | Thermo Fisher Scientific Inc., Waltham, MA, USA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmeškalová, A.; Popelová, A.; Exnerová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Cellular Signaling and Anti-Apoptotic Effects of Prolactin-Releasing Peptide and Its Analog on SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 6343. https://doi.org/10.3390/ijms21176343
Zmeškalová A, Popelová A, Exnerová A, Železná B, Kuneš J, Maletínská L. Cellular Signaling and Anti-Apoptotic Effects of Prolactin-Releasing Peptide and Its Analog on SH-SY5Y Cells. International Journal of Molecular Sciences. 2020; 21(17):6343. https://doi.org/10.3390/ijms21176343
Chicago/Turabian StyleZmeškalová, Anna, Andrea Popelová, Aneta Exnerová, Blanka Železná, Jaroslav Kuneš, and Lenka Maletínská. 2020. "Cellular Signaling and Anti-Apoptotic Effects of Prolactin-Releasing Peptide and Its Analog on SH-SY5Y Cells" International Journal of Molecular Sciences 21, no. 17: 6343. https://doi.org/10.3390/ijms21176343