Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Selection of Test Articles
2.2. Transformation Assay
2.3. Identification and Distribution of DMRs
2.4. Overlapping DMRs in Promoter Regions across NGTXCs
2.5. Enriched Functional Annotation and Canonical Pathway
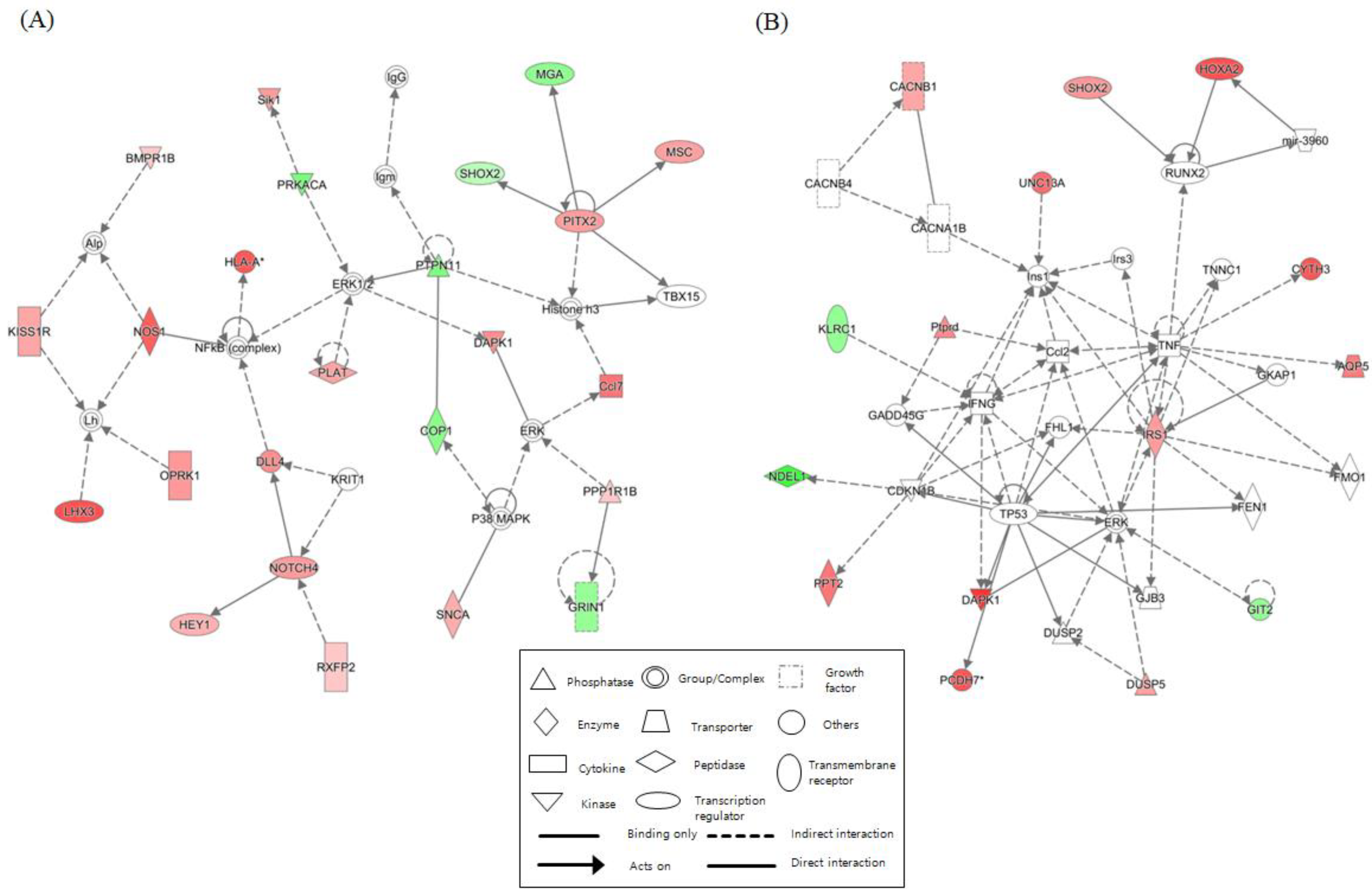
2.6. Network Identification
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and Cell Culture
4.3. Cell Growth Assay
4.4. In Vitro Bhas 42 CTA for the Promotion Test
4.5. Isolation of Transformed Foci
4.6. Genomic DNA Extraction and Reduced-Representation Bisulfite Sequencing (RRBS)
4.7. RRBS Data Analysis
4.8. Molecular Network, Pathway, and Functional Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CTA | In vitro cell transformation |
| NGTXC | Non-genotoxic carcinogen |
| DMR | Differentially methylated region |
| RRBS | Reduced-representation bisulfate sequencing |
| IATA | Integrated approach to testing and assessment |
| IPA | Ingenuity pathway analysis |
| QSAR | Quantitative structure–activity relationship |
| MH | Methapyrilene hydrochloride |
| DEHP | Bis(2-ethylhexyl) phthalate |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| OA | Okadaic acid |
| CA | Cholic acid |
| DEA | Diethanolamine |
| SA | Sodium saccharin |
References
- Hernández, L.G.; van Steeg, H.; Luijten, M.; van Benthem, J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res. 2009, 682, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Kakunaga, T. A quantitative system for assay of malignant transformation by chemical carcinogens using a clone derived from Balb 3T3. Int. J. Cancer 1973, 12, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.L. In vitro transformation assays for chemical carcinogens. Mutat. Res. 1983, 115, 323–338. [Google Scholar] [CrossRef]
- Hayashi, M.; Kojima, H.; Corvi, R.; Stokes, W.; Jacobs, A.; Morita, T.; Schechtman, L.; Suzuki, A. The Validation Management Team Report on the Bhas 42 CTA; EURL ECVAM Recommendations; EU Reference Laboratory for Alternatives to Animal Testing: Ispra, Italy, 2012. [Google Scholar]
- Sakai, A.; Sasaki, K.; Muramatsu, D.; Arai, S.; Endou, N.; Kuroda, S.; Hayashi, K.; Lim, Y.M.; Yamazaki, S.; Umeda, M.; et al. A Bhas 42 cell transformation assay on 98 chemicals: The characteristics and performance for the prediction of chemical carcinogenicity. Mutat. Res. 2010, 702, 100–122. [Google Scholar] [CrossRef]
- Tanaka, N.; Sasaki, K.; Hayashi, K.; Sakai, A.; Asada, S.; Muramatsu, D.; Kuroda, S.; Mizuhashi, F.; Nagai, M.; Suzuki, H.; et al. An international collaborative study on a cell transformation assay using Bhas 42 cells. AATEX 2009, 14, 831–848. [Google Scholar]
- Sakai, A.; Sasaki, K.; Hayashi, K.; Muramatsu, D.; Arai, S.; Endou, N.; Kuroda, S.; Poth, A.; Bohnenberger, S.; Kunkelmann, T.; et al. An international validation study of a Bhas 42 cell transformation assay for the prediction of chemical carcinogenicity. Mutat. Res. 2011, 725, 57–77. [Google Scholar] [CrossRef]
- OECD. Organisation for Economic and Cooperative Development. Guidance Document on the in vitro Bhas 42 cell transformation assay. Series on Testing and Assessment. No. 231. Paris. 2016. Available online: https://www.oecd.org/env/ehs/testing/ENV_JM_MONO(2016)1.pdf (accessed on 3 December 2016).
