The Roles of Immune Cells in the Pathogenesis of Fibrosis


1
Department of Pathology and Shenzhen Institute of Research and Innovation, The University of Hong Kong, Hong Kong, China
2
Department of Rheumatology and Immunology, the Second People’s Hospital of Three Gorges University, Yichang 443000, China
*
Authors to whom correspondence should be addressed.
†
E.H. and N.P. contributed equally to this work.
Int. J. Mol. Sci. 2020, 21(15), 5203; https://doi.org/10.3390/ijms21155203
Submission received: 29 June 2020
/
Revised: 19 July 2020
/
Accepted: 21 July 2020
/
Published: 22 July 2020
(This article belongs to the Special Issue Immune Cell Mediated Tissue Regeneration)
Abstract
:Tissue injury and inflammatory response trigger the development of fibrosis in various diseases. It has been recognized that both innate and adaptive immune cells are important players with multifaceted functions in fibrogenesis. The activated immune cells produce various cytokines, modulate the differentiation and functions of myofibroblasts via diverse molecular mechanisms, and regulate fibrotic development. The immune cells exhibit differential functions during different stages of fibrotic diseases. In this review, we summarized recent advances in understanding the roles of immune cells in regulating fibrotic development and immune-based therapies in different disorders and discuss the underlying molecular mechanisms with a focus on mTOR and JAK-STAT signaling pathways.
1. Introduction
Fibrosis is the abnormal formation of excess fibrous connective tissue during chronic inflammation and tissue repair. The excessive depositions of extracellular matrix (ECM) components especially collagens are the leading cause of fibrosis. A variety of stimuli such as allergic responses, autoimmune development, persistent infections, and tissue injury contribute to the initiation and progression of fibrogenesis in various tissues and organs including skin, liver, lung, heart, and kidney [1]. Fibrosis is a common pathway to organ injury and failure in many diseases including systemic sclerosis (SSc), idiopathic pulmonary fibrosis (IPF), and the recent pandemic Coronavirus disease 2019 [1,2]. Fibrosis is often associated with poor prognosis and accounts for substantial morbidity and mortality [1].
Lines of evidence from clinical observations and animal studies have demonstrated the involvement of various cell subsets during fibrogenesis. The activation of myofibroblasts, one of the primary producers of ECM, is the key fibrogenic event in fibrotic diseases. Furthermore, compelling evidence suggests that both innate and adaptive immune cell populations are critically involved in the regulation of myofibroblast activation and fibrogenic responses in many fibrotic diseases [3,4]. During inflammatory responses, activated immune cells orchestrate the cellular and molecular processes of fibrosis in responses to external stimuli and microenvironmental factors. The recruitment and activation of immune cells including macrophages, neutrophils, natural killer (NK) cells, T cells, and B cells regulate the progression and regression of fibrogenic development in various organs and tissues through different molecular mechanisms.
In this review, we described and summarized recent advances in the understanding of the key roles of immune cells in fibrosis pathogenesis and the underlying molecular mechanisms, which may shed new light on the development of novel anti-fibrotic strategies by modulating immune cell functions.
2. Innate Immune Cells in the Pathogenesis of Fibrosis
2.1. Macrophages
Macrophages are important regulators of tissue repair and fibrosis. It has been recognized that macrophages undergo differentiation with phenotypic and functional changes during initiation and progression of fibrosis development [5]. Macrophages are heterogeneous populations with multiple phenotypes and diverse plasticity under the influence of different microenvironmental factors in various organs [6]. Based on their differential cytokine profiles and effector functions, macrophages are divided into classically activated (M1) and alternatively activated (M2) macrophages subsets. Although this classification does not adequately describe the diverse phenotypes of macrophage subsets, previous studies have suggested an involvement of these macrophages in fibrogenesis [7]. Moreover, increasing evidence indicates that tissue resident macrophages including Kupffer cells are key players in tissue fibrosis.
Accumulated data have suggested the critical roles of macrophages in fibrogenic responses. Conditional depletion of macrophages using CD11b-DTR mice markedly decreased numbers of myofibroblasts and attenuated carbon tetrachloride (CCl4)-induced liver fibrosis, suggesting a pro-fibrogenic role of macrophages in the fibrotic model [8]. Consistently, macrophages contributed to NF-κB-induced liver fibrosis in a liver injury-independent manner, possibly through regulating myofibroblast survival [9,10]. Moreover, inflammatory Gr1+ monocytes were recruited into injured liver in a CCR2-dependent manner and differentiated into LY6Chi CD11b+ F4/80+ macrophages that strongly promoted liver fibrosis [11]. As heterogeneous populations, monocytes and macrophages comprise different functional subsets. A new subset of segregated-nucleus-containing atypical monocytes was identified as a critical pro-fibrotic player in bleomycin-induced lung fibrosis [12]. In addition, an expression quantitative trait locus analysis in monocyte-derived macrophages revealed that changes of macrophage transcriptome were closely associated with the susceptibility of SSc, an autoimmune disease with fibrosis in skin and internal organs [13].
Macrophages polarize to M1 phenotypes in response to IFN-γ and TNF-α, and M2 phenotypes in response to IL-4, IL-10, IL-13, and TGF-β [14]. Previous studies have suggested that both M1 and M2 macrophages are involved in fibrosis development. M1 macrophages produce massive amounts proinflammatory cytokines and chemokines that promote tissue inflammation and myofibroblast activation and differentiation [7]. However, M1 macrophages can also release matrix metalloproteinases (MMPs) including MMP12 that may degrade ECM and contribute to the resolution of fibrosis, suggesting an anti-fibrotic role of these cells [15].
M2 macrophages exhibit anti-inflammatory properties and play a pro-fibrotic role in fibrosis. F4/80+CD301+ M2 macrophages were found to be the dominant population in kidneys of mice with unilateral ureteral obstruction (UUO)-induced renal fibrosis. Depletion of these M2 macrophages by clodronate liposome administration ameliorated renal fibrosis and reduced epithelial-to-mesenchymal transition while transfer of M2 macrophages into kidney capsules increased the expressions of α-smooth muscle actin (α-SMA), an important indicator of fibrosis [16]. Similarly, significantly increased numbers of M2 macrophages in lung tissues were detected in mice with bleomycin-induced pulmonary fibrosis and patients with IPF, which was associated with the upregulation of macrophage alternative activation signature genes [17,18]. Depletion of alveolar macrophages or circulating monocytes by clodronate liposome treatment markedly reduced lung fibrosis [17]. Moreover, mice with IL-10-induced pulmonary fibrosis showed significantly increased numbers of M2 macrophages in both bronchoalveolar lavage and lung tissue than normal mice, which was associated with enhanced expression of C-C motif chemokine ligand 2 (CCL2). Notably, anti-CCL2 antibody treatment attenuated lung fibrosis in these mice, suggesting that targeting M2 macrophages may represent a promising therapeutic strategy for treating fibrosis [19]. Alveolar M2 cells promote myofibroblast differentiation through regulating Akt1 activation and mitophagy. Phosphorylation of Akt1 was observed in the alveolar macrophages of IPF patients and bleomycin-treated mice. Akt1 increased the production of ROS and induced mitophagy. Constitutive activation of Akt1 enhanced TGF-β expression which was regulated by Park2, an important E3 ubiquitin ligase in mitophagy. Thus, the Akt1–ROS–mitophagy pathway promoted the pro-fibrotic effects of M2 macrophages [20].
Tissue resident macrophages have been recognized as important players in fibrosis. As major resident macrophages in liver, Kupffer cells have a crucial role in sustaining liver immune homeostasis. The numbers of Kupffer cells were decreased during hepatic inflammation and fibrogenesis [4]. Moreover, Kupffer cells produced pro-fibrotic cytokines including TGF-β and platelet-derived growth factor (PDGF) that activated hepatic stellate cells (HSCs) and promoted fibrogenic responses [21]. Kupffer cells also secreted multiple MMPs that contributed to the resolution of fibrosis, suggesting the multiple functions of Kupffer cells in liver fibrosis [22]. Although tissue resident macrophages differ significantly from monocyte-derived macrophages, a population of alveolar macrophages with tissue resident phenotypes derived from monocytes promoted lung fibrosis and persisted in the lung over the life span, suggesting that targeting tissue resident alveolar macrophage differentiation may ameliorate pulmonary fibrosis [23].
Macrophages are important players in the development of steatosis, inflammation and fibrosis in nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). The polarization of inflammatory monocytes and activation of adipose tissue macrophages in the visceral compartment are both critically involved in the disease pathogenesis [24]. The portal infiltration of macrophages has been identified as an early event in human NAFLD, occurring before the development of inflammation or fibrosis, which may also serve as a predictive factor for disease progression [25]. Kupffer cells respond to hepatocyte injuries at early stage and produce massive amount of TNF -α, IL-1-β, and chemokines, which promote monocyte recruitment and inflammatory process during NASH/NAFLD development [26,27]. Mice with depletion of liver Kupffer cells by gadolinium chloride were protected from the development of diet-induced hepatic steatosis and insulin resistance [28]. Moreover, M2 Kupffer cells induced M1 cell apoptosis in culture and attenuated alcohol- and high fat diet-induced fatty liver diseases in mice, suggesting that M2 macrophages play a protective role in the development of NAFLD [29].
