Ostm1 from Mouse to Human: Insights into Osteoclast Maturation

1
Institut de Recherches Cliniques de Montreal (IRCM), Montreal, QC H2W 1R7, Canada
2
Departement de Medecine, Universite de Montreal, Montreal, QC H2W 1R7, Canada
3
Department of Medicine, Division of Experimental Medicine, McGill University, Montreal, QC H3A 1A3, Canada
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(16), 5600; https://doi.org/10.3390/ijms21165600
Submission received: 16 July 2020
/
Revised: 29 July 2020
/
Accepted: 4 August 2020
/
Published: 5 August 2020
(This article belongs to the Special Issue New Insights in Osteoclasts’ Biology)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The maintenance of bone mass is a dynamic process that requires a strict balance between bone formation and resorption. Bone formation is controlled by osteoblasts, while osteoclasts are responsible for resorption of the bone matrix. The opposite functions of these cell types have to be tightly regulated not only during normal bone development, but also during adult life, to maintain serum calcium homeostasis and sustain bone integrity to prevent bone fractures. Disruption of the control of bone synthesis or resorption can lead to an over accumulation of bone tissue in osteopetrosis or conversely to a net depletion of the bone mass in osteoporosis. Moreover, high levels of bone resorption with focal bone formation can cause Paget’s disease. Here, we summarize the steps toward isolation and characterization of the osteopetrosis associated trans-membrane protein 1 (Ostm1) gene and protein, essential for proper osteoclast maturation, and responsible when mutated for the most severe form of osteopetrosis in mice and humans.
1. Introduction
Osteoclasts derive from hematopoietic stem cells that are shared with early myeloid lineage precursors. Differentiation of osteoclast precursors is dependent on mature osteoblasts that produce macrophage colony-stimulating factor (M-CSF), receptor activator of NF-κB Ligand (RANKL), and osteoprotegerin (OPG) a soluble decoy receptor of RANKL [1,2,3,4,5]. Upon recruitment and attachment to bone, mononuclear pre-osteoclasts undergo a process of fusion and these newly-formed multinucleated cells are structurally and functionally induced to generate active osteoclasts [6]. Mature osteoclasts are large multinucleated cells with numerous mitochondria, vacuoles, and lysosomes, which resorb mineralized cartilage and bone [7].
The biochemical characterization of osteoclasts have been hampered by the fact that these giant cells are tightly attached to the bone matrix and are therefore difficult to isolate. Moreover, as these cells are terminally differentiated and non-proliferative, a large number of cells have to be isolated at once. However, despite these impediments, osteoclast specific markers have been defined and novel efficient tools have been developed to analyze osteoclast biology ex vivo and in vivo [8].
When osteoclasts are activated, a resorption cycle is induced causing several proteins to be relocalized along with cytoskeletal rearrangement. Active osteoclasts are polarized and show two cellular histo-morphologic characteristics: an actin ring and a ruffled border. The actin ring, devoided of organelles, is enriched in dynamic and adhesive projections of the cell membrane called podosomes and in αVβ3 integrins that allow spreading and tight attachment to the bone surface [9,10,11]. The plasma membrane in contact with the bone surface enlarges into the ruffled border that induces polarization of the osteoclast. Following this attachment, the osteoclast secretory lysosomes, also found in immune cells and melanocytes [12,13], will associate and move along the microtubules, fuse to the plasma membrane, and then participate in ruffled border formation [14,15]. The ruffled border is an infolded finger-like distortion of the plasma membrane adjacent to the bone surface that participates with lysosomal proton pump H+/V-ATPase and chloride exchanger ClC-7 in acidification of the extracellular resorbing lacunae to ensure bone matrix demineralization [16,17,18]. In the lacunae, release from secretory lysosomes of tartrate resistant acid phosphatase (Trap), matrix metallopeptidases (Mmp 9,14) [19], and cathepsin K (Ctsk) result in osteoid degradation which is principally type I collagen [20] whereas high acidity potentiates dissolution of hydroxyapatite, the bone mineral component. The protein and mineral degradation products are phagocytosed at the ruffled border into of the osteoclast as digestive vacuole. Thus, bone resorption involves exocytosis and endocytosis at the ruffled border and exocytosis on the contralateral side of osteoclasts [10,21]. Importantly, osteoclast bone resorption has been demonstrated to be critical for normal hematopoietic progenitors recruitment and proliferation that link bone remodeling to hematopoiesis regulation [22]. Furthermore, numerous fundamental bone–immune interactions through shared factors have been discovered and are the subject of the field of osteoimmunology [23,24,25,26].
Defective osteoclast differentiation or generation of inefficient osteoclasts leads to the severe bone pathology called osteopetrosis, a heterogenous inherited disease of bone metabolism [27,28]. This disease was first described by Albers-Schönberg [29] and results in accumulation of mineralized osteoid and cartilage due to loss of bone resorption [4,30]. Different forms of osteopetrosis have been characterized in various vertebrate species [31,32,33] and mouse models were essential toward our understanding of mammalian osteoclast formation and function [12,34]. In humans, three clinical groups have been defined:
- infantile-malignant autosomal recessive (ARO) which is fatal within the first few years of life;
- intermediate recessive (IRO) which appears during the first decade of life but does not mediate a malignant response;
- autosomal dominant (ADO), with full-life expectancy but with major bone malformations.
Each form of the disease is characterized by a reduced bone marrow compartment leading to hematopoietic defects including anemia and high susceptibility to infections [35,36]. Characterization of autosomal recessive osteopetrotic mutations in mouse models and in human patients defined ‘osteoclast-poor’ (impaired osteoclast differentiation) and ‘osteoclast-rich’ (inactive osteoclasts) osteopetrosis leading to more targeted therapies [37,38,39].
2. Osteopetrotic Grey-Lethal Mouse Model
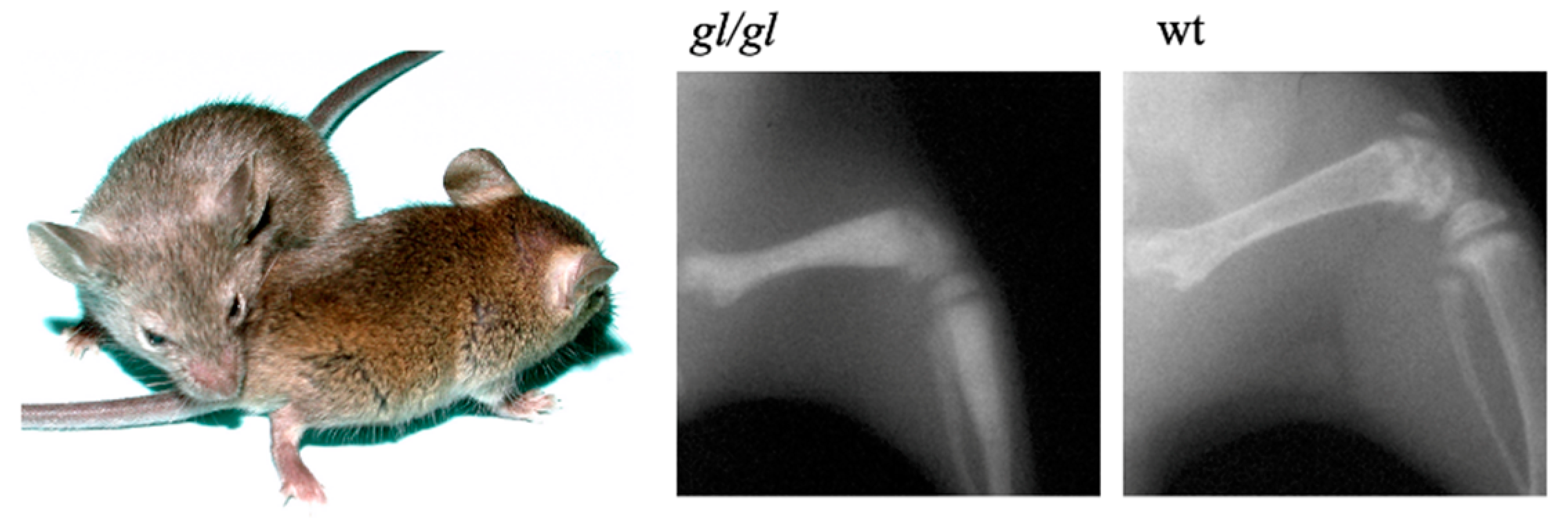
The spontaneous osteopetrotic grey-lethal (gl) mouse mutant was described by Gruneberg [40]. The homozygous gl/gl mice display a severe growth delay and a grey-coat color on an agouti background due to pheomelanin granule clumping [41]. Homozygous mice show a characteristic severe reduction of bone marrow space, lack of tooth eruption, and die around 3 weeks of age (Figure 1).
