Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction: Cancer Heterogeneity and Tumor-Initiation Models
2. Glioblastoma (GBM) and Putative Cells of Origin
3. Glioblastoma Stem Cells (GSCs)
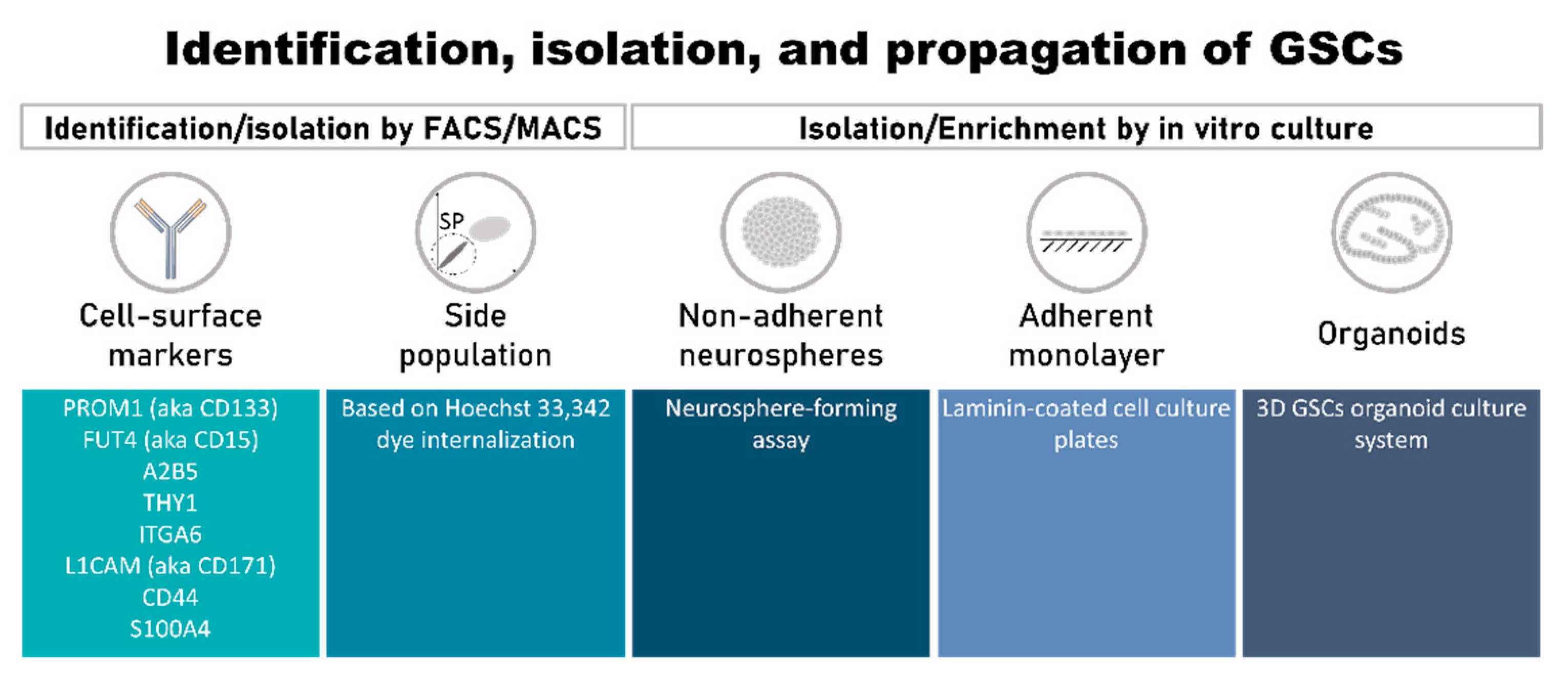
3.1. Identification, Isolation, and Propagation of GSCs
3.1.1. Cell-Surface Markers of GSCs
CD133 (Official Symbol: PROM1)
CD15 (Official Symbol: FUT4)
A2B5
CD90 (Official Symbol: THY1)
ITGA6
CD171 (Official Symbol: L1CAM)
CD44
S100A4
3.1.2. Side Population
3.1.3. Methods of GSC Isolation/Enrichment and Culture In Vitro
4. GSC Molecular Features Amenable for Therapeutic Intervention
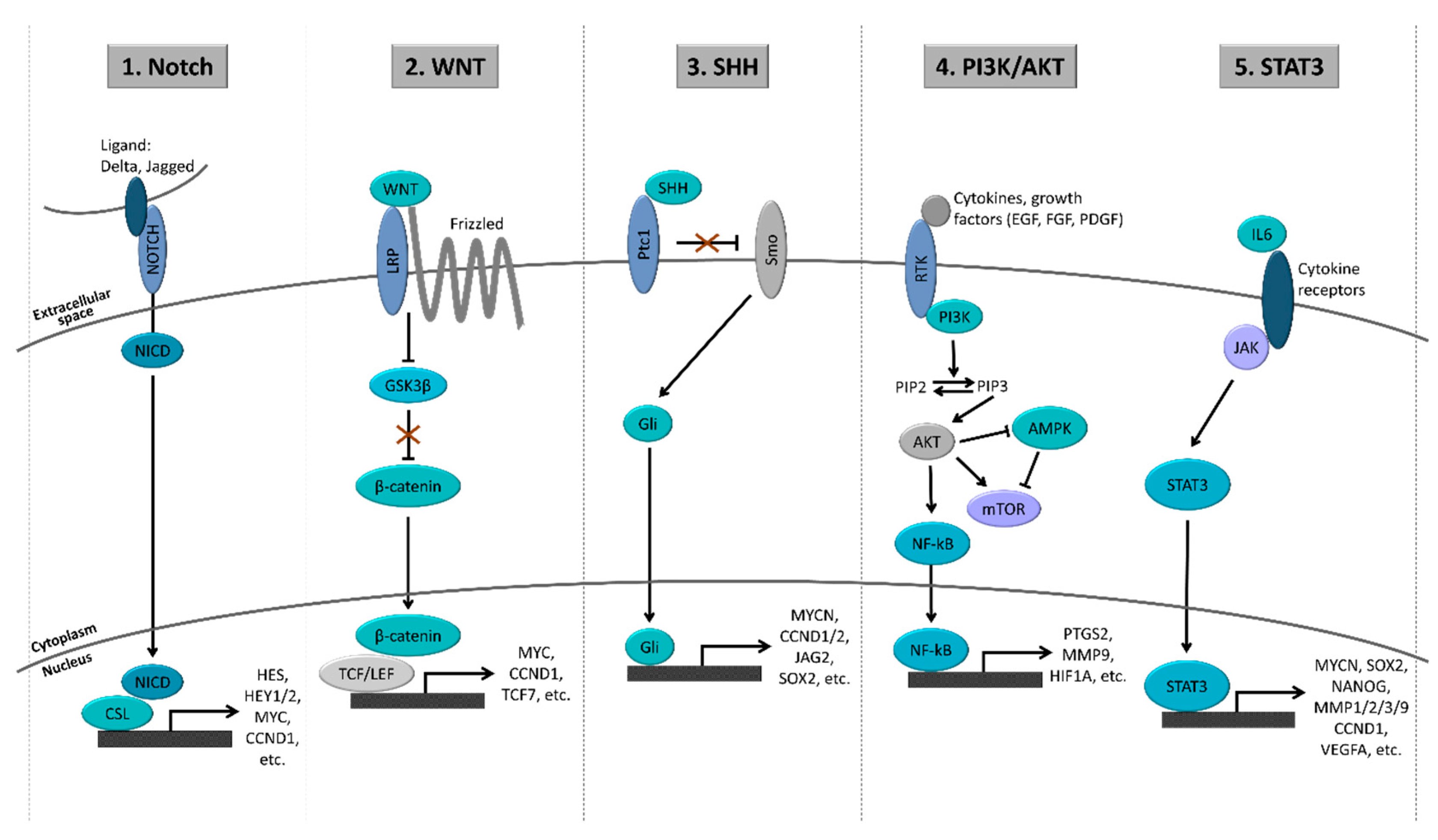
4.1. Major Signaling Pathways in GSCs
4.1.1. Notch Pathway
4.1.2. WNT Pathway
4.1.3. Sonic Hedgehog (SHH) Pathway
4.1.4. PI3K/AKT Pathway
4.1.5. STAT3 Pathway
4.2. Targeting GSCs Vascular and Hypoxic Microenvironment
4.3. Targeting GSCs by Inducing Differentiation
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Inda, M.M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Shen, M.M. The roots of cancer: Stem cells and the basis for tumor heterogeneity. Bioessays 2013, 35, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.; Lutz, C.; Van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.-L. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notta, F.; Mullighan, C.G.; Wang, J.C.; Poeppl, A.; Doulatov, S.; Phillips, L.A.; Ma, J.; Minden, M.D.; Downing, J.R.; Dick, J.E. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011, 469, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.P. Identifying cancer stem cells in solid tumors: Case not proven. Cancer Res. 2006, 66, 1891–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Ebben, J.D.; Treisman, D.M.; Zorniak, M.; Kutty, R.G.; Clark, P.A.; Kuo, J.S. The cancer stem cell paradigm: A new understanding of tumor development and treatment. Expert Opin. Ther. Targets 2010, 14, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Lamszus, K. The neurobiology of gliomas: From cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 2011, 12, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Alvarez-Buylla, A.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Jacques, T.S.; Swales, A.; Brzozowski, M.J.; Henriquez, N.V.; Linehan, J.M.; Mirzadeh, Z.; O’Malley, C.; Naumann, H.; Alvarez-Buylla, A.; Brandner, S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010, 29, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009, 15, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Zheng, H.; Tomasek, G.J.; Zhang, P.; McKeever, P.E.; Lee, E.Y.; Zhu, Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 2009, 15, 514–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243. [Google Scholar] [CrossRef]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.S.; James, D.; Iyer, R.; Falciatori, I.; Hambardzumyan, D.; Wang, S.; Butler, J.M.; Rabbany, S.Y.; Hormigo, A. Prominin 1/CD133 endothelium sustains growth of proneural glioma. PLoS ONE 2013, 8, e62150. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.S.; Lee, J.I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Investig. 2008, 88, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fine, H.A. Glioma stem cells: Not all created equal. Cancer Cell 2009, 15, 247–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunther, H.S.; Schmidt, N.O.; Phillips, H.S.; Kemming, D.; Kharbanda, S.; Soriano, R.; Modrusan, Z.; Meissner, H.; Westphal, M.; Lamszus, K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 2008, 27, 2897–2909. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sakariassen, P.O.; Tsinkalovsky, O.; Immervoll, H.; Boe, S.O.; Svendsen, A.; Prestegarden, L.; Rosland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Lottaz, C.; Beier, D.; Meyer, K.; Kumar, P.; Hermann, A.; Schwarz, J.; Junker, M.; Oefner, P.J.; Bogdahn, U.; Wischhusen, J.; et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010, 70, 2030–2040. [Google Scholar] [CrossRef] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.