2.2. Neutrophils
Neutrophils are recruited to inflammatory sites at early stages of wound healing and play an important pro-fibrotic role in fibrosis. Depletion of neutrophils significantly attenuated lung fibrosis, suggesting a critical pro-fibrotic role of neutrophils [30]. It has been shown that neutrophils are recruited to damaged lungs dependent on formyl peptide receptor 1 while failure in recruiting neutrophils or depletion of neutrophils protect mice from bleomycin-induced pulmonary fibrosis [31]. Moreover, neutrophil-secreted elastase (NE) has been shown to play a pro-fibrotic role in asbestos-induced lung fibrosis. Deficiency of NE or treatment of NE antagonists ONO-5046 resulted in significantly decreased myofibroblast cell numbers and collagen deposition in mice with asbestos-induced lung fibrosis. Moreover, endocytosis of NE by fibroblasts increased α-SMA expression, promoted cell proliferation and enhanced migration and contractility of these cells independent of PI3K/Akt activation [32]. A clinical study has reported higher levels of NE in lung parenchyma, bronchoalveolar lavage fluid (BALF) and sera from patients with IPF when compared with those in normal subjects, suggesting an involvement of NE in the pathogenesis of IPF [33]. Furthermore, pharmacological inhibition of neutrophil extracellular trap formation may represent a promising therapy for the treatment of various inflammatory diseases [34].
2.3. NK Cells
Numerous studies have suggested the anti-fibrotic roles of NK cells in liver fibrosis through regulating HSCs that can differentiate into myofibroblasts and contribute to collagen deposition. It was found that hepatic NK cells killed activated HSCs dependent on TNF-related apoptosis-inducing ligand (TRAIL) and NKG2D-mediated signaling in mice with CCl4-induced liver fibrosis [35]. IFN-γ stimulation upregulated NKG2D and TRAIL expressions and increased the cytotoxicity of hepatic NK cells [35]. NK cells can also eliminate senescent HSCs during fibrosis progression. NKG2D-deficient mice showed increased numbers of senescent cells around the fibrotic scar areas of liver with increased collagen depositions in mice with CCl4 challenge [36]. Consistently, curcumin increased NKG2D ligands expressions in HSCs and enhanced the cytotoxicity of NK cells from mice with CCl4-induced liver fibrosis, suggesting a role of NKG2D in regulating the killing capacities of NK cells [37]. Clinical observations showed that a subset of CD56dimKLRG1+ NK cells decreased in the peripheral blood and liver of patients with chronic HBV infection at advanced stages of fibrosis. These NK cells exhibited high level of IFN-γ expression and TRAIL-dependent cytotoxicity against HSCs, which could be further enhanced by IFN-γ and CD44 stimulations [38].
NK cells have been shown to suppress cardiac fibrosis in experimental autoimmune myocarditis. Eosinophil infiltration and collagen deposition were increased in the heart of mice with cardiac fibrosis. However, depletion of NK cells exacerbated cardiac fibrosis, which was dependent on eosinophils. Moreover, NK cells induced the apoptosis of eosinophils in culture, suggesting that NK cells play a protective role in cardiac fibrosis by regulating eosinophils [39].
NK cells also exhibited pro-fibrotic roles in some conditions. Depletion of NK cells ameliorated leukocyte infiltration and liver fibrosis in a low-dose rotavirus model [40]. Moreover, CD161+CD3− NK cells secreted massive amounts of IL-17 and promoted acute kidney injury-induced renal fibrosis in athymic mice while blockade of IL-17 attenuated renal inflammation and fibrosis, suggesting that these NK cells contributed to renal fibrosis by producing IL-17 [41].
2.4. Innate Lymphoid Cells
Innate lymphoid cells (ILCs) are heterogeneous innate immune populations that lack antigen-specific receptors and lineage markers, which play important roles in sustaining immune homeostasis and innate immune responses against pathogens. ILCs are mainly classified into three groups based on their signature cytokines and transcription factors. Indeed, three groups of ILCs, namely ILC1, ILC2, and ILC3, mirror the cytokine signatures of Th1, Th2, and Th17 cells, respectively [42]. Recent evidence indicates that ILCs exert significant functions in tissue repair and fibrosis development [43,44].
Clinical observations have shown that an ILC2 population was detected in the lungs of IPF patients [45]. Moreover, the percentages of CD69+ ILC2s were positively correlated with aggravation of liver fibrosis in patients with liver diseases, suggesting that ILC2s may contribute to the immunopathology of liver fibrosis [46]. The pro-fibrotic roles of ILC2 in pulmonary fibrosis were further supported by animal studies. IL-13 produced by IL-25-activated ILC2s was shown to drive collagen deposition in the lungs of mice with bleomycin-induced fibrosis [45]. Intranasal delivery of recombinant IL-25 also induced pulmonary inflammation and fibrosis, which was associated with connective tissue growth factor and TGF-β production [45]. Moreover, IL-33, a cytokine closely associated with organ fibrosis, also promoted IL-13 production by ILC2s, suggesting that IL-33 might drive fibrosis via regulating ILC2 functions [47].
ILC1s and ILC3s are also involved in organ fibrosis pathogenesis. It was recently reported that ILC1s accumulated in adipose tissues of patients with type 2 diabetes whereas these cells promoted adipose fibrogenesis through activation of TGF-β signaling [48]. Furthermore, inhibition of adipose accumulation of ILC1s attenuated adipose tissue fibrosis, suggesting that ILC1s might be promising therapeutic targets for treating adipose fibrosis [48]. ILC3s are characterized by the production of IL-17, which exhibited elevated levels in patients with IPF and SSc [44]. The frequencies of ILC3s with IL-17 production were increased in liver from CCl4-induced fibrotic mice than those from normal mice. Adoptive transfer of ILC3s into ILC-depleted mice increased ECM accumulation and promoted liver fibrosis, suggesting a pro-fibrotic role of ILC3s in liver fibrotic progression [49].
2.5. γδT Cells
γδT cells showed diverse functions during fibrosis development. It was reported that adoptively transferred γδT cells from CCl4-treated mice infiltrated into liver and reduced hepatic inflammation and fibrosis through inducing apoptosis of HSCs in recipient mice, suggesting a protective role of γδT cells in liver fibrosis [50]. A subset of Vγ6Vδ1 γδT cells actively secreted IL-17 and dramatically increased in mice with repeated B. subtilis infection. These Vγ6Vδ1 γδT cells were shown to be anti-fibrotic, because Vγ4/6 or δ chain deficiency led to severer collagen deposition while Vγ6Vδ1 transgenic mice exhibited alleviated pulmonary fibrosis [51]. Moreover, Vγ6 γδT cells and a subset of γδTCRlo γδT cells expressed IL-22 in an aryl hydrocarbon receptor (AhR)-dependent manner. Deficiency of γδTCR or inhibition of AhR signaling aggravated lung fibrosis, which was restored by nasal inhalation of recombinant IL-22 [52]. A subset of NK1.1+ γδT cells suppressed fibrosis in bleomycin model via IFN-γ secretion. Transfer of NK1.1+ γδT cells to δ chain deficient mice partially ameliorated lung fibrosis [53]. In contrast, depletion of Vγ2 γδT cells decreased collagen fiber in liver of mice with S. japonicum infection-induced liver fibrosis, suggesting a pro-fibrotic role of these cells [54].
2.6. Dendritic Cells
Growing evidence indicates that dendritic cells (DCs) are novel players in the pathogenesis of various fibrotic diseases [55,56]. DCs are potent antigen-presenting cells with key roles in modulating immune responses. Recent studies have revealed the involvement of different DCs subsets in the development of fibrosis. The frequencies of circulating conventional CD141+ and CD1c+ DCs, namely cDC1 and cDC2, and CD303+ plasmacytoid DCs (pDCs) were significantly reduced in patients with IPF when compared with those in age and sex matched healthy controls [57]. Moreover, a subset of BDCA1+ DCs were detected in the lungs of patients with IPF or hypersensitivity pneumonitis, suggesting a potential role of BDCA1+ DCs in lung fibrosis [58]. Consistently, accumulated DCs in lung tissue were observed in mice with pulmonary fibrosis. Selective depletion of lung DCs markedly exacerbated lung fibrosis in mice, suggesting a protective role of lung DCs in fibrogenesis [59]. Furthermore, increased mobilization of lung CD11b+ DC regulated pulmonary fibrosis development in mice [60]. These studies have suggested the potential of DC-based immunotherapy for the treatment of lung fibrosis.