Restoration of the capacity to resorb bone matrix following normal spleen and/or bone marrow cells transplantation in gl/gl mice suggested a hematopoietic cell-intrinsic defect [42]. It is now well established that osteoclasts derived from hematopoietic precursors [43]. Our characterization of hematopoiesis in gl/gl mice was associated with mild anemia, a significant expansion of granulocyte-macrophage progenitors (CFU-GM) that give rise to osteoclasts and consistent with an increase of splenic CD11b+/Lys6-G+ monocytic cell subpopulation. In addition to this myeloid defect, deregulation of lymphoid lineages in gl/gl mice resulted in a reduction of B cell populations and altered T cell distribution with thymus hypo-cellularity [44]. This result provides the first evidence of an intrinsic time and differentiation stage-dependent molecular role for the gl gene in lymphoid cell lineage.
Importantly, in situ histological characterization of gl/gl bone tissue demonstrated the presence of numerous mature multinucleated osteoclasts, suggesting an intrinsic osteoclast defect that excluded cell differentiation impairment due to environmental factors. Consistent with this, ultra-structural analysis of bone sections showed that gl/gl osteoclasts are in close contact to the bone matrix but display an underdeveloped ruffled border essential for proper bone matrix resorption [45]. Accordingly, ex vivo analysis of gl/gl osteoclasts in culture demonstrated normal spreading through formation of an intact actin ring but these cells were unable to resorb bone matrix. The presence of an inactive mature osteoclast population in these mice classified the gl/gl phenotype as an ‘osteoclast-rich’ osteopetrosis [46].
3. Mapping the gl Locus and Characterization of the Ostm1 Gene
We have successfully used a positional cloning strategy to isolate and characterize the gene responsible for the mouse osteopetrotic gl mutation that most closely resembles human recessive osteopetrosis. By generating—for the first time—two backcross panels penetrant for the gl mutation, we have produced a genetic map and reduced the genetic interval on murine chromosome 10 that included the gl locus from 5 cM to ~1 cM [47]. During our systematic genetic mapping of this region, identification of specific polymorphisms had a tremendous impact on the establishment of our physical and transcriptional map of 98 yeast artificial chromosomes (YAC) clones assembled in a ~8 Mb contig [48]. Additional recombination events further reduced our gl candidate region to a ~1000 kb genomic interval. This interval was then covered with a contig of 17 overlapping bacterial artificial chromosomes (BAC) and novel polymorphic markers narrowed our candidate interval to ~500 kb. BAC transgenic lines were produced and full functional rescue obtained with one BAC clone in transgenic homozygous gl/gl mice defined a 180kb genomic candidate segment for the localization of the gl locus. BAC sequencing and transcription sequence analysis defined a single gene, called Ostm1, which encodes a unique 3kb transcript highly expressed in osteoclasts and undetectable in homozygous gl/gl animals [49]. Of note, an additional allele of Ostm1 (Ostm1om) was detected in an N-ethyl-N-nitrosourea (ENU) screen and results in a mild osteopetrotic phenotype but the mutation still needs to be defined [50].
Ostm1 is widely expressed and detectable in embryonic hematopoietic, skeletal, and brain tissues which is maintained after birth along with an appearance in gut, kidney, and skin. Subsequently, full-length cDNA was isolated and we established that the Ostm1 gene is composed of six exons and five introns. In contrast to its wild-type counterpart, gl genomic DNA has a ~7.5 kb deletion. This deletion covers most of the promoter, the first exon, and a large portion of the first intron leading to a null allele [49]. Importantly, targeted early re-expression of Ostm1 in hematopoietic cells of transgenic mice with the regulatory sequences of the transcriptional factor gene PU.1 (PU.1-Ostm1) resulted in full rescue of osteopetrosis and hematopoietic defects [44]. This provided definitive evidence that Ostm1 is the gene responsible for the gl mutation. Subsequently, we isolated the human OSTM1 gene and a database search identified homologs only in metazoans. Interestingly, genomic sequence analysis of 19 osteopetrotic patients led us to characterize the first human OSTM1 mutation associated with the disease that results in exon 5 skipping. This result was confirmed at the RNA level and allowed us to design and apply the first prenatal diagnostic test for carriers [49,51]. Additional OSTM1 mutations with severe osteopetrosis also displayed neurological disorders [52,53] suggesting that Ostm1 activity can be essential in maintenance of neuronal cell homeostasis. In these cases, however, a secondary neuronal effect from compression of cervical nerve and foramina occlusion, due to an excess of bone, has been excluded [54,55].
In parallel, gene expression profile analyses of gl/gl hematopoietic tissue identified the Inositol polyphosphate 4-phosphatase type II (Inpp4b) transcript as constantly downregulated. First, we isolated and characterized the Inpp4b gene in the mouse [56]. Second, systemic loss of Inpp4b in the mouse was induced and we demonstrated that Inpp4b is a negative regulator of osteoclast differentiation ex vivo. These mice consistently develop an osteoporotic phenotype in vivo, linking lipid metabolism to a specific bone phenotype. In humans, we showed that specific INPP4B variants were associated with variable bone mineral density and established INPP4B as a susceptibility locus to osteoporosis in pre-menopausal women [57]. Nevertheless, while the direct link between Ostm1 and Inpp4b remains to be elucidated, these results indicate that additional genomic loci can be deregulated due to the absence of Ostm1 expression and may give rise to the discovery of novel regulators of bone mineral density in mouse and human.
4. Ostm1 Protein Structure and Partners
The structure of the Ostm1 protein was investigated by various biochemical approaches. The open reading frame of the mouse Ostm1 gene encodes a 338-amino acid protein while the 334 amino acid human OSTM1 protein is 83% homologous to the mouse protein. Our structural analysis defined a signal peptide and a unique trans-membrane domain that classified Ostm1 as a type I trans-membrane protein where the majority of Ostm1 is luminal with a short cytosolic 30 amino acid C-terminus (Figure 2).
Interestingly, loss of the unique transmembrane domain resulted in secretion of the protein in vitro [58,59]. The predicted mass of the mature Ostm1 protein without modifications is ~34 kDa and we established that Ostm1 protein has 10 N-glycosylation sites consistent with the apparent protein mass of ~60 kDa. Upon use of different inhibitors of glycosylation, we confirmed that all potential luminal sites in Ostm1 appear effectively glycosylated. This post-translational glycosylation is very rapid and occurs in the endoplasmic reticulum. Analysis of Ostm1 subcellular localization also detected Ostm1 in the Golgi apparatus and late endosome/lysosome compartment with a punctuated distribution in the cytosol [58,60].