-C.; Paris, F.; Pecqueur, C. Glioblastoma stem-like cells, metabolic strategy to kill a challenging target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Chandran, U.R.; Luthra, S.; Santana-Santos, L.; Mao, P.; Kim, S.-H.; Minata, M.; Li, J.; Benos, P.V.; DeWang, M.; Hu, B.; et al. Gene expression profiling distinguishes proneural glioma stem cells from mesenchymal glioma stem cells. Genom. Data 2015, 5, 333–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saga, I.; Shibao, S.; Okubo, J.; Osuka, S.; Kobayashi, Y.; Yamada, S.; Fujita, S.; Urakami, K.; Kusuhara, M.; Yoshida, K.; et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014, 16, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Tunici, P.; Bissola, L.; Lualdi, E.; Pollo, B.; Cajola, L.; Broggi, G.; Sozzi, G.; Finocchiaro, G. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol. Cancer 2004, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sanchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Wong, K.K.; Su, J.M.; Adesina, A.M.; Yu, L.T.; Tsang, Y.T.; Antalffy, B.C.; Baxter, P.; Perlaky, L.; Yang, J.; et al. Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 2008, 26, 1414–1424. [Google Scholar] [CrossRef] [Green Version]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fargeas, C.A.; Huttner, W.B.; Corbeil, D. Nomenclature of prominin-1 (CD133) splice variants–An update. Tissue Antigens 2007, 69, 602–606. [Google Scholar] [CrossRef]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef]
- Wang, C.H.; Chiou, S.H.; Chou, C.P.; Chen, Y.C.; Huang, Y.J.; Peng, C.A. Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine 2011, 7, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Emlet, D.R.; Gupta, P.; Holgado-Madruga, M.; Del Vecchio, C.A.; Mitra, S.S.; Han, S.Y.; Li, G.; Jensen, K.C.; Vogel, H.; Xu, L.W.; et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Kong, W.; Falk, A.; Hu, J.; Zhou, L.; Pollard, S.; Smith, A. CD133 (Prominin) negative human neural stem cells are clonogenic and tripotent. PLoS ONE 2009, 4, e5498. [Google Scholar] [CrossRef] [Green Version]
- Capela, A.; Temple, S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron 2002, 35, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Suva, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [Green Version]
- Tchoghandjian, A.; Baeza, N.; Colin, C.; Cayre, M.; Metellus, P.; Beclin, C.; Ouafik, L.; Figarella-Branger, D. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain Pathol. 2010, 20, 211–221. [Google Scholar] [CrossRef]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Waziri, A.E.; Lochhead, R.A.; Fusco, D.; Lopez, K.; Ellis, J.A.; Kang, J.; Assanah, M.; McKhann, G.M.; Sisti, M.B.; et al. Identification of A2B5+CD133- tumor-initiating cells in adult human gliomas. Neurosurgery 2008, 62, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Baeza-Kallee, N.; Bergès, R.; Soubéran, A.; Colin, C.; Denicolaï, E.; Appay, R.; Tchoghandjian, A.; Figarella-Branger, D. Glycolipids Recognized by A2B5 Antibody Promote Proliferation, Migration, and Clonogenicity in Glioblastoma Cells. Cancers 2019, 11, 1267. [Google Scholar] [CrossRef] [Green Version]
- Auvergne, R.M.; Sim, F.J.; Wang, S.; Chandler-Militello, D.; Burch, J.; Al Fanek, Y.; Davis, D.; Benraiss, A.; Walter, K.; Achanta, P.; et al. Transcriptional differences between normal and glioma-derived glial progenitor cells identify a core set of dysregulated genes. Cell Rep. 2013, 3, 2127–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, C.M.; Weissman, I.L.; Tsukamoto, A.S.; Buckle, A.M.; Peault, B. Isolation of a candidate human hematopoietic stem-cell population. Proc. Natl. Acad. Sci. USA 1992, 89, 2804–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. 2012, 11, M111 010744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fael Al-Mayhani, T.M.; Ball, S.L.; Zhao, J.W.; Fawcett, J.; Ichimura, K.; Collins, P.V.; Watts, C. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J. Neurosci. Methods 2009, 176, 192–199. [Google Scholar] [CrossRef]