Increasing evidence indicates that DCs are involved in cardiac fibrosis. The infiltrated CD209+ DCs and CD11c+ DCs in human infarcted heart were increased in patients with cardiac rupture, which were associated with impaired cardiac reparative fibrosis [61]. Moreover, the CD11b+CD11c+ tolerogenic DCs with low expression of MHC-II, CD86, CD80 and high level of IL-10 production reduced heart inflammation and fibrosis in a mouse model of chronic Chagas disease cardiomyopathy [62]. The protective roles of tolerogenic DCs in cardiac fibrosis appear to be associated with reduced expressions of pro-inflammatory cytokines and increased IL-10 production [62]. Recent studies have suggested that pDCs are involved in SSc pathogenesis [56]. The pDCs infiltrated into the skin of SSc patients and produced large amounts of CXCL4 and IFN-α [63]. CXCL4 and DNA formed liquid crystalline complexes and activated pDCs in a TLR-9-dependent manner, which promoted IFN-α production by pDCs [63]. Depletion of pDCs attenuated fibrosis of the skin and lung in the bleomycin-induced SSc mice, indicating a pathogenic role of pDCs in SSc pathogenesis [64]. A group of classical CD11b+ DCs played a profibrotic role in a mouse model of allergic eye disease (AED), which was dependent on activation of the retinoic acid pathway [65]. The classical CD11b+ DCs within ocular mucosa exhibited activation of aldehyde dehydrogenase (ALDH), a critical enzyme required for retinoic acid synthesis. The DCs-derived ALDH increased ligation of retinoic acid with conjunctival fibroblast retinoid X receptor (RXR) and induced rapid onset of ocular mucosal fibrosis [65].
2.7. NKT Cells and Mucosal-Associated Iinvariant T (MAIT) Cells
Recent studies have revealed a role of NKT cells in the development of fibrosis. In HBV-transgenic mice that resemble human HBV carriers, CCl4-induced liver fibrosis becomes more pronounced than that in wild type mice. Depletion of NK cells and NKT cells or blockade of CD1d reduces the levels of α-SMA expression in the liver, while depletion of NK cells alone shows no such effect. Moreover, blockade of IL-4 or IL-13 inhibits the effects of NKT cells on upregulating α-SMA in HSCs in vitro, suggesting that NKT cells promote liver fibrosis via Th2 cytokines in HBV-associated liver fibrosis [66]. In a diet-induced NAFLD mouse model, CD1d−/− mice showed severer T cell infiltration in the kidneys with increased renal expression of TLR4, TGF-β, and α-SMA. However, the renal pathology was ameliorated in TLR4-deficient mice, suggesting that CD1d-dependent NKT cells played a protective role in NAFLD-associated renal inflammation and fibrosis via suppressing TLR4-mediated signaling function [67]. The Rag2−/− mice overexpressing TCR Vα3.2 and Vβ9 chains showed increased generation of type II NKT cells and spontaneously developed hepatitis and liver fibrosis, in which type II NKT cells produced sufficient Th2 cytokines and contributed to the liver fibrosis [68].
Invariant NKT cells (iNKT) play a protective role in bleomycin-induced pulmonary fibrosis via secretion of IFN-γ. The increase of TGF-β and collagen deposition in the lung tissue of CD1d−/− diseased mice could be reversed by transfer of iNKT cells from wild type mice but not IFN-γ-deficient mice [69]. However, repeated intranasal administration of α-Galactosylceramide (α-Galcer) induced chronic obstructive pulmonary disease (COPD)-like symptoms in mice, with increased leukocyte infiltration in the lung tissues and fibrosis of airways. These COPD-like symptoms could be reversed by blockade of IL-4, suggesting that iNKT cells promoted airway inflammation and fibrosis via IL-4 [70]. In 2-OA-BSA-induced autoimmune cholangitis mice, activation of iNKT cells by α-Galcer treatment promoted lymphocyte infiltration and collagen deposition in the liver, which was ameliorated in CD1-deficient mice, indicating a profibrotic role of iNKT cells in primary biliary cholangitis (PBC) [71]. An early study of NASH using mice fed with high fat diet showed that Jα18-deficient mice with iNKT cell deficiency exhibited severer hepatic inflammation and ultimately extensive liver fibrosis, suggesting a protective role of iNKT cells in NASH [72]. However, NASH and liver fibrosis were ameliorated in the liver of CD1d-deficient mice fed with high-fat high-carbohydrate diet, suggesting that CD1d-dependent NKT cells were profibrotic in NASH [73]. Given that CD1d-dependent NKT cells comprise various subsets including iNKT cells, the different subsets may play opposite roles in the pathogenesis of NASH [74].
Mucosal-associated invariant T cells (MAIT cells) are decreased in the liver and blood of patients with autoimmune liver diseases (AILDs). MAIT cells could induce HSC proliferation and expression of collagen and proinflammatory cytokines in vitro [75]. Jiang et al. found decreased numbers of circulating MAIT cells in patients with PBC, a subset of AILDs. Activated MAIT cells promoted hepatic myofibroblast (HMF) proliferation in an MR-1 dependent manner, and induced IL-6 and IL-8 expression in HMF. CCl4-induced liver fibrosis was decreased in MR1−/− mice, while both CCl4-induced and bile duct ligation-induced liver fibrosis were exacerbated in Vα19 TCR transgenic mice with enhanced MAIT cell generation, demonstrating a profibrotic role of MAIT cells in liver fibrosis [76]. In patients with chronic HCV-infection, the liver hepatic inflammation and fibrosis scores were negatively correlated with the frequencies of MAIT cells in liver, suggesting that MAIT cells may be involved in HCV-associated liver fibrosis [77]. A study on chronic kidney disease showed that the numbers of MAIT cells in the tissues were positively correlated with the severity of renal fibrosis. MAIT cells were in close proximity with proximal tubular epithelial cells (PTECs) in the fibrotic kidneys, suggesting that MAIT cells may contribute to fibrogenesis via targeting PTECs in chronic kidney disease [78].
3. Adaptive Immune Cells in the Pathogenesis of Fibrosis
3.1. Th1 Cells
Th1 cells and their secreted cytokines including IFN-γ and IL-12 have been suggested to exert anti-fibrotic functions under various conditions. Administration of IL-12, a potent inducer of Th1 cells, attenuated bleomycin-induced pulmonary fibrosis through inducing IFN-γ production, suggesting a protective role of Th1-associated cytokines in fibrosis [79]. Furthermore, deficiency of T-bet, the signature transcription factor of Th1 cells, resulted in markedly increased expression of the profibrotic factor TGF-β and deposition of collagen in lungs from bleomycin-treated mice [80].
However, recent findings have revealed pro-fibrotic roles of Th1 cells and associated cytokines in fibrosis. S. epidermidis triggered IL-6-dependent Th1 expansion which activated STAT1 in peritoneal membrane and subsequent peritoneal fibrosis. IFN-γ deficiency resulted in significantly ameliorated S. epidermidis-induced peritoneal fibrosis while adoptive transfer of Th1 cells induced fibrotic progression in fibrosis-resistant IL-6 deficient mice [81]. Similarly, it was shown that Th1 cells induced TGF-β production in cardiac fibroblasts and selectively drove cardiac fibrosis in an IFNγ-dependent manner, suggesting a pro-fibrotic role of Th1 cells and IFN-γ in nonischemic heart failure [82].
3.2. Th2 Cells
Extensive evidence suggests that type 2 immunity contributes to the development of fibrosis in different organs [83]. The important roles of IL-4 and Th2 cells in bacterial infection-associated hepatic fibrosis have long been recognized [84]. The pro-fibrotic roles of Th2 cells were also demonstrated in pulmonary fibrosis. It has been shown that chronic asthma induces the thickening and fibrosis of bronchial basement membrane. Administration of house dust mites in the airways induced amphiregulin-producing memory Th2 cells, which further enhanced airway inflammation-induced fibrosis [85]. The pathogenic memory Th2 cells-derived amphiregulin promoted myofibroblast differentiation and sub-basement membrane fibrosis of the airway through the induction of osteopontin by infiltrated inflammatory eosinophils [85]. The pathogenic roles of amphiregulin-producing memory Th2 cells may be potential targets for the treatment of fibrosis induced by chronic allergic disorders.
3.3. Th17 Cells
In addition to their roles in immunity and inflammation, Th17 cells are actively involved in fibrogenic responses with different roles in various fibrotic diseases. Th17 cells produce massive amounts of IL-17 and IL-22, both of which are associated with tissue fibrosis. IPF patients showed increased levels of IL-17 in BALF than normal volunteers [86]. Animal studies indicated that Th17 cells and IL-17 promoted skin and lung fibrosis development in a bleomycin-induced murine model of SSc [87]. It has been reported that IL-17 exhibits dual roles in pulmonary fibrosis. Mice with IL-17RA deficiency showed impaired clearance of B. subtilis and increased lung fibrosis, suggesting a protective role of IL-17 [51]. However, IL-17 deficient mice exhibited attenuated bleomycin-induced pulmonary fibrosis [86]. Moreover, intratracheal administration of IL-17 was shown to induce collagen accumulation and fibrotic lesions while neutralization of IL-17 reduced tissue fibrosis, indicating a pro-fibrotic role of IL-17 in chemical-induced fibrosis [86]. The frequencies of circulating Th17 cells were increased in patients with HBV infection-induced liver cirrhosis [88]. Moreover, Th17 frequencies were higher in liver tissues with more advanced fibrosis [89,90], suggesting a role of Th17 cells in the development of liver fibrosis.