- Melnick, R.L.; Kohn, M.C.; Portier, C.J. Implications for risk assessment of suggested nongenotoxic mechanisms of chemical carcinogenesis. Environ. Health Perspect. 1996, 104 (Suppl. 1), 123–134. [Google Scholar]
- Hattis, D.; Chu, M.; Rahmioglu, N.; Goble, R.; Verma, P.; Hartman, K.; Kozlak, M. A preliminary operational classification system for nonmutagenic modes of action for carcinogenesis. Crit. Rev. Toxicol. 2009, 39, 97–138. [Google Scholar] [CrossRef]
- Waters, M.D.; Jackson, M.; Lea, I. Characterizing and predicting carcinogenicity and mode of action using conventional and toxicogenomics methods. Mutat. Res. 2010, 705, 184–200. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Herceg, Z.; Lambert, M.P.; van Veldhoven, K.; Demetriou, C.; Vineis, P.; Smith, M.T.; Straif, K.; Wild, C.P. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 2013, 34, 1955–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Reinhardt, R.; Jeltsch, A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res. 2006, 34, 1182–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greally, J.M.; Jacobs, M.N. In vitro and in vivo testing methods of epigenomic endpoints for evaluating endocrine disruptors. ALTEX 2013, 30, 445–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Liu, G.; Zhou, F.; Su, B.; Li, Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin. Exp. Med. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Van Gurp, T.P.; Wagemaker, N.C.; Wouters, B.; Vergeer, P.; Ouborg, J.N.; Verhoeven, K.J. epiGBS: Reference-free reduced representation bisulfite sequencing. Nat. Methods 2016, 13, 322–324. [Google Scholar] [CrossRef]
- Walker, D.L.; Bhagwate, A.V.; Baheti, S.; Smalley, R.L.; Hilker, C.A.; Sun, Z.; Cunningham, J.M. DNA methylation profiling: Comparison of genome-wide sequencing methods and the Infinium Human Methylation 450 Bead Chip. Epigenomics 2015, 7, 1287–1302. [Google Scholar] [CrossRef]
- Poburski, D.; Thierbach, R. Improvement of the BALB/c-3T3 cell transformation assay: A tool for investigating cancer mechanisms and therapies. Sci. Rep. 2016, 6, 32966. [Google Scholar] [CrossRef]
- Chang, C.; Xi, L.; Zhang, J.; Zhao, W.; Liu, Z.; Guo, J.; Xu, C. Roles of Cyclin A, Myc, Jun and Ppm1l in tumourigenic transformation of NIH3T3 cell. J. Genet. 2018, 97, 1155–1168. [Google Scholar] [CrossRef]
- Vaccari, M.; Mascolo, M.G.; Rotondo, F.; Morandi, E.; Quercioli, D.; Perdichizzi, S.; Zanzi, C.; Serra, S.; Poluzzi, V.; Angelini, P.; et al. Identification of pathway-based toxicity in the BALB/c 3T3 cell model. Toxicol. In Vitro 2015, 29, 1240–1253. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Yeom, H.; Eom, S.Y.; Lee, Y.-M.; Lee, M. Genome-wide DNA methylation changes in transformed foci induced by non-genotoxic carcinogens. Environ. Mol. Mutagen. 2019, 60, 576–587. [Google Scholar] [CrossRef]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic. Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Gu, H.; Bock, C.; Gnike, A.; Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.N.; Colacci, A.; Louekari, K.; Luijten, M.; Hakkert, B.C.; Paparella, M.; Vasseur, P. International regulatory needs for development of an IATA for non-genotoxic carcinogenic chemical substances. ALTEX 2016, 33, 359–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, D.; Kasper, P.; Martus, H.J.; Müller, L.; van Benthem, J.; Madia, F.; Corvi, R. Updated recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 795, 7–30. [Google Scholar] [CrossRef] [PubMed]