- Hall, P.E.; Lathia, J.D.; Caldwell, M.A.; Ffrench-Constant, C. Laminin enhances the growth of human neural stem cells in defined culture media. BMC Neurosci. 2008, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef]
- Izumoto, S.; Ohnishi, T.; Arita, N.; Hiraga, S.; Taki, T.; Hayakawa, T. Gene expression of neural cell adhesion molecule L1 in malignant gliomas and biological significance of L1 in glioma invasion. Cancer Res. 1996, 56, 1440–1444. [Google Scholar] [PubMed]
- Suzuki, T.; Izumoto, S.; Fujimoto, Y.; Maruno, M.; Ito, Y.; Yoshimine, T. Clinicopathological study of cellular proliferation and invasion in gliomatosis cerebri: Important role of neural cell adhesion molecule L1 in tumour invasion. J. Clin. Pathol. 2005, 58, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; Li, Z.; Sathornsumetee, S.; Wang, H.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008, 68, 6043–6048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813. [Google Scholar] [CrossRef]
- Naor, D.; Wallach-Dayan, S.B.; Zahalka, M.A.; Sionov, R.V. Involvement of CD44, a molecule with a thousand faces, in cancer dissemination. Semin. Cancer Biol. 2008, 18, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.G.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.A.; Neilson, E.G.; et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Little, M.H. A side order of stem cells: The SP phenotype. Stem Cells 2006, 24, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hadnagy, A.; Gaboury, L.; Beaulieu, R.; Balicki, D. SP analysis may be used to identify cancer stem cell populations. Exp. Cell Res. 2006, 312, 3701–3710. [Google Scholar] [CrossRef]
- Srivastava, V.K.; Nalbantoglu, J. Flow cytometric characterization of the DAOY medulloblastoma cell line for the cancer stem-like phenotype. Cytom. Part A J. Int. Soc. Anal. Cytol. 2008, 73, 940–948. [Google Scholar] [CrossRef]
- Dirks, P.B. Brain tumor stem cells: Bringing order to the chaos of brain cancer. J. Clin. Oncol. 2008, 26, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bez, A.; Corsini, E.; Curti, D.; Biggiogera, M.; Colombo, A.; Nicosia, R.F.; Pagano, S.F.; Parati, E.A. Neurosphere and neurosphere-forming cells: Morphological and ultrastructural characterization. Brain Res. 2003, 993, 18–29. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Venere, M.; Rao, M.S.; Rich, J.N. Seeing is believing: Are cancer stem cells the Loch Ness monster of tumor biology? Stem Cell Rev. 2011, 7, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Fine, H.A.; Dirks, P.B.; Rich, J.N. Cancer stem cells in gliomas: Identifying and understanding the apex cell in cancer’s hierarchy. Glia 2011, 59, 1148–1154. [Google Scholar] [CrossRef] [Green Version]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, B.; Mathew, R.K.; Polson, E.S.; Williams, J.; Wurdak, H. Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov 2018, 23, 862–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma model using human cerebral organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef]