Increased hepatic Th17 cells were detected in patients with advanced-stage HBV-related liver fibrosis [90]. Animal studies suggested a pro-fibrotic role of IL-17 in liver injury-induced fibrosis. IL-17 signaling facilitated the production of multiple cytokines including TGF-β, and directly induced production of collagen in HSCs through activation of signal transducers and activators of transcription (STAT) 3 signaling pathway [91]. Inhibition of Th17 cells by small molecules Halofugine and Magnolol significantly reduced the severity of Concanavalin A-induced liver fibrosis, suggesting that Th17 cells may represent novel therapeutic targets for treating liver fibrosis [92,93].
IL-22 has been shown to suppress pulmonary fibrosis. Bleomycin-treated mice exhibited markedly reduced levels of IL-22 in the lung [94]. Intraperitoneal injection of anti-IL-22 antibodies upregulated fibrosis-associated molecules including α-SMA, collagens and TGF-β in lung tissues of mice with bleomycin-induced fibrosis [94]. However, it has been suggested that IL-22 may contribute to HCV-associated liver fibrosis. Increased intrahepatic IL-22-producing cells were positively correlated with fibrotic severities in patients with HCV infection while IL-22 increased α-SMA expression and collagen production by HSCs in culture [95]. Thus, current data indicate a dual role of IL-22 in fibrosis development.
3.4. Regulatory T Cells
Increasing evidence indicates a pivotal role of regulatory T cells (Tregs) in fibrogenic responses. An imbalance between Tregs and Th17 cells was observed in IPF patients while the frequencies of Tregs were negatively correlated with the severities of IPF [96,97]. Moreover, depletion of Tregs by anti-CD25 antibodies at a late stage increased fibrotic scores and hydroxyproline content in the lungs of bleomycin-treated mice, suggesting a protective role of Tregs in pulmonary fibrosis [98]. Similarly, Tregs played a protective role against pneumococcus-induced lung fibrosis in mice [99]. Depletion of Tregs by diphtheria toxin resulted in increased lung collagen deposition, elevated Th1/Th2 cytokine levels and exacerbated infection-induced pulmonary fibrosis [99]. However, Tregs expansion markedly attenuated pneumococcus-induced fibrosis in mice [99]. Tregs suppressed TGF-β-induced pulmonary fibrosis through decreasing fibroblast growth factor 9 (FGF-9) expression by parenchymal cells and alveolar macrophages [100]. Depletion of Tregs in vivo increased inflammatory cytokine production and exacerbated hepatic fibrosis in mice with bile duct ligation (BDL) [101]. Moreover, Treg depletion resulted in increased Th2 cytokine production and skin fibroblast activation with upregulated profibrotic gene expressions in a bleomycin-induced murine model of skin sclerosis [102]. The conditional deletion of Gata3 in Tregs led to increased fibroblast activation and dermal fibrosis, suggesting an important role of Gata3 in modulating Tregs function during fibrosis development [102]. A recent study also revealed a protective role of Tregs in kidney injury and fibrosis [103].
It has been shown that the mammalian target of rapamycin (mTOR) signaling is critically involved in regulating the protective function of Tregs [104]. Adoptive transfer of rapamycin-treated Tregs ameliorated kidney fibrosis and improved renal functions in mice with acute kidney injury, suggesting an important role of mTOR signaling in regulating Treg functions [104]. Previous studies showed that activation of AhR signal preferentially promoted gut Tregs with enhanced suppressive activities in vivo [105]. A natural AhR agonist norisoboldine promoted Tregs differentiation through regulating glycolysis and NAD+/SIRT1/SUV39H1/H3K9me3 signaling pathway during colitis development [106]. A recent study indicated a role of AhR signal in regulating Tregs functions during fibrogenic responses [107]. Bleomycin-challenged mice with treatment of FICZ, a natural AhR ligand, exhibited increased number of Tregs with attenuated lung fibrosis, suggesting a therapeutic potential of targeting AhR for treating fibrotic diseases [107]. Tregs also exerted protective functions against coxsackievirus B3-induced cardiac fibrosis via secretion of IL-10, an important regulatory cytokine [108]. IL-10 activated STAT3 signaling, inhibited p38 mitogen-activated protein kinase (MAPK) activation and suppressed HuR expression, resulting in attenuated ventricular remodeling [109]. Moreover, IL-10 suppressed HuR transcription, inhibited renin-angiotensin-aldosterone system and reduced renal fibrosis in the UUO-induced fibrosis model [110]. Notably, IL-10 exerted an inhibitory function in fibrosis by activating PI3K/AKT and STAT3 signaling pathways which were downstream mediators of the IL-10 receptor in scar-forming fibroblasts [111]. However, depletion of Tregs at early stages reduced TGF-β expression, collagen depositions and fibrosis scores in lungs of bleomycin-treated mice, indicating that Tregs may promote fibrosis at early stages of disease progression [98]. Consistently, adoptive transfer of Tregs into Rag1−/− mice before intratracheal treatment of bleomycin exacerbated pulmonary fibrosis and increased mortality [112]. Moreover, substantial upregulation of IL-8 expression by Tregs was detected in patients with chronic hepatitis C viral infection, whereas the Tregs increased expressions of pro-fibrogenic markers by primary human HSCs [113]. Furthermore, Tregs isolated from the affected skin of SSc patients produced massive amounts of profibrotic cytokines including IL-4 and IL-13. The skin-localized Tregs expressed ST2 chain of the IL-33 receptor. IL-33, a cytokine abundantly produced in skin of SSc patients, skewed the differentiation of Treg cells into Th2-like cell phenotypes and contributed to tissue fibrosis [114].
In summary, available results have suggested the complex functions of Tregs in fibrogenic responses. Tregs may exert either protective or pathogenic roles at different stages of fibrosis development.
3.5. Follicular Helper T Cells
Follicular helper T (Tfh) cells are specialized CD4 helper T cells that provide help to B cells in germinal center reactions. Tfh cells produce a signature cytokine IL-21 that is critical for sustained B cell responses. It was reported that a Tfh-like cell subset infiltrated the skin of SSc patients and was associated with dermal fibrosis [115]. Moreover, an ICOS+ Tfh-like cell subset contributed to dermal fibrosis via producing IL-21 in the skin of graft-versus-host disease (GVHD)-SSc mice [115]. Either anti-ICOS treatment or IL-21 neutralization in GVHD-SSc mice inhibited inflammation and dermal fibrosis, suggesting that inhibition of ICOS and IL-21 might have therapeutic benefits for the treatment of SSc [115]. The proliferation and activation of Tfh cells were observed in patients with IPF [116]. It was shown that IL-21 contributed to pulmonary fibrosis through promoting the differentiation of naïve CD8 cell into pro-fibrotic CD8 T Cells in bleomycin-treated mice [117]. Both IL-21 deficient and IL-21 receptor deficient mice developed pulmonary inflammation but no fibrosis upon bleomycin challenge, suggesting an important role of IL-21 in fibrogenic development [117]. Since IL-21 is produced by various T cell subsets including Tfh cells and Th17 cells [118], the available data did not show the direct participation of Tfh cells in fibrosis. Thus, further studies are needed to delineate a role of Tfh cells in the development of fibrosis.
3.6. B Cells
B cells actively participate in tissue fibrosis among different organs. It was reported that deficiency of CD19 resulted in diminished B cell responses and significantly reduced susceptibility to bleomycin-induced lung fibrosis. In contrast, mice with CD19 overexpression exhibited exacerbated fibrosis, suggesting a profibrotic role of B cells in the development of pulmonary fibrosis [119]. It was suggested that B cells may promote fibrosis through regulating cytokines expression in a hyaluronan-TLR4-dependent manner [120]. Moreover, B cells were shown to promote skin fibrosis in an SSc model. B cells in TSK/+ mice that resemble human SSc symptoms showed lower stimulation thresholds with constitutive phosphorylation of CD19, increased Ca2+ release upon anti-CD19 activation, and enhanced IL-6 and IgG productions. CD19 deficiency inhibited B cell functions and attenuated skin fibrosis in TSK/+ mice [121]. Further studies suggested that the hyper-reactive B cell phenotypes in TSK/+ mice may partially result from dysregulated CD22 functions [122]. Moreover, deficiency of B cells markedly attenuated CCl4-induced fibrotic development in mice [123]. B cell-specific IL-6 deficient mice showed attenuated skin and lung fibrosis upon bleomycin challenge, suggesting a pathogenic role of B cell-derived IL-6 in the scleroderma model [124]. In culture, IL-6-producing effector B cells promoted collagen secretion by fibroblasts in a cell-cell contact-dependent manner. Notably, inhibition of B-cell activating factor (BAFF) ameliorated skin and lung fibrosis with a reduction of the effector B cells, suggesting a potential therapeutic strategy by targeting BAFF and B cells [124]. Clinical observations also suggested a role of CD19 in SSc. A case-control association study showed that functional CD19 polymorphism was associated with the susceptibility to SSc [125]. In co-culture experiments, B cells induced α-SMA and collagen expression by human dermal fibroblasts, which was further enhanced by BAFF [126]. Importantly, B cell depletion therapy with anti-CD20 treatment was well tolerated and ameliorated clinical symptoms of SSc patients [127,128].