Based on the protein structure of Ostm1, we designed a tandem affinity purification (TAP) screen using a tagged version of the C-terminus of Ostm1 [61] and identified by mass spectrometry (MS) analysis specific cytosolic partners within the EcR293 kidney cell line [58]. Interactions were validated by glutathione-s-transferase (GST) pull-down assays with the C-terminus of Ostm1 in the same cells and in RAW cell-derived osteoclasts. This screen identified proteins classified into four subgroups, several of which were confirmed to interact directly with Ostm1 by immunoprecipitation assays. This indicates that Ostm1 can have multiple interactions with cytosolic proteins and could participate in a multi-functional protein platform. Particularly, we demonstrated a direct cytosolic interaction of Ostm1 with the anterograde motor protein kinesin family member 5B (Kif5B). Co-localization experiments by live imaging showed the dynamic Ostm1/Kif5B complex re-localization and trafficking that conveyed an adaptor role for the trans-membrane Ostm1 protein. Depletion of Kif5B led to peri-nuclear clustering of Ostm1 and lysosomes, demonstrating that the Ostm1-Kif5B interaction is essential for late-endosome/lysosome organelle dispersion [58,62]. Significantly, this cellular function could elucidate some of the physiological mechanisms underlying the wide gl/gl phenotypic spectrum.
Directly related to osteoclast biology we, and others, have also detected the protein ClC-7, a Cl−-H+ exchanger, as a partner of Ostm1 [63]. The Ostm1/ClC-7 complex is localized to late endosome/lysosome membranes and is responsible for acidification of secretory lysosomes and osteoclast resorption lacunae. Similar to Ostm1, the loss of ClC-7 leads to osteopetrosis in mice and humans with neuronal defects and retinal degeneration [64,65]. However, ClC-7 null mice display a milder form of osteopetrosis compared to Ostm1, therefore suggesting that Ostm1 may have additional functions. The present model defined Ostm1 as an essential partner required for ClC-7 stabilization and protection from lysosomal degradation [63]. This complex is also essential for ClC-7 transport to the osteoclast ruffled border [66].
5. Ostm1 in Osteoclast Maturation and Activation
To analyze the role of Ostm1 in a cell specific manner, we generated an Ostm1lox allele to induce conditional ablation (cKO) of Ostm1 protein in any tissue. As the first human mutation, we described results from OSTM1 exon 5 skipping, loxP sites flanking exon 5 were introduced in the mouse locus to mimic the human mutation [67]. We validated functionality of the floxed allele to reproduce a similar osteopetrotic gl phenotype by crossing Ostm1lox/lox mice with systemic deletion via Meox-Cre+ deleter transgenic line [68]. Accordingly, all Ostm1lox/lox Meox-Cre+ progenies develop severe osteopetrosis and die ~3 weeks after birth. Thus, we generated the first engineered Ostm1 cKO mouse model.
Subsequently, cKO of Ostm1 was induced in mature osteoclasts with the Cathepsin K (Ctsk-Cre) deleter transgenic line [69] to generate Ostm1Δexon5 Ctsk-Cre+ homozygous mice. An in-frame exon 5 deletion was subsequently confirmed by sequencing and the recombination level in mature Ostm1lox/lox Cre+ osteoclasts approached 100%. Ostm1lox/lox Ctsk-Cre+ mice develop severe osteopetrosis similar to the gl/gl phenotype with a short lifespan of ~3 weeks [67]. We also generated compound heterozygous Ostm1Δexon5/+ Ctsk-Cre+ mice that display a normal phenotype, excluding a dominant negative effect for the truncated/secreted Ostm1 protein as proposed in cell culture systems [59]. Our results are in accordance with heterozygous OSTM1 patients that are asymptomatic.
Analogous to systemic Ostm1 loss of function, we first showed that conditional ablation of Ostm1 in osteoclasts does not affect osteoclast differentiation but results in formation of numerous oversized multinucleated osteoclasts in vivo and ex vivo [67] similar to mice deficient in ClC-7 [70]. Normally, upon recruitment and attachment to bone matrix in vivo, committed mononuclear pre-osteoclasts undergo a process of fusion that gives rise to multinucleated mature osteoclasts [71,72,73]. Expression of the main fusogenic genes dendrocyte expressed seven trans-membrane protein (DC-Stamp), osteoclast stimulatory trans-membrane protein (OC-Stamp; Tm7sf4), and the d2 isoform of vacuolar ATPase V0 domain (Atp6v0d2) is essential in this mechanism [74,75,76]. The disruption of these genes inhibits the osteoclast fusion processes, blocks osteoclast maturation, and prevents bone resorption [77]. The role of the trans-membrane protein DC-Stamp in osteoclast fusion was demonstrated in DC-Stamp null mice that exhibit formation of only mononucleated pre-osteoclasts less capable to resorbing bone matrix, whereas transgenic mice overexpressing DC-Stamp accumulate osteoclasts of greater size with accelerated fusion [78]. Similarly, inactivation of the Atp6v0d2 gene led to reduced osteoclast fusion and defective bone resorption [76]. Consistent with osteoclast multi-nucleation stimulation due to the absence of Ostm1, a significant increase in transcription upregulation of these fusogenic genes was quantified in the oversized multinucleated cKO osteoclasts. This result was further substantiated with the upregulated expression and nuclear relocalization of the transcription factor Nfatc1, the upstream master regulator that controls expression of these target genes [78]. Together, these data demonstrate that Ostm1 is a negative regulator of the Nfatc1 pathway essential for osteoclast multinucleation [67].
Secondly, we demonstrated in vitro that cKO osteoclasts isolated from Ostm1lox/lox Ctsk-Cre+ mice were larger in size and were able to form a peripheral actin ring and podosomes, critical for osteoclast tight attachment to bone matrix. However, these cKO cells are defective in bone resorption similar to osteoclasts with complete loss of Ostm1 in gl/gl mice. In normal conditions, upon bone matrix interaction, polarization of multinucleated osteoclasts induces cytoskeletal rearrangements and formation of a ruffled border essential for proper bone resorption [79]. This structure is highly dependent on osteoclast secretory lysosomes that traffic and fuse to the plasma membrane to create an acidic (pH ~4.5) resorption lacunae [14,20,80]. Interestingly, autophagy proteins LC3-II, Atg5, Atg7, and Atg4B were also reported to participate in the formation of this cellular structure [81,82] and deficiency in such proteins impaired ruffled border development, but the underlying process is still undefined. The resorption lacunae acidification relies on the proton pump (H+/V-ATPase) and chloride transporter ClC-7 that secrete hydrogen and chloride ions into the lacunae [83]. The importance of H+/V-ATPase in mouse and humans was characterized by mutations in the a3 (oc; Tcirg1), Atp6v1c1 and Atp6ap1 (Ac45) enzyme subunits that result in non-functional osteoclasts [84,85,86,87]. Likewise, ClC-7 deficiency in mice and in humans causes osteopetrosis with variable severity [64,88].