- Shih, A.H.; Holland, E.C. Notch signaling enhances nestin expression in gliomas. Neoplasia 2006, 8, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.M.; Jin, X.; Lee, J.S.; Oh, S.Y.; Sohn, Y.W.; Park, H.J.; Joo, K.M.; Park, W.Y.; Nam, D.H.; DePinho, R.A.; et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 2008, 22, 2028–2033. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.Y.; Zheng, M.H.; Cheng, G.; Li, L.; Liang, L.; Gao, F.; Wei, Y.N.; Fu, L.A.; Han, H. Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer 2011, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, C.; Meng, Q.; Li, S.; Sun, X.; Bo, Y.; Yao, W. siRNA targeting Notch-1 decreases glioma stem cell proliferation and tumor growth. Mol. Biol. Rep. 2012, 39, 2497–2503. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Fu, J.; Zheng, S.; Yao, J.; Wang, S.; Liu, D.D.; Yuan, Y.; Sulman, E.P.; Lang, F.F.; Colman, H. A High Notch Pathway Activation Predicts Response to γ Secretase Inhibitors in Proneural Subtype of Glioma Tumor-Initiating Cells. Stem Cells 2014, 32, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Yahyanejad, S.; King, H.; Iglesias, V.S.; Granton, P.V.; Barbeau, L.M.; van Hoof, S.J.; Groot, A.J.; Habets, R.; Prickaerts, J.; Chalmers, A.J. NOTCH blockade combined with radiation therapy and temozolomide prolongs survival of orthotopic glioblastoma. Oncotarget 2016, 7, 41251. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-B.; Jiang, H.; Zhan, R.-Y. Aberrant Notch signaling in glioblastoma stem cells contributes to tumor recurrence and invasion. Mol. Med. Rep. 2016, 14, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Mirzaei, R.; Zemp, F.J.; Wei, W.; Senger, D.L.; Robbins, S.M.; Yong, V.W. Activation of NOTCH Signaling by Tenascin-C Promotes Growth of Human Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 3231–3243. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Shimizu, F.; Hovinga, K.; Beal, K.; Karimi, S.; Droms, L.; Peck, K.K.; Gutin, P.; Iorgulescu, J.B.; Kaley, T. Molecular and clinical effects of Notch inhibition in glioma patients: A phase 0/I trial. Clin. Cancer Res. 2016, 22, 4786–4796. [Google Scholar] [CrossRef] [Green Version]
- Pan, E.; Supko, J.G.; Kaley, T.J.; Butowski, N.A.; Cloughesy, T.; Jung, J.; Desideri, S.; Grossman, S.; Ye, X.; Park, D.M. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J. Neurooncol. 2016, 130, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, K.; Mirzadeh, Z.; Sakaguchi, M.; Yamashita, T.; Nikolcheva, T.; Gotoh, Y.; Peltz, G.; Gong, L.; Kawase, T.; Alvarez-Buylla, A.; et al. Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 2007, 25, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Gotoh, Y. Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci. Res. 2005, 51, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Mix, E.; Rateitschak, K.; Rolfs, A. Wnt signal pathways and neural stem cell differentiation. Neurodegener. Dis. 2006, 3, 76–86. [Google Scholar] [CrossRef]
- McCord, M.; Mukouyama, Y.-s.; Gilbert, M.R.; Jackson, S. Targeting WNT signaling for multifaceted glioblastoma therapy. Front. Cell. Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, C.J.; Altschuler, G.; Jeong, J.; Stromme, K.K.; Stangeland, B.; Murrell, W.; Grasmo-Wendler, U.H.; Myklebost, O.; Helseth, E.; Vik-Mo, E.O.; et al. Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt- signaling and a fingerprint associated with clinical outcome. Exp. Cell Res. 2013, 319, 2230–2243. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, C.S.; Vieira de Castro, J.; Pojo, M.; Martins, E.P.; Queirós, S.; Chautard, E.; Taipa, R.; Pires, M.M.; Pinto, A.A.; Pardal, F.; et al. WNT6 is a Novel Oncogenic Prognostic Biomarker in Human Glioblastoma. Theranostics 2018, 8, 4805–4823. [Google Scholar] [CrossRef]