- ICH. M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk; ICH: Geneva, Switzerland, 2015. [Google Scholar]
- Van Leeuwen, K.; Schultz, T.W.; Henry, T.; Diderich, B.; Veith, G.D. Using chemical categories to fill data gaps in hazard assessment. SAR QSAR Environ. Res. 2009, 20, 207–220. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C.; Tcheremenskaia, O. In vitro cell transformation assays for an integrated, alternative assessment of carcinogenicity: A data-based analysis. Mutagenesis 2013, 28, 107–116. [Google Scholar] [CrossRef] [PubMed]
- NTP. NTP Technical Report on the Hepatotoxicity Studies of the Liver Carcinogen: Methapyrilene Hydrochloride; Toxicity Report Series 46; National Institute of Environmental Health Sciences: Durham, UK, 2000. [Google Scholar]
- NTP. NTP Technical Report on the Toxicology and Carcinogenesis Studies of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin (TCDD) (CAS No. 1746-01-6) in Female Harlan Sprague-Dawley Rats (Gavage Studies); Natl. Toxicol. Program Tech. Rep. 521; National Institute of Environmental Health Sciences: Durham, UK, 2006. [Google Scholar]
- Aonuma, S.; Ushijima, T.; Nakayasu, M.; Shima, H.; Sugimura, T.; Nagao, M. Mutation induction by okadaic acid, a protein phosphatase inhibitor, in CHL cells, but not in S. typhimurium. Mutat. Res. 1991, 250, 375–381. [Google Scholar] [CrossRef]
- Kindrat, I.; Dreval, K.; Shpyleva, S.; Tryndyak, V.; de Conti, A.; Mudalige, T.K.; Chen, T.; Erstenyuk, A.M.; Beland, F.A.; Pogribny, I.P. Effect of methapyrilene hydrochloride on hepatic intracellular iron metabolism in vivo and in vitro. Toxicol. Lett. 2017, 281, 65–73. [Google Scholar] [CrossRef]
- Lehman-McKeeman, L.D.; Rodriguez, P.A.; Takigiku, R.; Caudill, D.; Fey, M.L. d-Limonene-induced male rat-specific nephrotoxicity: Evaluation of the association between d-limonene and alpha 2u-globulin. Toxicol. Appl. Pharmacol. 1989, 99, 250–259. [Google Scholar] [CrossRef]
- Ito, Y.; Nakajima, T. PPARalpha- and DEHP-Induced Cancers. PPAR Res. 2008, 2008, 759716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B 2005, 175, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Suganuma, M.; Yoshizawa, S.; Nishiwaki, S.; Winyar, B.; Sugimura, T. Mechanisms of action of okadaic acid class tumor promoters on mouse skin. Environ. Health Perspect. 1991, 93, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Baijal, P.K.; Fitzpatrick, D.W.; Bird, R.P. Modulation of colonic xenobiotic metabolizing enzymes by feeding bile acids: Comparative effects of cholic, deoxycholic, lithocholic and ursodeoxycholic acids. Food Chem. Toxicol. 1998, 36, 601–607. [Google Scholar] [CrossRef]
- Zeisel, S.H. Choline. A nutrient that is involved in the regulation of cell proliferation, cell death, and cell transformation. Adv. Exp. Med. Biol. 1996, 399, 131–141. [Google Scholar]
- AstraZeneca. Crestor (rosuvastatin calcium) Tablets; Full Prescribing Information” (PDF); AstraZeneca: Cambridge, UK, 2016. [Google Scholar]
- Cohen, S.M.; Ellwein, L.B. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 1991, 51, 6493–6505. [Google Scholar]