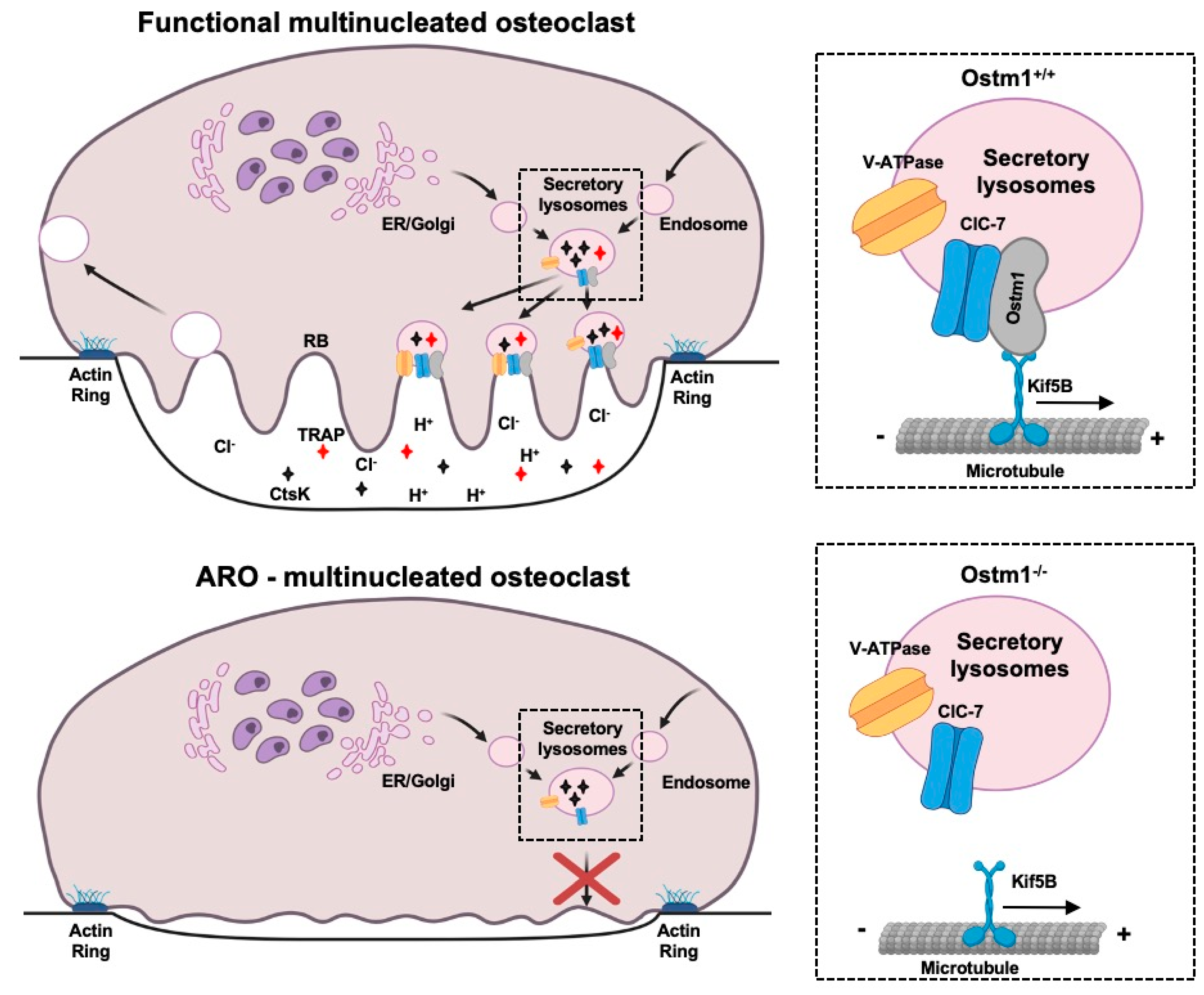
In Ostm1 cKO osteoclasts, we also demonstrated that the acidic luminal pH of the secretory lysosomes, essential for their function, was not altered [67] as described in gl/gl fibroblasts and neurons [63]. However, lysosomes showed a more disperse repartition and localization in resorbing cKO osteoclasts [67]. This result demonstrated that loss of Ostm1 directly affects intracellular dispersion and relocalization of the acidic endolysosomal compartment possibly through an interaction with the motor protein Kif5B, a partner of Ostm1 [58] (Figure 3).
Further evidence for a trafficking defect in absence of Ostm1 was obtained by analysis of the Acp5 (Trap) and Ctsk genes expression levels and proteins release. We showed that transcription levels of Acp5 and Ctsk were significantly enhanced in Ostm1 cKO osteoclasts. This response was consistent with concomitant upregulation of the Nfatc1 transcriptional factor expression since both Acp5 and Ctsk gene promotors are targets of Nfatc1. We then determined if the loss of Ostm1 can affect osteoclast protease release through biochemical quantification of extracellular levels of Trap and Ctsk enzymes. In Ostm1 cKO osteoclast cultures, Trap release was strongly reduced whereas the secreted Ctsk protein was undetectable. Impaired release of these proteases confirms that Ostm1 plays a role in osteoclast secretory lysosome trafficking and possibly in exocytosis (Figure 3). Additional support for a major role of Ostm1 in endosome/lysosome trafficking was described in the context of B cell lymphoma drug sensitivity [89] as well as in lysosome formation in osteoclasts [90]. These results demonstrate an osteoclast cell-intrinsic role for Ostm1 as a positive regulator of secretory lysosome dispersion, independent of other bone cells such as osteoblasts.
6. Ostm1 in Non-Bone Tissues
The gl/gl hematopoietic multi-lineage defects were functionally rescued by enabling Ostm1 cDNA expression under the control of the PU.1 transcription factor regulatory sequences [44]. This was accomplished by replacing the coding sequence of PU.1 with that of Ostm1 using homologous recombination in a PU.1 BAC. Several PU.1-Ostm1 BAC transgenic lines were produced with no phenotype and few were successively crossed to gl/+ mice to obtain PU.1-Ostm1-gl/gl homozygous animals. All PU.1-Ostm1 BAC gl/gl progenies from one line were rescued of gl/gl osteoclast defects, including osteopetrosis, but also of the altered myeloid and lymphoid lineages. This study provided evidence that Ostm1 is required independently for osteoclast and hematopoietic lineages.
However, PU.1-Ostm1-gl/gl transgenic mice still have a limited extended lifespan of ~7–8 weeks and still undergo premature death [65]. We investigated the cause of death of PU.1-Ostm1-gl/gl BAC mice and we first determined that Ostm1 was highly expressed in neurons and to a lesser extent in microglia and astrocytes. Consistent with the neurological disorders observed in some patients [52], the PU.1-Ostm1-gl/gl BAC mice develop brain inflammation with astrogliosis and microgliosis. Simultaneously, a rapidly progressive neurodegeneration affects all parts of the brain including cortex, hippocampus, cerebellum, and retinal degeneration that was associated with loss of photoreceptors [65]. This latter phenotype was most likely not a secondary effect due to excess bone accumulation and cranial nerve compression described in some osteopetrosis forms [55] but rather suggests an intrinsic neuronal role of Ostm1 that will be further analyzed.
The massive neuronal cell loss in PU.1-Ostm1-gl/gl BAC mice progressed swiftly from 3 to 7–8 weeks and cytosolic ubiquitin accumulation in osmophilic inclusions in neurons, suggested a storage-autophagy disorder. Demyelination was also occurring as previously observed in Ostm1 null gl/gl mice [53,91]. Through functional rescue using a series of targeted Ostm1 transgenic mice to individual brain cell-types, we demonstrated that neuronal death in these mice was specifically due to Ostm1 loss in neurons, excluding a direct implication of astrocyte or microglia cells. These mice represent the first in vivo model to analyze the neurological function of Ostm1.