- Gonçalves, C.S.; Xavier-Magalhães, A.; Martins, E.P.; Pinto, A.A.; Pires, M.M.; Pinheiro, C.; Reis, R.M.; Sousa, N.; Costa, B.M. A novel molecular link between HOXA9 and WNT6 in glioblastoma identifies a subgroup of patients with particular poor prognosis. Mol. Oncol. 2020, 14, 1224–1241. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauve, C.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295.e1218. [Google Scholar] [CrossRef] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. WNT5a drives an invasive phenotype in human glioblastoma stem-like cells. Cancer Res. 2017, 77, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Salaroglio, I.C.; Caldera, V.; Campia, I.; Kopecka, J.; Mellai, M.; Annovazzi, L.; Bosia, A.; Ghigo, D.; Schiffer, D. Temozolomide downregulates P-glycoprotein expression in glioblastoma stem cells by interfering with the Wnt3a/glycogen synthase-3 kinase/β-catenin pathway. Neuro Oncol. 2013, 15, 1502–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, N.; Chettiar, S.; Rathod, S.; Rath, P.; Muzumdar, D.; Shaikh, M.L.; Shiras, A. WNT3a mediated activation of WNT/beta-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell. Neurosci. 2013, 54, 44–57. [Google Scholar] [CrossRef]
- Jin, X.; Jeon, H.Y.; Joo, K.M.; Kim, J.K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 regulates stemness and invasiveness of migrating glioma cells established by serial intracranial transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.H.; Zhang, H.; Xiao, Y.; Perry, S.R.; et al. PLAGL2 regulates WNT signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, Y.; Gao, R.; Xiu, Z.; Sun, T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/β-catenin signaling pathway. Oncogene 2019, 39, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Xu, H.; Huang, M.; Ma, W.; Saxena, D.; Lustig, R.A.; Alonso-Basanta, M.; Zhang, Z.; O’Rourke, D.M.; Zhang, L. Circulating glioma cells exhibit stem cell-like properties. Cancer Res. 2018, 78, 6632–6642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/beta-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjörnsson, B.; Sandén, E.; Darabi, A.; Siesjö, P.; Kool, M.; et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 8904. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [Green Version]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.-C. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell 2018, 33, 874–889.e877. [Google Scholar] [CrossRef] [Green Version]
- Van Brocklyn, J.R.; Wojton, J.; Meisen, W.H.; Kellough, D.A.; Ecsedy, J.A.; Kaur, B.; Lehman, N.L. Aurora-A inhibition offers a novel therapy effective against intracranial glioblastoma. Cancer Res. 2014, 74, 5364–5370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, O.A.; Özcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- DuBois, S.G.; Mosse, Y.P.; Fox, E.; Kudgus, R.A.; Reid, J.M.; McGovern, R.; Groshen, S. Phase II Trial of Alisertib in Combination with Irinotecan and Temozolomide for Patients with Relapsed or Refractory Neuroblastoma. Clin. Cancer Res. 2018, 24, 6142–6149. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falchook, G.; Coleman, R.L.; Roszak, A.; Behbakht, K.; Matulonis, U.; Ray-Coquard, I.; Sawrycki, P.; Duska, L.R.; Tew, W.; Ghamande, S.; et al. Alisertib in Combination With Weekly Paclitaxel in Patients With Advanced Breast Cancer or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA Oncol. 2019, 5, e183773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wen, Y.L.; Ma, J.W.; Ye, J.C.; Wang, X.; Huang, J.X.; Meng, C.Y.; Xu, X.Z.; Wang, S.X.; Zhong, X.Y. Tetrandrine inhibits glioma stem-like cells by repressing β-catenin expression. Int. J. Oncol. 2017, 50, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Joyner, A.L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 2005, 437, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Pahlman, S.; Stockhausen, M.T.; Fredlund, E.; Axelson, H. Notch signaling in neuroblastoma. Semin. Cancer Biol. 2004, 14, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Pitz, M.; Hamai, A.; Siemianowicz, K.; Likus, W.; Wiechec, E.; Toyota, B.D. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol. Ther. 2018, 184, 13–41. [Google Scholar] [CrossRef]
- Shahi, M.; Farheen, S.; Mariyath, M.; Castresana, J. Potential role of Shh-Gli1-BMI1 signaling pathway nexus in glioma chemoresistance. Tumor Biol. 2016, 37, 15107–15114. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar]