Regulatory B cells (Bregs) exert immunosuppressive functions with secretion of anti-inflammatory cytokines, which are shown to participate in the pathogenesis of various diseases. A protective role of Bregs was supported by the evidence that B cell-specific IL-10-deficient mice exhibited significantly increased dermal thickness and exacerbated lung fibrosis in a bleomycin-induced scleroderma model [124]. However, anti-CD22 treatment preferentially depleted Bregs and attenuated lung fibrosis in mice with silica instillation-induced pulmonary fibrosis, suggesting that Bregs may promote pulmonary fibrosis [129]. These contradictory results might be attributed to different models and experimental methods for fibrosis induction. Recent evidence indicates that plasma cells possess immunomodulatory functions via IL-10 production [130]. Further studies may provide new insight in understanding the role of IL-10-producing plasma cells during the development of fibrosis in chronic autoimmune diseases [131].
4. The Molecular Mechanisms of Immune-Mediated Signaling Pathways in Fibrosis
4.1. The Roles of mTOR Signaling Pathway in Fibrosis
The mTOR protein is a key regulator of many cellular activities including proliferation, metabolism and protein synthesis. The mTOR interacts with adaptor proteins to form mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), both of which are involved in the pathogenesis of fibrosis.
A number of stimuli including cytokines, growth factors and mitogens activate mTOR signaling. The activation of PI3K/AKT suppresses Tuberous Sclerosis Complex (TSC), a major negative regulator of mTORC through the GTP binding protein Ras homolog enriched in brain (Rheb). It has been shown that dysregulated mTOR signaling is closely associated with pulmonary fibrosis, liver fibrosis and SSc. Genome-wide association study suggested that mTOR signaling was associated with susceptibility to IPF [132]. The overactivation of mTOR in mesenchymal cells by conditional deletion of TSC1 exacerbated CCl4-induced liver fibrosis, which was reversed by mTOR inhibitor rapamycin [133]. Moreover, inhibition of mTOR by rapamycin significantly reduced the expression of pro-inflammatory cytokines and fibrogenic mediators including IL-4, IL-6, IL-17, and TGF-β, which finally resulted in attenuated skin fibrosis in both TSK/+ and bleomycin-induced SSc model mice, suggesting a pivotal role of mTOR signaling in promoting fibrosis development [134].
The mTOR signaling is critically involved in regulating the functions of immune cells, including DCs, macrophages, NK cells and T cells, which are key players in fibrosis pathogenesis. Inhibition of mTOR promoted IL-12 but suppressed IL-10, TNF, and IL-6 production, suggesting a proinflammatory role of mTOR in regulating immune responses [135]. It has been well-recognized that mTOR signaling regulates Tregs differentiation and functions. The protective roles of Tregs were regulated by mTOR in the repair of acute kidney injury [104]. Moreover, deletion of mTORC1 activity in CD4+ T cells resulted in increased inflammation, accelerated fibrosis development and increased mortality, which was associated with IL-17 production derived from γδT cells [136].
TGF-β, a central mediator of fibrogenesis, interacts with TGF-β receptor on fibroblasts and activates Smad proteins, which finally regulates the genes associated with EMT and fibroblast transdifferentiation [137]. TGF-β-activated mTORC1/4E-BP1 signaling through Smad phosphorylation was critical for collagen production in lung fibroblasts derived from IPF patients. Inhibition of TGF-β-induced PI3K/Akt activation showed undetectable effects on collagen production while mTOR inhibition and mTORC1 deficiency markedly reduced collagen I deposition in primary human lung fibroblasts, suggesting a role of PI3K/Akt independent activation of mTOR signaling in fibrogenesis [138]. Further analysis suggested that mTORC1/4E-BP1 axis represented a common fibrogenic signaling pathway during the development of fibrosis in different organs including lung, liver and skin [138]. Moreover, it was shown that Smad-independent TGF-β signaling also contributed to fibrosis pathogenesis by modulating p38, ERK, MAPK, and mTOR signaling [137]. TGF-β-mediated PI3K/AKT signaling may further induce activation of mTOR and subsequent fibrosis-related gene expressions [139].
4.2. The Roles of JAK-STAT Signaling Pathway in Fibrosis
Increasing evidence indicates that the JAK-STAT signaling pathway activation is involved in the development of many human diseases including fibrotic disorders. The binding of extracellular ligands such as cytokines, growth factors and hormones to their respective receptors activates JAKs including JAK1, JAK2, JAK3, and TYK2. Activated JAKs add phosphates at specific tyrosine residues of the receptors which serve as docking sites for the STATs [140]. The subsequent phosphorylation, dimerization and translocation of STATs regulate downstream gene expressions. Up to date, seven mammalian STAT family members have been identified, namely STAT1, STAT2, STAT3, STAT4, STAT5 (STAT5A and STAT5B), and STAT6. Recent studies have revealed that activation of the JAK-STAT signaling pathway by multiple cytokines including IL-6, IL-17, and IFNs exerts important functions in fibrosis pathogenesis.
Activated JAK-STAT signaling has been detected in various fibrotic diseases. The SSc patients showed significantly elevated phosphorylation levels of JAK1/JAK2/JAK3 and STAT3 in both skin and lung tissues when compared with healthy controls. Furthermore, inhibition of JAK by tofacitinib markedly ameliorated fibrosis development in both bleomycin-induced SSc mice and TSK1/+ mice, suggesting a critical role of JAK-STAT signaling in fibrotic changes of SSc [141]. Likewise, IPF patients exhibited enhanced levels of phosphorylated JAK2-STAT3 in lung tissues while dual inhibition of phosphorylated JAK2-STAT3 reduced lung fibrosis in mice with bleomycin challenge [142]. Moreover, both JAK2 inhibitor and STAT3 inhibitor attenuated left-atrial fibrosis in an atrial fibrillation model, suggesting a therapeutic potential of targeting JAK-STAT pathway in treating fibrosis [143].
Many cytokines derived from immune cells can activate JAK-STAT signaling pathway, which contribute to fibrosis development through various mechanisms. Type I and type II IFNs, mainly produced by pDCs, macrophages and T cells, strongly activate STAT1 phosphorylation. IFN-γ induced Smad7 expression and impaired TGF-β signaling through activating STAT1 in activated HSCs, indicating anti-fibrotic roles of IFN-γ in liver fibrosis [144]. Moreover, IFN-α reduced collagen expression and suppressed liver fibrosis though regulating STAT1 and p300 [145]. Recent evidence suggested that STAT1 activation prevented renal fibrosis by regulating macrophages differentiation and renal infiltration upon chronic kidney injury, indicating a protective role of STAT1 in renal fibrosis [146]. However, forkhead box O1, a prominent member of the forkhead box family, suppressed STAT1 activation and inhibited tubulointerstitial fibrosis in mice with diabetic kidney disease [147]. The oxidative hepatic environment in obesity increased STAT1 activation through suppressing T cell protein tyrosine phosphatase (TCPTP), which promoted hepatic fibrosis. Consistently, inhibition of the enhanced STAT1 signaling prevented T cell infiltration and liver fibrosis, suggesting a dual role of STAT1 in fibrotic development [148]. STAT3 has been suggested to integrate several profibrotic signals and serves as a core mediator of fibrosis [149]. Fibroblast-specific deficiency of STAT3 resulted in ameliorated TBRact-induced skin fibrosis in mice [149]. The JAK, JNK, SRC, and c-ABL kinases jointly activated STAT3 phosphorylation in fibroblasts with TGF-β stimulation [149]. SHP2 is an important regulator of TGF-β-induced STAT3 activation. TGF-β promoted recruitment of SHP2 to JAK2 in fibroblasts, which resulted in subsequent activation of STAT3 [150]. The inactivation of SHP2 reduced JAK2-STAT3 signaling and ameliorated dermal and pulmonary fibrosis in mice, suggesting that SHP2 and STAT3 might be molecular checkpoints for tissue fibrosis [149,150]. IL-6 is produced by epithelial cells and activated innate and adaptive immune cells including DCs, macrophages and T cells. IL-6 serves as an important activator of STAT3 and is critically involved in fibrosis. IL-6 was shown to enhance TGF-β/Smad3 signaling and collagen production, which was dependent on STAT3 activation [151]. Blocking IL-6 trans-signaling protected against kidney fibrosis by suppressing STAT3 activation in the UUO-induced renal fibrosis model [152]. Rilpivirine, a widely used anti-HIV drug, has been shown to ameliorate liver fibrosis though suppressing STAT3 and promoting STAT1-mediated HSC apoptosis [153]. Moreover, propylene glycol alginate sodium sulfate, a natural extract from brown algae, significantly reduced hepatic injury and fibrosis partially through suppressing JAK2-STAT3 activation [154].