Our cellular analysis in these mice unraveled a marked accumulation of autophagosomes in neurons from the cortex and hippocampus, indicative of an impaired autophagy mechanism. This phenotype results in axonal swelling consistent with a trafficking defect [65]. In this PU.1-Ostm1-gl/gl BAC model, the neuronal pathology features an autophagy mechanism independent of Beclin1 signaling, but reliant on downregulation of the mTOR (Mechanistic Target of Rapamycin) pathway and downstream targets [65]. These phenotypic studies on the loss of Ostm1 in the central nervous system (CNS) showed some similarities with known lysosomal storage diseases like Parkinson’s and Alzheimer’s [92,93,94]. More importantly, these findings were correlated with neuropathologic defects observed in OSTM1 patients further supporting an essential primary role of Ostm1 in the CNS, independent of the hematopoietic lineage [52].
7. OSTM1 and Human Osteopetrotic Patients
OSTM1 mutations are responsible for the most severe form of infantile type 3 autosomal recessive osteopetrosis with neuropathy (OMIM no. 259720) [52,95,96]. Presently, very few patients with OSTM1 loss of function mutations have been characterized (which include splice site variants, frameshift, and nonsense) and they represent around 5% of spontaneous human ARO [49,51,53,97,98]. Additional patients with OSTM1 micro-deletions defined a new class of mutations to be considered in diagnostic screening [99]. In parallel, new available molecular technologies such as high-throughput exome sequencing greatly facilitate identification and characterization of new mutations in carrier families [100].
Until now, allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment for ARO but the success rate is not optimal with engraftment being a limiting factor and overall outcomes remain disappointing [101,102,103,104]. However, significant improvements in transplantation success rates were obtained with safer regimens and reduced drug conditioning [105] as well as the use of non-invasive magnetic resonance imaging (MRI) of post-transplantation skeletal remodeling [106].
Early in utero interventions were also successfully designed to restore osteoclast activity in oc (Tcirg1−/−) mice [107]. Alternatively, a novel protocol tested in mice consists in transfusion of monocytic cells that can rescue bone marrow development in an early onset of osteopetrosis in the absence of HSCT [108]. As validation of this concept, microglial engraftment through single in utero transplant in the mouse can improve some of the brain phenotype described in lysosomal storage disease [109] and could possibly alleviate neuronal defects due to osteopetrosis. In fact, based on the implication of Ostm1 in neuronal homeostasis, curative treatment of OSTM1 patients is still challenging and consensus guidelines are being established [110]. Despite successful HSCT, neuronal pathology progresses in some OSTM1 patients [111]. However, the possible use of less invasive monocytic transfusions can give promise of less painful, more humane, and versatile therapies for OSTM1 patients.
8. Concluding Remarks
Our successful quest to understand the mouse grey-lethal osteopetrotic mutation was a long journey but it has allowed us to characterize the previously unknown Ostm1 locus responsible for the most severe form of osteopetrosis in mice and humans. All of these studies were made possible through generation of mouse models as well as molecular and cellular protocols to analyze osteoclast phenotypes. In osteoclasts, Ostm1 is a negative modulator of cell multi-nucleation, an essential step toward cell maturation. In osteoclast activation, Ostm1 along with specific partners is a positive regulator of intracellular trafficking of secretory lysosomes responsible for ruffled border formation, extracellular acidification, and bone matrix degradation. Therefore, Ostm1 expression in mature osteoclasts is absolutely required to prevent osteopetrosis. For OSTM1 patients, major progress has been made by the design of prenatal screens in carrier families, however more studies are needed to develop efficient curative treatments since these patients can frequently relapse independently from the hematopoietic cell lineage itself. The characterization of the molecular mechanisms of Ostm1 was of fundamental importance for our understanding of osteoclast biology, but also of high clinical relevance for bone diseases like osteopetrosis. Further studies in mice are still required, including discovery of additional Ostm1 partners to get insights in the complex bone cell crosstalk responsible for maintenance of bone matrix homeostasis. The osteoclast remains a fascinating cell to study and a better knowledge of its biology and multiple cellular interactions will characterize novel therapeutic targets for major bone diseases.
Author Contributions
Conceptualization, J.V.; Writing—original draft preparation, J.V., M.B., and M.P.; Writing—review and editing, J.V., M.B., and M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Canadian Institutes of Health Research MOP 86655 to J.V.
Conflicts of Interest
The authors state that they have no conflict of interest.
References
- Lacey, D.L.; Timms, E.; Tan, H.-L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Scully, S.; Hsu, H.; Sullivan, J.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Schwarz, E.M.; Boyce, B.F. Osteoclasts precursors, RANKL/RANK, and immunology. Immunol. Rev. 2005, 208, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L.; Ross, P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Petretto, E.; Gordon, S.; Duncan Bassett, J.H.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131, jcs216267. [Google Scholar] [CrossRef] [Green Version]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Vacher, J.; Saad, L.; Pata, M. Technologies, Tools and Genetic models to study Osteoclast. In Encyclopedia of Bone Biology; Zaidi, M., Ed.; Academic Press: Oxford, UK, 2020; Volume 1, pp. 329–339. [Google Scholar]
- Jurdic, P.; Saltel, F.; Chabadel, A.; Destaing, O. Podosome and sealing zone: Specificity of the osteoclast model. Eur. J. Cell Biol. 2006, 85, 195–202. [Google Scholar] [CrossRef]
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell Sci. 2000, 113, 377–381. [Google Scholar]
- Zou, W.; Teitelbaum, S.L. Absence of Dap12 and the αvβ3 integrin causes severe osteopetrosis. J. Cell Biol. 2015, 208, 125–136. [Google Scholar] [CrossRef]
- Edwards, J.R.; Mundy, G.R. Advances in osteoclast biology: Old findings and new insights from mouse models. Nat. Rev. Rheumatol. 2011, 7, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Karsenty, G.; Ferron, M. Regulation of lysosome biogenesis and functions in osteoclasts. Cell Cycle 2013, 12, 2744–2752. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Roux, S. Rab GTPases in Osteoclastic Endomembrane Systems. BioMed. Res. Int. 2018, 2018, 4541538. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Neutzsky-Wulff, A.V.; Karsdal, M.A.; Henriksen, K. Characterization of the bone phenotype in ClC-7-deficient mice. Calcif. Tissue Int. 2008, 83, 425–437. [Google Scholar] [CrossRef]
- Nishi, T.; Forgac, M. The vacuolar (H+)-ATPases-nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [Google Scholar] [CrossRef]