- Xu, Q.; Yuan, X.; Liu, G.; Black, K.L.; Yu, J.S. Hedgehog signaling regulates brain tumor-initiating cell proliferation and portends shorter survival for patients with PTEN-coexpressing glioblastomas. Stem Cells 2008, 26, 3018–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro Oncol. 2013, 15, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Niu, M.; Xie, P.; Yue, C.; Liu, N.; Qi, Z.; Gao, S.; Liu, H.; Shi, Q.; Yu, R.; et al. Smoothened is a poor prognosis factor and a potential therapeutic target in glioma. Sci. Rep. 2017, 7, 42630. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Jin, X.; Kim, L.J.Y.; Dixit, D.; Jeon, H.-Y.; Kim, E.-J.; Kim, J.-K.; Lee, S.Y.; Yin, J.; Rich, J.N.; et al. Inhibition of ID1-BMPR2 Intrinsic Signaling Sensitizes Glioma Stem Cells to Differentiation Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Filbin, M.G.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef] [Green Version]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffero, F.; Daga, A.; Marubbi, D.; Capra, M.C.; Melotti, A.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Porcile, C.; Barbieri, F.; et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J. Biol. Chem. 2009, 284, 7138–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Jiang, Y.; Zou, F.; Liu, Y.; Wang, S.; Xu, N.; Xu, W.; Cui, C.; Xing, Y.; Liu, Y.; et al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6829–6834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeda, A.; Inagaki, A.; Oka, N.; Ikegame, Y.; Aoki, H.; Yoshimura, S.; Nakashima, S.; Kunisada, T.; Iwama, T. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J. Biol. Chem. 2008, 283, 10958–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyler, C.E.; Foo, W.C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; He, Y.-Y. Targeting the AMP-activated protein kinase for cancer prevention and therapy. Front. Oncol. 2013, 3, 175. [Google Scholar] [CrossRef] [Green Version]
- Chhipa, R.R.; Fan, Q.; Anderson, J.; Muraleedharan, R.; Huang, Y.; Ciraolo, G.; Chen, X.; Waclaw, R.; Chow, L.M. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018, 20, 823–835. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef]
- Wang, W.J.; Long, L.M.; Yang, N.; Zhang, Q.Q.; Ji, W.J.; Zhao, J.H.; Qin, Z.H.; Wang, Z.; Chen, G.; Liang, Z.Q. NVP-BEZ235, a novel dual PI3K/mTOR inhibitor, enhances the radiosensitivity of human glioma stem cells in vitro. Acta Pharmacol. Sin. 2013, 34, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, K.; Wang, S.; Balasubramaniyan, V.; Conrad, C.; Lang, F.F.; Aldape, K.; Szymanski, S.; Fokt, I.; Dasgupta, A.; Madden, T.; et al. Induction of cell-cycle arrest and apoptosis in glioblastoma stem-like cells by WP1193, a novel small molecule inhibitor of the JAK2/STAT3 pathway. J. Neurooncol. 2012, 107, 487–501. [Google Scholar] [CrossRef]
- Almiron Bonnin, D.A.; Havrda, M.C.; Lee, M.C.; Liu, H.; Zhang, Z.; Nguyen, L.N.; Harrington, L.X.; Hassanpour, S.; Cheng, C.; Israel, M.A. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 2018, 37, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.; Lahtz, C.; Song, J.; Aftabizadeh, M.; Cherryholmes, G.A.; Xin, H.; Adamus, T.; Lee, H.; Grunert, D.; Armstrong, B.; et al. Integrin α6 signaling induces STAT3-TET3-mediated hydroxymethylation of genes critical for maintenance of glioma stem cells. Oncogene 2019. [Google Scholar] [CrossRef]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.-G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated From Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/gp130/STAT3 Pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Yu, T.; Dong, N.; Wang, B.; Sun, F.; Jiang, D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J. Exp. Clin. Cancer Res. CR 2019, 38, 289. [Google Scholar] [CrossRef]