It has been well recognized that STAT4 is activated in immune cells in responses to IL-12 and type I IFNs. The interaction of IL-12 and its receptor activates JAK2-TYK2 and STAT4, which regulates the expression of downstream cytokines such as IFN-γ. STAT4 plays essential roles in Th1 cells differentiation and functions [155]. It has been shown that STAT4 is a genetic risk factor for SSc and SSc related fibrosing alveolitis [156,157]. STAT4 deficient mice were protected against dermal fibrosis upon bleomycin challenge. These mice with STAT4 deficiency showed significantly decreased T cells infiltration and cytokines production in skin lesions, suggesting that STAT4 exerted pro-fibrotic functions by modulating T cell responses [158]. Moreover, a STAT4 variant was shown to be associated with increased hepatic inflammation and fibrosis, which might be partially attributed to the aberrant STAT4-dependent IFN-γ production by NK cells [159]. IL-12-induced STAT4 activation also contributed to cigarette smoke-induced airway fibrosis though regulating fibroblasts [160]. However, it has also been noted that IL-12 and IFN-γ show anti-fibrotic effects in bleomycin-induced pulmonary fibrosis, suggesting possible dual roles of STAT4 in regulating fibrotic diseases [79]. STAT5 proteins are activated by growth factors and cytokines. Loss of hepatic STAT5 increased TGF-β and STAT3 activation in mice upon CCl4 treatment, which resulted in exacerbated liver fibrosis [161]. STAT5 could directly bind to TGF-β through its N-terminal sequences and decreased TGF-β protein stability, suggesting a role of STAT5-TGF-β-STAT3 axis in liver fibrosis [161]. STAT6 is primarily activated by Th2 cytokines, including IL-13 and IL-4. IL-13 increased collagen production by activating STAT6 and promoted S. mansoni infection-induced liver fibrosis in a TGF-β-independent manner [162,163]. IL-4 also played a profibrotic role because deficiency of IL-4Rα signaling suppressed inflammatory monocyte infiltration and liver fibrogenesis, which was associated with reduced MMPs expression by macrophages through IL-4 and IL-13-mediated STAT6 activation [164].
5. Current Progress on Immune-Based Anti-Fibrotic Therapies
Recent findings on the critical involvement of immune cells in fibrogenesis have facilitated the development of novel therapeutic strategies for treating fibrotic diseases. Both pre-clinical and clinical investigations have shown a promising therapeutic potential by modulating immune cell differentiation and function for the treatment of fibrosis.
Pentraxin 2, also known as serum amyloid P, is a potent inhibitor of macrophage activation and differentiation [165]. Patients with IPF showed significantly reduced plasma levels of pentraxin 2, which was correlated with disease severities [166]. Recent studies have reported that a recombinant human pentraxin 2, named PRM-151, is well tolerated and slows the decline of lung function in IPF patients, suggesting that modulation of macrophages differentiation and activation may serve as potential therapies for treating lung fibrosis in the future [167,168].
Kupffer cells, a major resident macrophage population in liver, produce pro-fibrotic factors and contribute to liver fibrosis. Current studies have suggested therapeutic benefits by suppressing the activation of Kupffer cells [169,170]. It has been shown that TLR4-dependent activation of Kupffer cells is reduced by broad-spectrum antibiotics, which is associated with the reduction of liver fibrosis progression [169,171]. Selonsertib, an inhibitor of apoptosis signal-regulating kinase 1 (ASK1), has been shown to modulate the activation of macrophages including Kupffer cells. Pre-treatment of selonsertib reduced TNF-α expression and suppressed inflammasome activation in isolated Kupffer cells [172]. In a phase 2 clinical trial, selonsertib reduced liver fibrosis in a substantial proportion of patients with nonalcoholic steatohepatitis and stage 2–3 fibrosis, which was associated with reductions in liver stiffness, collagen content and lobular inflammation [173]. Thus, therapeutic intervention through modulating Kupffer cell activation may represent a potential therapeutic strategy for treating liver fibrosis [174].
Monocyte-derived macrophages are recruited by Kupffer cells into the liver, which promote tissue inflammation and fibrosis. Inhibition of inflammatory monocyte recruitment into liver by targeting chemokines or chemokine receptors has shown promising therapeutic benefits in various liver diseases, including liver fibrosis [175]. The serum and hepatic C-C motif chemokine ligand (CCL) 5 levels were increased in drug-induced liver injury (DILI) patients. Moreover, inhibition of CCL5 greatly alleviated liver injury and improved survival in mice, suggesting that CCL5 blockage might be a promising therapeutic strategy for the treatment of DILI patients [176]. Pharmacological inhibition of CCL2 suppressed the migration of Ly6C+ monocytes and accelerated regression of liver fibrosis in mice with both CCl4 challenge and methionine-choline-deficient diet treatment [177]. Cenicriviroc, an oral inhibitor of CCR2 and CCR5, significantly reduced the recruitment of hepatic Ly6C+ monocytes, inhibited alcohol-induced steatohepatitis and ameliorated liver fibrosis in mice [177,178]. A randomized, controlled clinical trial has revealed that cenicriviroc treatment is well-tolerated and achieves anti-fibrotic benefits in patients with nonalcoholic steatohepatitis, particularly in those with advanced fibrosis [179,180].
B cells have been suggested to play important roles in fibrogenesis. B cell depletion therapy by rituximab is well tolerated and has achieved clinical improvements in SSc patients [127,128,181]. Rituximab treatment led to a decrease in disease activity index and disease severity index. Moreover, IL-6 levels were decreased during the follow up [128]. Mechanistically, it has been shown that rituximab-mediated B cell depletion improves skin fibrosis regression through regulating TGFβ-Dkk-1 axis in SSc patients [127]. Moreover, treatment with rituximab may serve as an effective, potentially life-saving, therapeutic intervention for patients with severe interstitial lung disease associated with connective tissue disease [182]. Up to date, several clinical trials have been initiated to investigate the safety and efficacy of rituximab in combination with other therapies in treating IPF patients (NCT03584802, NCT01969409, and NCT03286556).
Immune cell targeted therapies for fibrotic diseases have been developing rapidly during recent years. Preclinical studies in fibrotic animal models have provided important information on novel immune-based therapeutic strategies by targeting different immune cells. Moreover, the molecular insights into fibrogenesis have suggested clinical therapeutic potential by targeting key molecular components, including PI3K/mTOR and JAK-STAT pathways [183,184,185,186]. Future controlled clinical trials are essential to evaluate the safety and efficacy of potential therapies in patients with fibrotic diseases.
6. Conclusions and Future Perspectives
Fibrosis is a common pathway to organ injury and failure in a variety of diseases. Many immune cell populations are involved in the pathogenesis of fibrosis with diverse functions (Figure 1, Table 1). The recruitment and activation of both innate and adaptive immune cells orchestrate the fibrotic process. The interactions between immune cells and myofibroblast are key events in fibrogenic responses. Activated immune cells produce multiple cytokines that modulate the differentiation, proliferation, survival, and collagen production of myofibroblasts. Moreover, macrophages and other cells also secrete massive amounts of TGF-β that directly contributes to fibrosis. Current studies have suggested multifaceted functions of immune cells in fibrotic diseases, possibly due to dynamic changes in microenvironment during disease development. Moreover, most immune cell types are heterogeneous with functional plasticity modulated by both systemic and microenvironmental factors. Thus, the cellular identities and local niches are of key significance for their functions in fibrosis. The functions of both immune cells and fibroblasts are tightly regulated by molecular network. The perturbed mTOR and JAK-STAT signaling pathways contribute to immune dysregulation and subsequent fibrosis development. Multiple cytokines and growth factors activate these critical molecular mediators in immune cells and fibroblasts, which exert diverse functions during fibrosis. Further studies on the regulation and functions of immune cells in fibrosis will facilitate the development of immune cell-based therapies for the treatment of fibrotic diseases.
Author Contributions
Conceptualization, X.W. and L.L.; writing—original draft preparation, E.H., N.P., and F.X.; writing—review and editing, N.P., D.H., X.W., and L.L.; funding acquisition, N.P., X.W. and L.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the National Natural Science Foundation of China (No. 91842304 and 81771761), Health Research Fund from Yichang Science and Technology Bureau (A20-2-035), Hong Kong Health and Medical Research Fund (17160832), and Hong Kong Croucher Foundation (260960116).