- Zhu, L.; Tang, Y.; Li, X.-Y.; Keller, E.T.; Yang, J.; Cho, J.-S.; Feinberg, T.Y.; Weiss, S.J. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci. Trans. Med. 2020, 12, eaaw6143. [Google Scholar] [CrossRef]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The great beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Stenbeck, G. Formation and function of the ruffled border in osteoclasts. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 13, pp. 285–292. [Google Scholar]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Aaron, J.R.; Choi, Y. Osteoimmunology: Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Takegahara, N.; Kim, H.S.; Choi, Y. Updating osteoimmunology: Regulation of bone cells by innate and adaptative immunity. Nat. Rev. Rheumatol. 2018, 14, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. New immune connections in osteoclast formation. Ann. Rev. N. Y. Acad. Sci. 2009, 1192, 117–123. [Google Scholar] [CrossRef]
- Janssens, K.; Van Hul, W. Molecular genetics of too much bone. Hum. Mol. Genet. 2002, 11, 2385–2393. [Google Scholar] [CrossRef] [Green Version]
- Helfrich, M.H. Osteoclast diseases. Microsc. Res. Tech. 2003, 61, 514–532. [Google Scholar] [CrossRef]
- Albers-Schönberg, H.E. Röntgenbilder einer seltenen Knock-enerkrankung. Munch. Med. Wochenschr. 1904, 5, 365–368. (In German) [Google Scholar]
- Tolar, J.; Teitelbaum, S.L.; Orchard, P.J. Osteopetrosis. N. Engl. J. Med. 2004, 351, 2839–2849. [Google Scholar] [CrossRef]
- Van Wesenbeek, L.; Odgren, P.R.; MacKay, C.A.; D’Angelo, M.; Safadi, F.F.; Popoff, S.N.; Van Hul, W.; Marks, S.C. The osteopetrotic mutation toothless (tl) is a loss-of-function frameshift mutation in the rat Csf1 gene: Evidence of a crucial role for CSF-1 in osteoclastogenesis and endochondral ossification. Proc. Natl. Acad. Sci. USA 2002, 99, 14303–14308. [Google Scholar] [CrossRef] [Green Version]
- Van Wesenbeeck, L.; Odgren, P.R.; Coxon, F.P.; Frattini, A.; Moens, P.; Perdu, B.; MacKay, C.A.; Van Hul, E.V.; Timmermans, J.-P.; Vanhoenacker, F.; et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J. Clin. Investig. 2007, 117, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Meyers, S.N.; McDaneld, T.G.; Swist, S.L.; Marron, B.M.; Steffen, D.J.; O’Toole, D.; O’Connell, J.R.; Beever, J.E.; Sonstegard, T.S.; Smith, T.P. A deletion mutation in bovine SLC4A2 is associated with osteopetrosis in Red Angus cattle. BMC Genom. 2010, 11, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia-Silvestre, T.; Neutzsky-Wulff, A.V.; Sorensen, M.G.; Christiansen, C.; Blollerslev, J.; Karsdal, M.A.; Henriksen, K. Advances in osteoclast biology resulting from the study of osteopetrotic mutations. Hum. Genet. 2009, 124, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Van Wesenbeeck, L.; Van Hul, W. A clinical and molecular overview of the human ostopetroses. Calcif. Tissue Int. 2005, 77, 263–274. [Google Scholar] [CrossRef]
- Reeves, J.D.; August, C.S.; Humbert, J.R.; Weston, W.L. Host defense in infantile osteopetrosis. Pediatrics 1979, 64, 202–206. [Google Scholar] [PubMed]
- Teti, A.; Econs, M.J. Osteopetroses, emphasizing potential approaches to treatment. Bone 2017, 102, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.; Guerrini, M.M.; Cassani, B.; Pangrazio, A.; Sobacchi, C. Infantile malignant, autosomal recessive osteopetrosis: The rich and the poor. Cacif. Tissue Int. 2009, 84, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pangrazio, A.; Cassani, B.; Guerrini, M.M.; Crockett, J.C.; Marella, V.; Zammataro, L.; Strina, D.; Schulz, A.; Schlack, C.; Kornak, U.; et al. RANK-dependent autosomal recessive osteopetrosis: Characterization of five new cases with novel mutations. J. Bone Miner. Res. 2012, 27, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Gruneberg, H. A new sub-lethal colour mutation in the house mouse. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1935, 118, 321–342. [Google Scholar]
- Gruneberg, H. Grey-lethal, a new mutation in the house mouse. J. Hered. 1936, 27, 105–109. [Google Scholar] [CrossRef]
- Walker, D.G. Bone resorption restored in osteopetrotic mice by transplants of normal bone marrow and spleen cells. Science 1975, 190, 784–785. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [PubMed] [Green Version]
- Pata, M.; Héraud, C.; Vacher, J. OSTM1 bone defect reveals an intercellular hematopoietic crosstalk. J. Biol. Chem. 2008, 283, 30522–30530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapurohitam, V.; Chalhoub, N.; Benachenhou, N.; Neff, L.; Baron, R.; Vacher, J. The mouse osteopetrotic grey-lethal mutation induces a defect in osteoclast maturation/function. Bone 2001, 28, 513–523. [Google Scholar] [CrossRef]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Vacher, J.; Bernard, H. Genetic localization and transmission of the osteopetrotic grey-lethal mutation. Mamm. Genome 1999, 10, 239–243. [Google Scholar] [CrossRef]
- Chalhoub, N.; Benachenhou, N.; Vacher, J. Physical and transcriptional map of the mouse Chromosome 10 region syntenic to human 6q16-q21. Mamm. Genome 2001, 12, 887–892. [Google Scholar] [CrossRef]
- Chalhoub, N.; Benachenhou, N.; Rajapurohitam, V.; Pata, M.; Ferron, M.; Frattini, A.; Villa, A.; Vacher, J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat. Med. 2003, 9, 399–406. [Google Scholar] [CrossRef]
- Bosman, E.A.; Estabel, J.; Ismail, O.; Podrini, C.; White, J.K.; Steel, K.P. Omi, a recessive mutation on chromosome 10, is a novel allele of Ostm1. Mamm. Genome 2012, 24, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Quarello, P.; Forni, M.; Barbereis, L.; Defilippi, C.; Campagnoli, M.F.; Frattini, A.; Chalhoub, N.; Vacher, J.; Ramenghi, U. Severe malignant osteopetrosis due to a Gl gene mutation. J. Bone Miner. Res. 2004, 19, 1194–1199. [Google Scholar] [CrossRef]
- Maranda, B.; Chabot, G.; Décarie, J.-C.; Pata, M.; Azeddine, B.; Moreau, A.; Vacher, J. Clinical and cellular manifestations of OSTM1 related infantile osteopetrosis. J. Bone Miner. Res. 2008, 23, 296–300. [Google Scholar] [CrossRef]
- Pangrazio, A.; Poliani, P.L.; Megarbane, A.; Lefranc, G.; Lanino, E.; Di Rocco, M.; Rucci, F.; Lucchini, F.; Ravanini, M.; Facchetti, F.; et al. Mutations in OSTM1 (Grey Lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement. J. Bone Miner. Res. 2006, 21, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ramaker, J.M.; Radcliff, A.B.; Baldassari, S.; Mayer, J.A.; Ver Hoeve, J.N.; Zhang, C.-L.; Chiu, S.-Y.; Cloleddo, R.J.; Duncan, I.D. Spontaneous optic nerve compression in the osteopetrotic (op/op) mouse: A novel model of myelination failure. J. Neurosci. 2013, 33, 3514–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steward, C.G. Neurological aspects of osteopetrosis. Neuropathol. Appl. Neurobiol. 2003, 29, 87–97. [Google Scholar] [CrossRef]