- Luo, D.; Fraga-Lauhirat, M.; Millings, J.; Ho, C.; Villarreal, E.M.; Fletchinger, T.C.; Bonfiglio, J.V.; Mata, L.; Nemesure, M.D.; Bartels, L.E. Phospho-valproic acid (MDC-1112) suppresses glioblastoma growth in preclinical models through the inhibition of STAT3 phosphorylation. Carcinogenesis 2019, 40, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Iwamaru, A.; Szymanski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.Y.; Wu, A.S.; Doucette, T.; Wei, J.; Priebe, W.; Fuller, G.N.; Qiao, W.; Sawaya, R.; Rao, G.; Heimberger, A.B. Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clin. Cancer Res. 2010, 16, 5722–5733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med 2015, 4, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-target JAK2/STAT3 inhibition slows disease progression in orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol. 2013, 15, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endaya, B.B.; Lam, P.Y.P.; Meedeniya, A.C.B.; Neuzil, J. Transcriptional profiling of dividing tumor cells detects intratumor heterogeneity linked to cell proliferation in a brain tumor model. Mol. Oncol. 2016, 10, 126–137. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [Green Version]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell 2008, 3, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Fessler, E.; Borovski, T.; Medema, J.P. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol. Cancer 2015, 14, 157. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.N.; Yang, L.; Lv, Y.F.; Shi, Y.; Shen, L.L.; Yao, X.H.; Guo, Q.N.; Zhang, P.; Cui, Y.H.; Zhang, X. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.S.; Huang, P. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, T.; Langlois, B.; Koczorowska, M.M.; Radwanska, A.; Sun, Z.; Hussenet, T.; Lefebvre, O.; Murdamoothoo, D.; Arnold, C.; Klein, A.; et al. Tenascin-C Orchestrates Glioblastoma Angiogenesis by Modulation of Pro- and Anti-angiogenic Signaling. Cell Rep. 2016, 17, 2607–2619. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Rebollo, M.; Garrido, C.; Sánchez-Cid, L.; Soler-Botija, C.; Meca-Cortés, O.; Rubio, N.; Blanco, J. Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci. Rep. 2019, 9, 1–3. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Janic, B.; Guo, A.M.; Iskander, A.S.; Varma, N.R.; Scicli, A.G.; Arbab, A.S. Human cord blood-derived AC133+ progenitor cells preserve endothelial progenitor characteristics after long term in vitro expansion. PLoS ONE 2010, 5, e9173. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Niu, C.S.; Li, M.W. Correlation between glioblastoma stem-like cells and tumor vascularization. Oncol. Rep. 2012, 27, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Chiao, M.T.; Yang, Y.C.; Cheng, W.Y.; Shen, C.C.; Ko, J.L. CD133+ glioblastoma stem-like cells induce vascular mimicry in vivo. Curr. Neurovasc. Res. 2011, 8, 210–219. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [Green Version]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [Green Version]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- Tafani, M.; Di Vito, M.; Frati, A.; Pellegrini, L.; De Santis, E.; Sette, G.; Eramo, A.; Sale, P.; Mari, E.; Santoro, A.; et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J. Neuroinflammation 2011, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Chen, C.; Shi, Y.; Wu, Q.; Gimple, R.C.; Fang, X.; Huang, Z.; Zhai, K.; Ke, S.Q.; Ping, Y.-F.; et al. Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell 2017, 21, 591–603.e594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.L.; Panchision, D.M. Concise review: Bone morphogenetic protein pleiotropism in neural stem cells and their derivatives--alternative pathways, convergent signals. Stem Cells 2007, 25, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Panchision, D.M.; Pickel, J.M.; Studer, L.; Lee, S.H.; Turner, P.A.; Hazel, T.G.; McKay, R.D. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 2001, 15, 2094–2110. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.K.; Miller, R.H. Emerging roles for bone morphogenetic proteins in central nervous system glial biology. J. Neurosci. Res. 2004, 76, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Mabie, P.C.; Zhu, G.; Gokhan, S.; Kessler, J.A. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev. Neurosci. 2000, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gomez, P.; Anselmo, N.P.; Mira, H. BMPs as therapeutic targets and biomarkers in astrocytic glioma. BioMed Res. Int. 2014, 2014, 549742. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Pistollato, F.; Chen, H.L.; Rood, B.R.; Zhang, H.Z.; D’Avella, D.; Denaro, L.; Gardiman, M.; te Kronnie, G.; Schwartz, P.H.; Favaro, E.; et al. Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high-grade glioma. Stem Cells 2009, 27, 7–17. [Google Scholar] [CrossRef]
- Tate, C.M.; Pallini, R.; Ricci-Vitiani, L.; Dowless, M.; Shiyanova, T.; D’Alessandris, G.Q.; Morgante, L.; Giannetti, S.; Larocca, L.M.; di Martino, S.; et al. A BMP7 variant inhibits the tumorigenic potential of glioblastoma stem-like cells. Cell Death Differ. 2012, 19, 1644–1654. [Google Scholar] [CrossRef]
- Chirasani, S.R.; Sternjak, A.; Wend, P.; Momma, S.; Campos, B.; Herrmann, I.M.; Graf, D.; Mitsiadis, T.; Herold-Mende, C.; Besser, D.; et al. Bone morphogenetic protein-7 release from endogenous neural precursor cells suppresses the tumourigenicity of stem-like glioblastoma cells. Brain 2010, 133, 1961–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169, 1342–1356.e1316. [Google Scholar] [CrossRef] [Green Version]
- Rieth, J.; Subramanian, S. Mechanisms of Intrinsic Tumor Resistance to Immunotherapy. Int. J. Mol. Sci. 2018, 19, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tian, T.; Kalland, K.-H.; Ke, X.; Qu, Y. Targeting Wnt/b-Catenin Signaling for Cancer Immunotherapy. Trends Pharmacol. Sci. 2018, 39, 648–658. [Google Scholar] [CrossRef]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019, 121, 666–678. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, H.J.; Stead, L.F.; Nsengimana, J.; Reilly, K.A.; Droop, A.; Wurdak, H.; Mathew, R.K.; Corns, R.; Newton-Bishop, J.; Melcher, A.A.; et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin. Exp. Immunol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kokubu, Y.; Tabu, K.; Muramatsu, N.; Wang, W.; Murota, Y.; Nobuhisa, I.; Jinushi, M.; Taga, T. Induction of protumoral CD 11chigh macrophages by glioma cancer stem cells through GM-CSF. Genes Cells 2016, 21, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 2017, 20, 450–461.e454. [Google Scholar]
- Hide, T.; Komohara, Y.; Miyasato, Y.; Nakamura, H.; Makino, K.; Takeya, M.; Kuratsu, J.-I.; Mukasa, A.; Yano, S. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine 2018, 30, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Ping, Y.-F.; Zhou, W.; He, Z.-C.; Chen, C.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.-C.; Zhou, K. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nature communications 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira de Castro, J.; Gonçalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278. https://doi.org/10.3390/ijms21155278
Vieira de Castro J, Gonçalves CS, Hormigo A, Costa BM. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. International Journal of Molecular Sciences. 2020; 21(15):5278. https://doi.org/10.3390/ijms21155278
Chicago/Turabian StyleVieira de Castro, Joana, Céline S. Gonçalves, Adília Hormigo, and Bruno M. Costa. 2020. "Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting" International Journal of Molecular Sciences 21, no. 15: 5278. https://doi.org/10.3390/ijms21155278