Acknowledgments
We are thankful to the technical support from Otis Ko and the Medical Faculty Core Facility at The University of Hong Kong.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| α-SMA | α-smooth muscle actin |
| α-Galcer | α-Galactosylceramide |
| AhR | aryl hydrocarbon receptor |
| AILD | autoimmune liver diseases |
| ASK1 | apoptosis signal-regulating kinase 1 |
| BAFF | B-cell activating factor |
| BALF | bronchoalveolar lavage fluid |
| BDL | Bile duct ligation |
| Bregs | regulatory B cells |
| CCl4 | carbon tetrachloride |
| CCL2 | C-C motif chemokine ligand 2 |
| CKD | chronic kidney disease |
| COPD | chronic obstructive pulmonary disease |
| DC | Dendritic cell |
| DILI | drug-induced liver injury |
| ECM | extracellular matrix |
| FGF-9 | fibroblast growth factor 9 |
| GVHD | graft-versus-host disease |
| NASH | nonalcoholic steatohepatitis |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HMF | hepatic myofibroblast |
| HSCs | hepatic stellate cells |
| ICOS | inducible co-stimulator |
| ILCs | innate lymphoid cells |
| iNKT cells | invariant natural killer T cells |
| IPF | idiopathic pulmonary fibrosis |
| JAK | Janus kinase |
| MAIT cells | mucosal-associated invariant T cells |
| MAPK | mitogen-activated protein kinase |
| MMPs | matrix metalloproteinases |
| mTOR | mammalian target of rapamycin |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NE | neutrophil elastase |
| NK cells | natural killer cells |
| NKT cells | natural killer T cells |
| PBC | primary biliary cholangitis |
| PDGF | platelet-derived growth factor |
| PTECs | proximal tubular epithelial cells |
| SSc | systemic sclerosis |
| STAT | signal transducers and activators of transcription |
| Tfh cells | T follicular helper cells |
| TGF-β | transforming growth factor-β |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Tregs | regulatory T cells |
| UUO | unilateral ureteral obstruction |
References
- Urban, M.L.; Manenti, L.; Vaglio, A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 373, 95–96. [Google Scholar] [CrossRef]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020. [Google Scholar] [CrossRef]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages during the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.-P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Sunami, Y.; Leithäuser, F.; Gul, S.; Fiedler, K.; Güldiken, N.; Espenlaub, S.; Holzmann, K.-H.; Hipp, N.; Sindrilaru, A.; Luedde, T.; et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology 2012, 56, 1117–1128. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 2017, 541, 96–101. [Google Scholar] [CrossRef]
- Moreno-Moral, A.; Bagnati, M.; Koturan, S.; Ko, J.-H.; Fonseca, C.; Harmston, N.; Game, L.; Martin, J.; Ong, V.; Abraham, D.J.; et al. Changes in macrophage transcriptome associate with systemic sclerosis and mediate GSDMA contribution to disease risk. Ann. Rheum. Dis. 2018, 77, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Pellicoro, A.; Aucott, R.L.; Ramachandran, P.; Robson, A.J.; Fallowfield, J.A.; Snowdon, V.K.; Hartland, S.N.; Vernon, M.; Duffield, J.S.; Benyon, R.C.; et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012, 55, 1965–1975. [Google Scholar] [CrossRef]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages regulate renal fibrosis through modulating TGFβ superfamily signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.Y.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. Orlando Fla 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef] [Green Version]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Li, Z.; Zhang, Q.; Qu, Y.; Xu, M.; Wan, X.; Lu, L. CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J. Cell. Mol. Med. 2018, 22, 5050–5061. [Google Scholar] [CrossRef]
- Feng, M.; Ding, J.; Wang, M.; Zhang, J.; Zhu, X.; Guan, W. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int. J. Biol. Sci. 2018, 14, 1033–1040. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [Green Version]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.-C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Hasan, S.A.; Eksteen, B.; Reid, D.; Paine, H.V.; Alansary, A.; Johannson, K.; Gwozd, C.; Goring, K.-A.R.; Vo, T.; Proud, D.; et al. Role of IL-17A and neutrophils in fibrosis in experimental hypersensitivity pneumonitis. J. Allergy Clin. Immunol. 2013, 131, 1663–1673. [Google Scholar] [CrossRef]
- Leslie, J.; Millar, B.J.M.; del Pons, A.C.; Burgoyne, R.A.; Frost, J.D.; Barksby, B.S.; Luli, S.; Scott, J.; Simpson, A.J.; Gauldie, J.; et al. FPR-1 is an important regulator of neutrophil recruitment and a tissue-specific driver of pulmonary fibrosis. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.-H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol. 2015, 98, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Obayashi, Y.; Yamadori, I.; Fujita, J.; Yoshinouchi, T.; Ueda, N.; Takahara, J. The role of neutrophils in the pathogenesis of idiopathic pulmonary fibrosis. Chest 1997, 112, 1338–1343. [Google Scholar] [CrossRef] [Green Version]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Jia, Y.; Yao, Z.; Huang, J.; Hao, M.; Yao, S.; Lian, N.; Zhang, F.; Zhang, C.; Chen, X.; et al. Hepatic stellate cell interferes with NK cell regulation of fibrogenesis via curcumin induced senescence of hepatic stellate cell. Cell. Signal. 2017, 33, 79–85. [Google Scholar] [CrossRef]
- Wijaya, R.S.; Read, S.A.; Schibeci, S.; Eslam, M.; Azardaryany, M.K.; El-Khobar, K.; van der Poorten, D.; Lin, R.; Yuen, L.; Lam, V.; et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J. Hepatol. 2019, 71, 252–264. [Google Scholar] [CrossRef]
- Ong, S.; Ligons, D.L.; Barin, J.G.; Wu, L.; Talor, M.V.; Diny, N.; Fontes, J.A.; Gebremariam, E.; Kass, D.A.; Rose, N.R.; et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 2015, 185, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Squires, J.E.; Shivakumar, P.; Mourya, R.; Bessho, K.; Walters, S.; Bezerra, J.A. Natural killer cells promote long-term hepatobiliary inflammation in a low-dose rotavirus model of experimental biliary atresia. PLoS ONE 2015, 10, e0127191. [Google Scholar] [CrossRef]
- Mehrotra, P.; Collett, J.A.; McKinney, S.D.; Stevens, J.; Ivancic, C.M.; Basile, D.P. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: Compensatory role of natural killer cells in athymic rats. Am. J. Physiol. Renal Physiol. 2017, 312, F385–F397. [Google Scholar] [CrossRef] [Green Version]
- Ebbo, M.; Crinier, A.; Vély, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef]
- Horsburgh, S.; Todryk, S.; Ramming, A.; Distler, J.H.W.; O’Reilly, S. Innate lymphoid cells and fibrotic regulation. Immunol. Lett. 2018, 195, 38–44. [Google Scholar] [CrossRef]
- Mikami, Y.; Takada, Y.; Hagihara, Y.; Kanai, T. Innate lymphoid cells in organ fibrosis. Cytokine Growth Factor Rev. 2018, 42, 27–36. [Google Scholar] [CrossRef]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Polo, V.; Pucci-Molineris, M.; Cervera, V.; Gambaro, S.; Yantorno, S.E.; Descalzi, V.; Tiribelli, C.; Gondolesi, G.E.; Meier, D. Group 2 innate lymphoid cells exhibit progressively higher levels of activation during worsening of liver fibrosis. Ann. Hepatol. 2019, 18, 366–372. [Google Scholar] [CrossRef]
- Li, D.; Guabiraba, R.; Besnard, A.-G.; Komai-Koma, M.; Jabir, M.S.; Zhang, L.; Graham, G.J.; Kurowska-Stolarska, M.; Liew, F.Y.; McSharry, C.; et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J. Allergy Clin. Immunol. 2014, 134, 1422–1432. [Google Scholar] [CrossRef]
- Wang, H.; Shen, L.; Sun, X.; Liu, F.; Feng, W.; Jiang, C.; Chu, X.; Ye, X.; Jiang, C.; Wang, Y.; et al. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat. Commun. 2019, 10, 3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, J.; Wu, S.; Cheng, L.; Shen, Y.; Ma, W.; She, W.; Yang, C.; Wang, J.; Jiang, W. Type 3 innate lymphoid cell: A new player in liver fibrosis progression. Clin. Sci. Lond. Engl. 1979 2018, 132, 2565–2582. [Google Scholar] [CrossRef]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Simonian, P.L.; Roark, C.L.; Wehrmann, F.; Lanham, A.M.; Born, W.K.; O’Brien, R.L.; Fontenot, A.P. IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. J. Immunol. 2009, 182, 6540–6549. [Google Scholar] [CrossRef] [Green Version]
- Simonian, P.L.; Wehrmann, F.; Roark, C.L.; Born, W.K.; O’Brien, R.L.; Fontenot, A.P. γδ T cells protect against lung fibrosis via IL-22. J. Exp. Med. 2010, 207, 2239–2253. [Google Scholar] [CrossRef]
- Segawa, S.; Goto, D.; Iizuka, A.; Kaneko, S.; Yokosawa, M.; Kondo, Y.; Matsumoto, I.; Sumida, T. The regulatory role of interferon-γ producing gamma delta T cells via the suppression of T helper 17 cell activity in bleomycin-induced pulmonary fibrosis. Clin. Exp. Immunol. 2016, 185, 348–360. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Hu, Y.; Wang, Y.; Huang, X.; Xu, Y.; Shen, Y.; Cao, J. Recruitment of Neutrophils Mediated by Vγ2 γδ T Cells Deteriorates Liver Fibrosis Induced by Schistosoma japonicum Infection in C57BL/6 Mice. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.T. Dendritic Cells: Novel Players in Fibrosis and Scleroderma. Curr. Rheumatol. Rep. 2012, 14, 30–38. [Google Scholar] [CrossRef]