- Ferron, M.; Vacher, J. Characterization of the murine Inpp4b gene and identification of a novel isoform. Gene 2006, 376, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Boudiffa, M.; Arsenault, M.; Rached, M.; Pata, M.; Giroux, S.; Elfassihi, L.; Kisseleva, M.V.; Majerus, P.W.; Rousseau, F.; et al. Inositol Polyphosphate 4-phosphatase b as a regulator of bone mass in mice and humans. Cell Metab. 2011, 14, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandruvada, S.N.; Beauregard, J.; Benjannet, S.; Pata, M.; Lazure, C.; Seidah, N.G.; Vacher, J. Role of Ostm1 Cytosolic Complex with Kinesin 5B in Intracellular Dispersion and Trafficking. Mol. Cell. Biol. 2016, 36, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Yu, J.; Park, E.-S.; Choi, S.-W.; Hwang, J.M.; Yun, H.; Chung, Y.-H.; Hong, K.S.; Choi, J.-S.; Takami, M.; et al. Secretion of a truncated osteopetrosis-associated transmembrane protein 1 (Ostm1) mutant inhibits osteoclastogenesis through downregulation of the B lymphocyte-induced maturation protein (Blimp1)-nuclear factor of activated T cells c1 (NFATc1) axis. J. Biol. Chem. 2014, 289, 35868–35881. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Lewis, B.; Krueger, C.C.; Sargeant, T.J.; D’Angelo, M.E.; Deery, M.J.; Feret, R.; Howard, J.A.; Lilley, K.S.; Watson, C.J. Stat3-mediated alterations in lysosomal membrane protein composition. J. Biol. Chem. 2018, 293, 4244–4261. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, B.; Jeronimo, C.; Langelier, M.F.; Cojocaru, M.; Bergeron, D. Interaction networks of the molecular machines that decode, replicate, and maintain the integrity of the human genome. Mol. Cell. Proteom. 2004, 3, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef]
- Lange, P.; Wartosch, L.; Jentsch, T.; Fuhrmann, J. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 2006, 440, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the CIC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Héraud, C.; Griffiths, A.; Pandruvada, S.N.M.; Kilimann, M.W.; Pata, M.; Vacher, J. Severe neurodegeneration with impaired autophagy mechanism triggered by Ostm1 deficiency. J. Biol. Chem. 2014, 289, 13912–13925. [Google Scholar] [CrossRef] [Green Version]
- Leisle, L.; Ludwig, C.F.; Wagner, F.A.; Jentsch, T.J.; Stauber, T. ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J. 2011, 30, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Pata, M.; Vacher, J. Ostm1 Bifunctional Roles in Osteoclast Maturation: Insights From a Mouse Model Mimicking a Human OSTM1 Mutation. J. Bone Miner. Res. 2018, 33, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallquist, M.D.; Soriano, P. Epiblast-restricted Cre expression in MORE mice: A tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 2000, 26, 113–115. [Google Scholar] [CrossRef]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen prevents bone loss via Estrogen Receptor α and induction of Fas ligand in osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Neutzsky-Wulff, A.V.; Sims, N.A.; Supanchart, C.; Kornak, U.; Felsenberg, D.; Poulton, I.J.; Martin, T.J.; Karsdal, M.A.; Henriksen, K. Severe developmental bone phenotype in ClC-7 deficient mice. Dev. Biol. 2010, 344, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Søe, K.; Hobolt-Pedersen, A.-S.; Delaisse, J.-M. The elementary fusion modalities of osteoclasts. Bone 2015, 73, 181–189. [Google Scholar] [CrossRef]
- Iwasaki, R.; Ninomiya, K.; Miyamoto, K.; Suzuki, T.; Sato, Y.; Kawana, H.; Nakagawa, T.; Suda, T.; Miyamoto, T. Cell fusion in osteoclasts plays a critical role in controlling bone mass and osteoblastic activity. Biochem. Biophys. Res. Commun. 2008, 377, 899–904. [Google Scholar] [CrossRef]
- Vignery, A. Macrophage fusion: The making of osteoclasts and giant cells. J. Exp. Med. 2008, 202, 337–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, M.; Miyamoto, T.; Sawatani, Y.; Iwamoto, K.; Hosogane, N.; Fujita, N.; Morita, K.; Ninomiya, K.; Suzuki, T.; Miyamoto, K.; et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 2005, 202, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Birnbaum, M.J.; Mackay, C.A.; Mason-Savas, A.; Thompson, B.; Odgren, P.R. Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protiern induced by Rankl that promotes osteoclast differentiation. J. Cell. Physiol. 2010, 215, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-H.; Rho, J.; Jeong, D.; Sul, J.-Y.; Kim, T.; Kim, N.; Kang, J.-S.; Miyamoto, T.; Suda, T.; Lee, S.-K.; et al. v-ATPase V0 subunit d2–deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Oursler, M.J. Recent advances in understanding the mechanisms of osteoclast precursor fusion. J. Cell. Biochem. 2010, 110, 1058–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, S.-H.; Kim, J.H.; Choi, Y.; Kim, N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the Dendritic Cell-Specific Transmembrane Protein (DC-STAMP). Mol. Endocrinol. 2008, 222, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Mulari, M.T.; Zhao, H.; Lakkakorpi, P.T.; Väänänen, H.K. Osteoclast ruffled border has distinct subdomains for secretion and degraded matrix uptake. Traffic 2003, 4, 113–125. [Google Scholar] [CrossRef]
- Coxon, F.P.; Taylor, A. Vesicular trafficking in osteoclasts. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2008; Volume 19, pp. 424–433. [Google Scholar]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, H.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; RTeitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [Green Version]
- McManus, S.; Roux, S. The adaptor protein p62/SQSTM1 in osteoclast signaling pathways. J. Mol. Signal. 2012, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Manolson, M.F.; Yu, H.; Chen, W.; Yao, Y.; Li, K.; Less, R.L.; Heersche, J.N. The a3 isofom of the 100-kDa V-ATPase subunit is highly but differentialy expressed in large (≥10 nuclei) and small (≤5 nuclei) osteoclasts. J. Biol. Chem. 2003, 278, 49271–49278. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, J.-C.; Franchi, A.; Trojani, C.; Parrinello, H.; Grosgeorge, J.; Robert, C.; Jaillon, O.; Poirier, C.; Gaudray, P.; Carle, G.F. The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 2000, 26, 207–213. [Google Scholar] [CrossRef]
- Voronov, I.; Ochotny, N.; Jaumouillé, V.; Owen, C.; Manolson, M.F.; Aubin, J.E. The R400S mutation in the V-ATPase a3 subunit increases lysosomal pH, impairs NFATc1 translocation, and decreases in vitro osteoclastogenesis. J. Bone Miner. Res. 2013, 28, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Kornak, U.; Schulz, A.; Friedrich, W.; Uhlhaas, S.; Kremens, B.; Voit, T.; Hasan, C.; Bode, U.; Jentsch, T.J.; Kubisch, C. Mutation in the a3 subunit of the vacular H+-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9, 2059–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimeca, J.-C.; Quincey, D.; Parinello, H.; Romatet, D.; Grogeorges, J.; Gaudray, P.; Philip, N.; Fisher, A.; Carle, G.F. Novel mutations in the TCIRG1 gene encoding the a3 subunit of the vacular proton pump in patients affected by infantile malignant osteopetrosis. Hum. Mutat. 2003, 21, 151–157. [Google Scholar] [CrossRef]
- Frattini, A.; Pangrazio, A.; Susani, L.; Sobacchi, C.; Mirolo, M.; Abinun, M.; Andolina, M.; Flanagan, A.; Horwitz, E.M.; Mihci, E.; et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and Intermediate osteopetrosis. J. Bone Miner. Res. 2003, 18, 1740–1747. [Google Scholar] [CrossRef]
- Gayle, S.; Landrette, S.; Becharry, N.; Conrad, C.; Hernandez, M.; Beckett, P.; Ferguson, S.M.; Mandelkern, T.; Zheng, M.; Xu, T.; et al. Identification of aplimod as a first-in-class PIKfyve kinase inhibitor for treatment of B-cell non-Hodgkin lymphoma. Blood 2017, 129, 1768–1778. [Google Scholar] [CrossRef]