- Chappel, C.I. A review and biological risk assessment of sodium saccharin. Regul. Toxicol. Pharmacol. 1992, 15, 253–270. [Google Scholar] [CrossRef]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Mazor, T.; Olshen, A.B.; Geng, H.; Gelston, L.C.; Akutagawa, J.; Lipka, D.B.; Plass, C.; Flotho, C.; Chehab, F.F.; et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2127. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kang, T.W.; Haam, K.; Kim, M.; Kim, S.K.; Kim, S.Y.; Lee, S.I.; Song, K.S.; Jeong, H.Y.; Kim, Y.S. Whole genome MBD-seq and RRBS analyses reveal that hypermethylation of gastrointestinal hormone receptors is associated with gastric carcinogenesis. Exp. Mol. Med. 2018, 50, 156. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive Changes in DNA Methylation are Associated with Expression of Mutant Huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervin, K.; Andreassen, B.K.; Hjorthaug, H.S.; Carlsen, K.C.L.; Carlsen, K.-H.; Undlien, D.E.; Lyle, R.; Munthe-Kaas, C. Intra-individual Changes in DNA Methylation Not Mediated by Cell-Type Composition Are Correlated with Aging During Childhood. Clin. Epigenerics 2016, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Caution, K.; Pan, A.; Krause, K.; Badr, A.; Hamilton, K.; Vaidya, A.; Gosu, H.; Daily, K.; Estfanous, S.; Gavrilin, M.A.; et al. Methylomic Correlates of Autophagy Activity in Cystic Fibrosis. J. Cyst. Fibros. 2019, 18, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, L.R.; Saavedra-López, E.; Romarate, L.; Mitxitorena, I.; Casanova, P.V.; Cribaro, G.P.; Gallego, J.M.; Pérez-Vallés, A.; Forteza-Vila, J.; Alfaro-Cervello, C.; et al. Imbalance of immunological synapse-kinapse states reflects tumor escape to immunity in glioblastoma. JCI Insight 2018, 3, e120757. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Sakurai, T.; Kakudo, K. Retinoid, Retinoic Acid Receptor Beta and Breast Cancer. Breast Cancer Res. Treat. 2002, 76, 167–173. [Google Scholar] [CrossRef]
- Parris, T.Z.; Vizlin-Hodzic, D.; Salmela, S.; Funa, K. Tumorigenic effects of TLX overexpression in HEK 293T cells. Cancer Rep. 2019, 2, e1204. [Google Scholar] [CrossRef]
- Lanceta, L.; O’Neill, C.; Lypova, N.; Li, X.; Rouchka, E.; Waigel, S.; Gomez-Gutierrez, J.G.; Chesney, J.; Imbert-Fernandez, Y. Transcriptomic Profiling Identifies Differentially Expressed Genes in Palbociclib-Resistant ER+ MCF7 Breast Cancer Cells. Genes 2020, 11, 467. [Google Scholar] [CrossRef]
- Sasaki, K.; Mizusawa, H.; Ishidate, M. Isolation and characterization of ras-transfected BALB/3T3 clone showing morphological transformation by 12-O-tetradecanoyl-phorbol-13-acetate. Jpn. J. Cancer Res. 1988, 79, 921–930. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Zabet, N.; Tsang, J. DMRcaller: Differentially Methylated Regions Caller. R Package Version 1.18.0. 2015. Available online: https://bioconductor.org/news/bioc_3_1_release/ (accessed on 2 March 2020).
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, B.C.; Maintainer, B.P. TxDb.Mmusculus.UCSC.mm10.knownGene: Annotation Package for TxDb Object(s). R Package Version 3.10.0. 2020. Available online: http://10.18129/B9.bioc.TxDb.Mmusculus.UCSC.mm10.knownGene (accessed on 2 March 2020).



{kind=link}
{kind=link}
{kind=link}
| Test Articles | Genotoxicity | Carcinogenicity | ICH M7 Prediction (Derek/Sarah) | OECD QSAR Toolbox Prediction |
|---|---|---|---|---|
| Methapyrilene hydrochloride (MH) | Equivocal in Ames test, Negative in in vitro CA and SCE, in vivo CA, SCE and MN | Potent hepatocarcinogen in rats | -/- | No alert found |
| D-limonene | Negative in Ames test, in vivo comet, in vitro CA | Male rat kidney tumors | -/- | No alert found |
| bis(2-ethylhexyl) phthalate (DEHP) | Negative in Ames test, in vivo MN, in vitro MN, MLA | IARC Group 2B | -/- | Structural alert for nongenotoxic carcinogenicity |
| TCDD | Negative in Ames test, in vitro MLA, CA, SCE, in vivo CA | IARC Group 1 | -/- | Structural alert for nongenotoxic carcinogenicity |
| Okadaic acid (OA) | Negative in Ames test, in vitro CHO/HGPRT, but mutagenic to CHL cells | Tumor promoter on mouse skin | -/- | No alert found |
| Cholic acid (CA) | Equivocal in Ames test, non-significant mutagenic activity in a battery of in vitro genotoxicity tests | Colon cancer promoter | -/- | No alert found |
| Diethanolamine (DEA) | Negative in Ames test, in vivo MN, in vitro CA | IARC Group 2B | -/- | No alert found |
| Rosuvastatin | Negative in Ames test, in vivo MN, in vitro CA, MLA | Positive response in mice and rats | -/- | Structural alert for nongenotoxic carcinogenicity |
| Melamine | Negative in Ame test, in vivo MN, in vitro CA | Bladder carcinoma | -/- | Structural alert for genotoxic carcinogenicity |
| Sodium saccharin (SA) | Negative in Ames test, in vivo CA and comet, in vitro MLA | Rat and mouse bladder tumors | -/equivocal | Structural alert for nongenotoxic carcinogenicity |
| Test Articles | Mode of Actions | Reference |
|---|---|---|
| Methapyrilene hydrochloride | The dysregulation of hepatic iron metabolism | [35] |
| D-limonene | α2 μ-globulin nephropathy | [36] |
| Bis(2-ethylhexyl) phthalate (DEHP) | Peroxisome proliferators | [37] |
| TCDD | Receptor binding | [38] |
| Okadaic acid | Inhibitors of protein serine/threonine phosphatases | [39] |
| Cholic acid | Inhibition of xenobiotic metabolizing enzyme | [40] |
| Diethanolamine | Choline deficiency | [41] |
| Rosuvastatin | Inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase | [42] |
| Melamine | Calculus formation | [43] |
| Sodium saccharin | Calculus formation | [44] |
| Conc. | % Viability | Foci/Well | Conc. | % Viability | Foci/Well |
|---|---|---|---|---|---|
| Sodium saccharin (128-44-9)—Positive | Methapyrilene HCl (135-23-9)—Positive | ||||
| 0 (μg/mL) | 100 | 3.5 ± 1.38 (a) | 0 (μg/mL) | 100 | 7.2 ± 0.75 |
| 500 | 107 | 9.3 ± 1.97 **,(b) | 3 | 107 | 14.5 ± 2.59 ** |
| 750 | 113 | 8.8 ± 1.94 ** | 3.5 | 110 | 16.0 ± 1.26 ** |
| 1000 | 93 | 9.3 ± 1.03 ** | 4 | 101 | 13.8 ± 1.60 ** |
| 1250 | 106 | 9.7 ± 1.21 ** | 4.5 | 100 | 15.0 ± 2.00 ** |
| 1500 | 94 | 8.2 ± 1.94 ** | 5 | 96 | 14.5 ± 2.43 ** |
| 1750 | 101 | 7.3 ± 2.16 ** | 5.5 | 99 | 10.5 ± 3.27 |
| DEHP (117-81-7)—Positive | D-limonene (5989-27-5)—Negative | ||||
| 0 (μg/mL) | 100 | 11.0 ± 1.79 | 0 (μg/mL) | 100 | 12.7 ± 3.01 |
| 2.5 | 93 | 13.8 ± 2.48 | 12.5 | 103 | 14.7 ± 3.72 |
| 5 | 92 | 19.5 ± 2.17 ** | 15 | 103 | 12.2 ± 3.19 |
| 10 | 91 | 20.5 ± 4.76 ** | 17.5 | 102 | 14.0 ± 2.00 |