- Sultana, F.; Morse, L.R.; Picotto, G.; Liu, W.; Jha, P.K.; Odgren, P.R.; Battaglino, R.A. Snx10 and PIKfyve are required for lysosome formation in osteoclasts. J. Cell. Biochem. 2019, 121, 2927–2937. [Google Scholar] [CrossRef]
- Prinetti, A.; Rocchetta, F.; Costantino, E.; Frattini, A.; Caldana, E.; Rucci, F.; Bettiga, A.; Poliani, P.L.; Chigorno, V.; Sonnino, S. Brain lipid composition in grey-lethal mutant mouse characterized by severe malignant osteopetrosis. Glycoconj. J. 2009, 26, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, J.P. Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzolari, E.; Forino, C.; Razza, A.; Porta, F.; Villa, A.; Notarangelo, L.D. A single-center experience in 20 patients with infantile malignant osteopetrosis. Am. J. Hematol. 2009, 84, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Castellano Chiodo, D.; DiRocco, M.; Gandolfo, C.; Morana, G.; Buzzi, D.; Rossi, A. Neuroimaging findings in malignant infantile osteopetrosis due to OSTM1 mutations. Neuropediatrics 2007, 38, 154–156. [Google Scholar] [CrossRef]
- Ramirez, A.; Faupel, J.; Goebel, I.; Stiller, A.; Beyer, S.; Stöckle, C.; Hasan, C.; Bode, U.; Kornak, U.; Kubish, C. Identification of a novel mutation in the coding region of grey-lethal gene OSTM1 in human malignant infantile osteopetrosis. Hum. Mutat. 2004, 23, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Souraty, N.; Noun, P.; Djambas-Khayat, C.; Chouery, E.; Pangrazio, A.; Villa, A.; Lefranc, G.; Frattini, A.; Mégarbané, A. Molecular study of six families originating from the middle-east and presenting with autosomal recessive osteopetrosis. Eur. J. Med. Genet. 2007, 50, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.E.; Fischer, B.; Schröter, P.; Richter, R.; Gupta, N.; Verma, N.; Kabra, M.; Mundlos, S.; Rajab, A.; Neitzel, H.; et al. Severe neuronopathic autosomal recessive osteopetrosis due to homozygous deletions affecting OSTM1. Bone 2013, 55, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Sui, W.; Ou, M.; Liang, J.; Ding, M.; Chen, J.; Liu, W.; Xiao, R.; Meng, X.; Wang, L.; Pan, X.; et al. Rapid gene identification in a Chinese osteopetrosis family by whole exome sequencing. Bone 2013, 516, 311–315. [Google Scholar] [CrossRef]
- Orchard, P.J.; Fasth, A.L.; Le Radmacher, J.; He, W.; Boelens, J.J.; Horwitz, E.M.; Al-Seraihy, A.; Ayas, M.; Binfim, C.M.; Boulad, F.; et al. Hematopoietic stem cell transplantation for infantile osteopetrosis. Blood 2015, 126, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Pronk, C.J.; Turkiewicz, D.; Vult von Steyern, K.; Ehinger, M.; Dykes, J.; Toporski, J. Transplantation of Haploidentical TcRaß-Depleted Hematopoietic Cells Allows for Optimal Timing and Sustained Correction of the Metabolic Defect in Children With Infantile Osteopetrosis. J. Bone Miner. Res. 2017, 32, 82–85. [Google Scholar] [CrossRef]
- Steward, C.G. Hematopoietic stem cell transplantation for osteopetrosis. Pediatr. Clin. N. Am. 2010, 57, 171–180. [Google Scholar] [CrossRef]
- Coccia, P.; Krivit, W.; Cervenka, J.; Clawson, C.; Kersey, J.; Kim, T.; Nesbit, M.; Ramsay, N.; Warkentin, P.; Teitelbaum, S.; et al. Successful bone-marrow transplantation for infantile malignant osteopetrosis. N. Engl. J. Med. 1980, 302, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Shadur, B.; Zaidman, I.; NaserEddin, A.; Lokshin, E.; Hussein, F.; Cohen Oron, H.; Avni, B.; Grisariu, S.; Stepensky, P. Successful hematopoietic stem cell transpalntation for osteopetrosis using reduced intensity conditionning. Pediatr. Blood Cancer 2018, 65, e27010. [Google Scholar] [CrossRef] [PubMed]
- Maximova, N.; Zennaro, F.; Gregori, M.; Boz, G.; Zanon, D.; Mbalaviele, G. Hematopoietic stem cell transplantation induced bone remodeling in autosomal recessive osteopetrosis: Interaction between skeleton and hematopoietic and sensory nervous systems. Bone 2020, 130, 115144. [Google Scholar] [CrossRef] [PubMed]
- Frattini, A.; Blai, H.C.; Sacco, M.G.; Cerisoli, F.; Faggioli, F.; Cato, E.M.; Pangrazio, A.; Musio, A.; Rucci, F.; Sobacchi, C.; et al. Rescue of ATPa3-deficient murine malignant osteopetrosis by hematopoietic stem cell transplantation in utero. Proc. Natl. Acad. Sci. USA 2005, 102, 14629–14634. [Google Scholar] [CrossRef] [Green Version]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Witt, R.G.; Wang, B.; Eikani, C.; Shea, J.; Smith, L.K.; Boyle, G.; Cadaoas, J.; Sper, R.; MacKenzie, J.D.; et al. Tolerance induction and microglial engraftment after fetal therapy without conditioning in mice with Mucopolysaccharidosis type VII. Sci. Transl. Med. 2020, 12, eaay8980. [Google Scholar] [CrossRef]
- Wu, C.C.; Econs, M.J.; DiMeglio, I.A.; Insigna, K.L.; Levine, M.A.; Orchard, P.J.; Miller, W.P.; Petryk, A.; Rush, E.T.; Shoback, D.M.; et al. Diagnosis abd management of osteopetrosis: Consensus guidelines from the osteopetrosis group. J. Clin. Endodrinol. Metab. 2017, 103, 3111–3123. [Google Scholar] [CrossRef]
- Overholt, K.M.; Rose, M.J.; Joshi, S.; Herman, G.E.; Bajwa, R.; Abu-Arja, R.; Rangarajan, H.G.; Horwitz, E.M. Hematopoietic cell transplantation for a child with OSTM1 osteopetrosis. Blood Adv. 2017, 1, 279–281. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Grey coat color in gl/gl mice (left) and representative X-rays of osteopetrotic gl/gl bone compared to agouti wild-type (wt) littermate.
Figure 1.
Grey coat color in gl/gl mice (left) and representative X-rays of osteopetrotic gl/gl bone compared to agouti wild-type (wt) littermate.

Figure 2.
Structure of the murine Ostm1 protein and conservation between the mouse and human proteins. SP: Signal peptide; TM: Trans membrane domain; |: Glycosylation site.
Figure 2.
Structure of the murine Ostm1 protein and conservation between the mouse and human proteins. SP: Signal peptide; TM: Trans membrane domain; |: Glycosylation site.

Figure 3.
Proposed working model of Ostm1-mediated osteoclast dysfunction leading to autosomal recessive osteopetrosis. RB: Ruffled border; ER: Endoplasmic Reticulum In ARO osteoclast, impaired secretory lysosome trafficking results in lack of ruffled border formation and ineffective bone matrix resorption.
Figure 3.
Proposed working model of Ostm1-mediated osteoclast dysfunction leading to autosomal recessive osteopetrosis. RB: Ruffled border; ER: Endoplasmic Reticulum In ARO osteoclast, impaired secretory lysosome trafficking results in lack of ruffled border formation and ineffective bone matrix resorption.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vacher, J.; Bruccoleri, M.; Pata, M. Ostm1 from Mouse to Human: Insights into Osteoclast Maturation. Int. J. Mol. Sci. 2020, 21, 5600. https://doi.org/10.3390/ijms21165600
AMA Style
Vacher J, Bruccoleri M, Pata M. Ostm1 from Mouse to Human: Insights into Osteoclast Maturation. International Journal of Molecular Sciences. 2020; 21(16):5600. https://doi.org/10.3390/ijms21165600
Chicago/Turabian StyleVacher, Jean, Michael Bruccoleri, and Monica Pata. 2020. "Ostm1 from Mouse to Human: Insights into Osteoclast Maturation" International Journal of Molecular Sciences 21, no. 16: 5600. https://doi.org/10.3390/ijms21165600
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.