| 25 | 95 | 18.5 ± 3.73 ** | 20 | 101 | 12.8 ± 3.60 |
| 50 | 93 | 18.5 ± 1.64 ** | 22.5 | 83 | 12.3 ± 2.07 |
| 100 | 90 | 13.0 ± 3.52 | 25 | 78 | 9.8 ± 2.14 |
| Melamine (108-78-1)—Negative | Rosuvastatin (287714-41-4)—Negative | ||||
| 0 (μM) | 100 | 7.5 ± 1.76 | 0 (μg/mL) | 100 | 8.0 ± 0.89 |
| 0.25 | 92 | 7.8 ± 2.23 | 0.1 | 103 | 8.8 ± 1.83 |
| 0.5 | 96 | 8.2 ± 2.48 | 0.25 | 100 | 7.8 ± 1.83 |
| 1 | 95 | 9.0 ± 2.68 | 0.5 | 102 | 8.7 ± 1.51 |
| 2.5 | 98 | 9.7 ± 3.08 | 0.75 | 117 | 8.2 ± 1.94 |
| 5 | 96 | 10.8 ± 2.23 | 1 | 125 | 9.8 ± 3.13 |
| 10 | 103 | 9.3 ± 3.20 | 2 | 116 | 8.7 ± 1.63 |
| Diethanolamine (111-42-2)—Positive | Okadaic acid (78111-17-8)—Weak positive | ||||
| 0 (μg/mL) | 100 | 7.8 ± 1.94 | 0 (ng/mL) | 100 | 8.8 ± 1.17 |
| 10 | 99 | 14.0 ± 3.22 * | 0.25 | 100 | 11.7 ± 2.25 |
| 25 | 117 | 16.7 ± 2.66 ** | 0.5 | 109 | 13.0 ± 3.35 * |
| 50 | 114 | 12.8 ± 3.87 | 0.75 | 97 | 11.2 ± 3.49 |
| 75 | 110 | 17.5 ± 2.07 ** | 1 | 102 | 12.8 ± 2.14 * |
| 100 | 88 | 16.8 ± 3.66 ** | 2 | 118 | 6.2 ± 0.75 |
| 150 | 112 | 17.8 ± 4.71 ** | 3 | 117 | 5.0 ± 1.41 |
| TCDD (1746-01-6)—Positive | Cholic acid (81-25-4)—Positive | ||||
| 0 (nM) | 100 | 11.3 ± 1.37 | 0 (μg/mL) | 100 | 5.0 ± 2.28 |
| 10 | 77 | 20.0 ± 3.35 ** | 10 | 124 | 8.8 ± 2.48 |
| 25 | 86 | 19.3 ± 3.14 ** | 25 | 119 | 9.0 ± 1.79 * |
| 50 | 81 | 21.2 ± 0.75 ** | 50 | 107 | 9.3 ± 1.97 * |
| 75 | 85 | 18.8 ± 3.43 ** | 100 | 122 | 9.0 ± 3.22 * |
| 100 | 89 | 19.7 ± 2.25 ** | 250 | 103 | 10.0 ± 2.68 ** |
| 200 | 79 | 17.3 ± 1.86 ** | 500 | 131 | 11.8 ± 2.99 ** |
| Associated Gene | Description | DMR Position | DMR Distance from TSS (1) | DMCs Number | Methylation | NGTXC |
|---|---|---|---|---|---|---|
| Genes harboring overlapping DMRs in 4 NGTXCs, -induced transformed foci | ||||||
| Asic1 | Acid-sensing (proton-gated) ion channel 1 | chr15: 99,691,202–99,691,300 | −933 | 8 | hypomethylated | DEHP/OA/TCDD/MH |
| Gm14169 | Predicted gene 14169 | chr2: 156,613,303–156,613,401 | 21 | 18 | hypermethylated | CA/DEHP/OA/TCDD |
| Gm7337 | DAZ interacting protein 1 pseudogene | chr5: 87,851,102–87,851,200 | 584 | 10 | hypomethylated | CA/DEHP/OA/MH |
| Gpsm2 | G-protein signaling modulator 2 | chr3: 108,721,702–108,721,900 | 216 | 26 | hypomethylated | CA/DEHP/OA/TCDD |
| Irs1 | Insulin receptor substrate 1 | chr1: 82,290,404–82,290,502 | 914 | 18 | hypermethylated | OA/TCDD/DEA/SS |
| Mpv17 | MpV17 mitochondrial inner membrane protein | ch5: 31,154,402–31,154,500 | −151 | 8 | hypermethylated | CA/DEHP/OA/TCDD |
| Nexn | Nexilin | chr3: 152,265,802–152,265,900 | −7 | 22 | hypomethylated | DEHP/DEA/MH/SS |
| Nrip2 | Nuclear receptor interacting protein 2 | chr6: 128,401,302–128,401,400 | 1009 | 2 | hypomethylated | CA/DEHP/TCDD/SS |
| Nsun7 | NOL1/NOP2/Sun domain family, member 7 | chr5: 66,261,102–66,261,200 | 790 | 10 | hypomethylated | CA/TCDD/DEA/SS |
| Pcdh7 | Protocadherin 7 | chr5: 57,720,202–57,720,300 | 79 | 14 | hypermethylated | CA/OA/DEA/MH |
| Pou6f1 | POU domain, class 6, transcription factor 1 | chr15: 100,599,102–100,599,200 | 664 | 6 | hypermethylated | CA/DEHP/TCDD/SS |
| Unc13a | Unc-13 homolog A | chr8: 71,668,402–71,668,500 | −320 | 12 | hypermethylated | CA/DEHP/OA/TCDD |
| Zfp882 | Zinc finger protein 882 | chr8: 71,908,855–71,908,954 | 0 | 14 | hypermethylated | TCDD/DEA/MH/SS |
| Genes harboring overlapping DMRs in 5 NGTXCs-induced transformed foci | ||||||
| H2-D1 | Histocompatibility 2, D region locus 1 | chr17: 35,263,302–35,263,400 | 206 | 23 | hypomethylated | CA/DEHP/DEA/MH/SS |
| Mir705 | MicroRNA 705 | chr6: 85,337,102–85,337,200 | −729 | 6 | hypomethylated | DEHP/OA/DEA/MH/SS |
| NGTXC | Categories | Disease or Functions Annotation | p-Value | Number of Molecules |
|---|---|---|---|---|
| Cholic acid | Cellular assembly and organization | Release of vesicles | 0.00605 | 2 |
| Lipid metabolism | Concentration of malonyl-coenzyme A | 0.00913 | 1 | |
| Embryonic development | Attachment of chorioallantoic membrane | 0.00913 | 1 | |
| Cancer, cellular growth and proliferation | Proliferation of mammary tumor cells | 0.0182 | 1 | |
| DEHP | Behavior | Learning | 7.93 × 10−6 | 25 |
| Cell-to-cell signaling and interaction | Uptake of neurotransmitter | 1.85 × 10−5 | 5 | |
| Neurological Disease | Cognitive impairment | 0.00009 | 16 | |
| Cell death and survival | Apoptosis of tumor cell lines | 0.0053 | 13 | |
| Okadaic aicd | Gastrointestinal disease | Meteorism | 0.000468 | 2 |
| Cell-to-cell signaling and interaction | Developmental process of synapse | 0.000555 | 4 | |
| Respiratory disease | Abnormal secretion of pulmonary surfactant | 0.00135 | 2 | |
| TCDD | Cellular movement | Cellular infiltration by macrophages | 7.68 × 10−6 | 9 |
| Behavior | Long-term recognition memory | 5.16 × 10−5 | 4 | |
| Cardiovascular system development and function | Systolic pressure | 0.00014 | 7 | |
| cancer | Cell transformation | 0.00125 | 9 | |
| Cell death and survival | Apoptosis | 0.00531 | 17 | |
| Diethanol-amine | Embryonic development | Formation of visceral endoderm | 0.000195 | 2 |
| Cell cycle | Aneuploidy of embryonic cell lines | 0.000291 | 2 | |
| Cell cycle | Aneuploidy of fibroblast cell lines | 0.000407 | 2 | |
| Cell-to-cell signaling and interaction | Developmental process of synapse | 0.00116 | 4 | |
| Cancer | Hyperplasia of urothelium | 0.00446 | 1 | |
| Methapyrilene HCl | Connective tissue development and function | Osteogenesis | 0.00316 | 2 |
| Cell-to-cell signaling and interaction | Synaptic transmission | 0.00793 | 6 | |
| Reproductive system development and function | Gestation | 0.00814 | 3 | |
| Sodium saccharin | Connective tissue development and function | Thickness of bone | 1.93 × 10−5 | 7 |
| Skeletal and muscular system development and function | Mineralization of bone | 3.44 × 10−5 | 8 | |
| Connective tissue disorders | Synostosis of cranium | 6.38 × 10−5 | 2 | |
| Behavior | Social behavior | 9.72 × 10−5 | 6 |
| NGTXC | Ingenuity Canonical Pathways | −log (p-value) | Ratio | Molecules |
|---|---|---|---|---|
| Cholic acid | Notch signaling | 2.37 | 0.0833 | CNTN1, DTX1, LFNG |
| Axonal guidance signaling | 1.97 | 0.0214 | ADAM19, ADAMTS1, GNG2, MYL12A, PRKAR1B, SEMA4A, SEMA4G, SEMA6C, SRGAP1, SRGAP3 | |
| Thyroid hormone biosynthesis | 1.57 | 0.333 | IYD | |
| RAR activation | 1.02 | 0.0211 | Akr1b10, NRIP2, PRKAR1B, SMARCD3 | |
| Gustation pathway | 0.863 | 0.0214 | GNG2, PDE1B, PRKAR1B | |
| Salvage pathways of pyrimidine ribonucleotides | 0.695 | 0.022 | DAPK1, GRK4 | |
| DEHP | Dopamine-DARPP32 feedback in cAMP Signaling | 3.54 | 0.0692 | GNAI1, GRIN1, GRIN3B, KCNJ12, NOS1, PPP1R1B, PPP2R5A, PRKACA, PRKAR1B, PRKCZ, PRKG2 |
| Notch signaling | 2.3 | 0.111 | DLL4, DTX1, HEY1, NOTCH4 | |
| nNOS signaling in neurons | 1.95 | 0.0889 | GRIN1, GRIN3B, NOS1, PRKCZ | |
| Gustation pathway | 1.25 | 0.0429 | ASIC1, GNG2, P2RY12, PDE6B, PRKACA, PRKAR1B | |
| Salvage pathways of pyrimidine ribonucleotides | 0.585 | 0.033 | AK1, DAPK1, GRK4 | |
| Okadaic aicd | Glutamate degradation II | 1.96 | 0.333 | GOT2 |
| L-Serine degradation | 1.96 | 0.333 | SDS | |
| Aspartate biosynthesis | 1.96 | 0.333 | GOT2 | |
| RAR activation | 1.49 | 0.0158 | Akr1b10, MAP2K5, NSD1 | |
| Salvage pathways of pyrimidine ribonucleotides | 1.36 | 0.022 | DAPK1, GRK4 | |
| Gustation pathway | 1.03 | 0.0143 | ASIC1, CACNB1 | |
| TCDD | Pyridoxal 5′-phosphate salvage pathway | 2.48 | 0.0625 | GRK4, MAP2K6, PDXK, PKN1 |
| PPARα/RXRα activation | 2.1 | 0.0331 | APOA1, GOT2, IRS1, MAP2K6, PPARA, PRKAR1A | |
| Apelin liver signaling pathway | 1.6 | 0.0769 | APLNR, IRS1 | |
| Salvage pathways of pyrimidine ribonucleotides | 1.25 | 0.033 | GRK4, MAP2K6, PKN1 | |
| Gustation pathway | 0.824 | 0.0214 | ASIC1, CACNA1D, PRKAR1A | |
| Diethanolamine | Regulation of the epithelial–mesenchymal Transition pathway | 1.99 | 0.0211 | CLDN3, FGF19, FGFR2, NOTCH1 |
| Glutamate degradation II | 1.88 | 0.333 | GOT2 | |
| Aspartate Biosynthesis | 1.88 | 0.333 | GOT2 | |
| Cancer drug resistance by drug efflux | 1.56 | 0.0345 | miR-133, PTEN | |
| Gustation pathway | 0.889 | 0.0143 | CACNB1, PDE8B | |
| PPARα/RXRα activation | 0.714 | 0.011 | GOT2, IRS1 | |
| Methapyrilene HCl | γ-glutamyl cycle | 4.14 | 0.3 | GGCT, GGT6, GGT7 |
| Leukotriene biosynthesis | 2.34 | 0.167 | GGT6, GGT7 | |
| Valine degradation I | 1.99 | 0.111 | ABAT, SDS | |
| Autophagy | 1.05 | 0.0345 | CTSF, ULK1 | |
| Phagosome maturation | 0.921 | 0.0214 | CTSF, HLA-A, TCIRG1 | |
| VDR/RXR activation | 0.839 | 0.0256 | BGLAP, IGFBP5 | |
| Salvage pathways of pyrimidine Ribonucleotides | 0.264 | 0.011 | CDK5 | |
| Sodium saccharin | Gustation pathway | 3.03 | 0.0429 | P2RX7, P2RY12, PDE1B, PDE8B, PDE9A, PRKACA |
| tRNA splicing | 2.37 | 0.0732 | PDE1B, PDE8B, PDE9A | |
| G-protein coupled receptor signaling | 1.68 | 0.0225 | P2RY12, PDE1B, PDE8B, PDE9A, PRKACA, PRKCB | |
| VDR/RXR activation | 1.61 | 0.0385 | PRKCB, RUNX2, SPP1 | |
| Phagosome maturation | 1.58 | 0.0286 | ATP6V1G3, HLA-A, LPO, TCIRG1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.-H.; Yeom, H.; Han, B.-I.; Ham, B.-J.; Lee, Y.-M.; Han, M.-R.; Lee, M. Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation. Int. J. Mol. Sci. 2020, 21, 5387. https://doi.org/10.3390/ijms21155387
Hwang S-H, Yeom H, Han B-I, Ham B-J, Lee Y-M, Han M-R, Lee M. Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation. International Journal of Molecular Sciences. 2020; 21(15):5387. https://doi.org/10.3390/ijms21155387
Chicago/Turabian StyleHwang, Sung-Hee, Hojin Yeom, Byeal-I Han, Byung-Joo Ham, Yong-Moon Lee, Mi-Ryung Han, and Michael Lee. 2020. "Predicting Carcinogenic Mechanisms of Non-Genotoxic Carcinogens via Combined Analysis of Global DNA Methylation and In Vitro Cell Transformation" International Journal of Molecular Sciences 21, no. 15: 5387. https://doi.org/10.3390/ijms21155387