- Carvalheiro, T.; Zimmermann, M.; Radstake, T.R.D.J.; Marut, W. Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clin. Exp. Immunol. 2020, 201, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galati, D.; Zanotta, S.; Polistina, G.E.; Coppola, A.; Capitelli, L.; Bocchino, M. Circulating dendritic cells are severely decreased in idiopathic pulmonary fibrosis with a potential value for prognosis prediction. Clin. Immunol. 2020, 215, 108454. [Google Scholar] [CrossRef]
- Greer, A.M.; Matthay, M.A.; Kukreja, J.; Bhakta, N.R.; Nguyen, C.P.; Wolters, P.J.; Woodruff, P.G.; Fahy, J.V.; Shin, J.-S. Accumulation of BDCA1+ Dendritic Cells in Interstitial Fibrotic Lung Diseases and Th2-High Asthma. PLoS ONE 2014, 9, e99084. [Google Scholar] [CrossRef] [Green Version]
- Tarrés, M.T.; Maus, R.; Stolper, J.; Aschenbrenner, F.; Welte, T.; Gauldie, J.; Kolb, M.; Maus, U.A. Role of dendritic cells in pulmonary fibrosis in mice. Eur. Respir. J. 2016, 48. [Google Scholar] [CrossRef]
- Tort Tarrés, M.; Aschenbrenner, F.; Maus, R.; Stolper, J.; Schuette, L.; Knudsen, L.; Lopez Rodriguez, E.; Jonigk, D.; Kühnel, M.P.; DeLuca, D.; et al. The FMS-like tyrosine kinase-3 ligand/lung dendritic cell axis contributes to regulation of pulmonary fibrosis. Thorax 2019, 74, 947–957. [Google Scholar] [CrossRef]
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 2014, 3, e000839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza Santos, E.; de Aragão-França, L.S.; Meira, C.S.; Cerqueira, J.V.; Vasconcelos, J.F.; Nonaka, C.K.V.; Pontes-de-Carvalho, L.C.; Soares, M.B.P. Tolerogenic Dendritic Cells Reduce Cardiac Inflammation and Fibrosis in Chronic Chagas Disease. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Lande, R.; Lee, E.Y.; Palazzo, R.; Marinari, B.; Pietraforte, I.; Santos, G.S.; Mattenberger, Y.; Spadaro, F.; Stefanantoni, K.; Iannace, N.; et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nat. Commun. 2019, 10, 1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafaja, S.; Valera, I.; Divekar, A.A.; Saggar, R.; Abtin, F.; Furst, D.E.; Khanna, D.; Singh, R.R. pDCs in lung and skin fibrosis in a bleomycin-induced model and patients with systemic sclerosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Ahadome, S.D.; Mathew, R.; Reyes, N.J.; Mettu, P.S.; Cousins, S.W.; Calder, V.L.; Saban, D.R. Classical dendritic cells mediate fibrosis directly via the retinoic acid pathway in severe eye allergy. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Sun, R.; Wei, H.; Gao, X.; Chen, Y.; Tian, Z. Accelerated liver fibrosis in hepatitis B virus transgenic mice: Involvement of natural killer T cells. Hepatology 2011, 53, 219–229. [Google Scholar] [CrossRef]
- Alhasson, F.; Dattaroy, D.; Das, S.; Chandrashekaran, V.; Seth, R.K.; Schnellmann, R.G.; Chatterjee, S. NKT cell modulates NAFLD potentiation of metabolic oxidative stress-induced mesangial cell activation and proximal tubular toxicity. Am. J. Physiol. Renal Physiol. 2016, 310, F85–F101. [Google Scholar] [CrossRef] [Green Version]
- Fransén-Pettersson, N.; Duarte, N.; Nilsson, J.; Lundholm, M.; Mayans, S.; Larefalk, Å.; Hannibal, T.D.; Hansen, L.; Schmidt-Christensen, A.; Ivars, F.; et al. A New Mouse Model That Spontaneously Develops Chronic Liver Inflammation and Fibrosis. PLoS ONE 2016, 11, e0159850. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, H.Y.; Kim, S.; Chung, J.-H.; Park, W.S.; Chung, D.H. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-gamma. Am. J. Pathol. 2005, 167, 1231–1241. [Google Scholar] [CrossRef]
- Tsao, C.-C.; Tsao, P.-N.; Chen, Y.-G.; Chuang, Y.-H. Repeated Activation of Lung Invariant NKT Cells Results in Chronic Obstructive Pulmonary Disease-Like Symptoms. PLoS ONE 2016, 11, e0147710. [Google Scholar] [CrossRef]
- Chang, C.-H.; Chen, Y.-C.; Zhang, W.; Leung, P.S.C.; Gershwin, M.E.; Chuang, Y.-H. Innate immunity drives the initiation of a murine model of primary biliary cirrhosis. PLoS ONE 2015, 10, e0121320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagi, T.; Takehara, T.; Uemura, A.; Nishio, K.; Shimizu, S.; Kodama, T.; Hikita, H.; Li, W.; Sasakawa, A.; Tatsumi, T.; et al. Absence of invariant natural killer T cells deteriorates liver inflammation and fibrosis in mice fed high-fat diet. J. Gastroenterol. 2010, 45, 1247–1254. [Google Scholar] [CrossRef]
- Bhattacharjee, J.; Kirby, M.; Softic, S.; Miles, L.; Salazar-Gonzalez, R.-M.; Shivakumar, P.; Kohli, R. Hepatic Natural Killer T-cell and CD8+ T-cell Signatures in Mice with Nonalcoholic Steatohepatitis. Hepatol. Commun. 2017, 1, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Boddupalli, C.S.; Verma, R.; Liu, J.; Yang, R.; Pastores, G.M.; Mistry, P.K.; Dhodapkar, M.V. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015, 125, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, K.; Rombouts, K.; Saffioti, F.; Roccarina, D.; Rosselli, M.; Hall, A.; Luong, T.; Tsochatzis, E.A.; Thorburn, D.; Pinzani, M. MAIT cells are chronically activated in patients with autoimmune liver disease and promote profibrogenic hepatic stellate cell activation. Hepatology 2018, 68, 172–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, P.; Weiss, E.; Paradis, V.; Wan, J.; Mabire, M.; Sukriti, S.; Rautou, P.-E.; Albuquerque, M.; Picq, O.; Gupta, A.C.; et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat. Commun. 2018, 9, 2146. [Google Scholar] [CrossRef] [Green Version]
- Bolte, F.J.; O’Keefe, A.C.; Webb, L.M.; Serti, E.; Rivera, E.; Liang, T.J.; Ghany, M.; Rehermann, B. Intra-Hepatic Depletion of Mucosal-Associated Invariant T Cells in Hepatitis C Virus-Induced Liver Inflammation. Gastroenterology 2017, 153, 1392–1403. [Google Scholar] [CrossRef] [Green Version]
- Law, B.M.P.; Wilkinson, R.; Wang, X.; Kildey, K.; Giuliani, K.; Beagley, K.W.; Ungerer, J.; Healy, H.; Kassianos, A.J. Human Tissue-Resident Mucosal-Associated Invariant T (MAIT) Cells in Renal Fibrosis and CKD. J. Am. Soc. Nephrol. 2019, 30, 1322–1335. [Google Scholar] [CrossRef]
- Keane, M.P.; Belperio, J.A.; Burdick, M.D.; Strieter, R.M. IL-12 attenuates bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L92–L97. [Google Scholar] [CrossRef]
- Xu, J.; Mora, A.L.; LaVoy, J.; Brigham, K.L.; Rojas, M. Increased bleomycin-induced lung injury in mice deficient in the transcription factor T-bet. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L658–L667. [Google Scholar] [CrossRef] [Green Version]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.-T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Nevers, T.; Salvador, A.M.; Velazquez, F.; Ngwenyama, N.; Carrillo-Salinas, F.J.; Aronovitz, M.; Blanton, R.M.; Alcaide, P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 2017, 214, 3311–3329. [Google Scholar] [CrossRef] [Green Version]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Cheever, A.W.; Williams, M.E.; Wynn, T.A.; Finkelman, F.D.; Seder, R.A.; Cox, T.M.; Hieny, S.; Caspar, P.; Sher, A. Anti-IL-4 treatment of Schistosoma mansoni-infected mice inhibits development of T cells and non-B, non-T cells expressing Th2 cytokines while decreasing egg-induced hepatic fibrosis. J. Immunol. 1994, 153, 753–759. [Google Scholar]
- Morimoto, Y.; Hirahara, K.; Kiuchi, M.; Wada, T.; Ichikawa, T.; Kanno, T.; Okano, M.; Kokubo, K.; Onodera, A.; Sakurai, D.; et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity 2018, 49, 134–150. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Zhao, C.; Qin, F.; He, Z.-Y.; Wang, X.; Zhong, X.-N. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol. 2016, 34 Suppl 100, 14–22. [Google Scholar]
- Fenoglio, D.; Bernuzzi, F.; Battaglia, F.; Parodi, A.; Kalli, F.; Negrini, S.; De Palma, R.; Invernizzi, P.; Filaci, G. Th17 and regulatory T lymphocytes in primary biliary cirrhosis and systemic sclerosis as models of autoimmune fibrotic diseases. Autoimmun. Rev. 2012, 12, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhang, Z.; Wang, F.-S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell. Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Hua, X.; Xie, C.; Liu, J.; Huang, Y.; Zhou, L.; Zhang, M.; Li, X.; Gao, Z. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zhang, B.; Shen, R.-W.; Liu, J.-B.; Gao, M.-H.; Geng, X.; Li, Y.; Li, Y.-Y.; Zhang, W. The effect of antifibrotic drug halofugine on Th17 cells in concanavalin A-induced liver fibrosis. Scand. J. Immunol. 2014, 79, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ju, B.; Zhang, X.; Zhu, Y.; Nie, Y.; Xu, Y.; Lei, Q. Magnolol Attenuates Concanavalin A-induced Hepatic Fibrosis, Inhibits CD4+ T Helper 17 (Th17) Cell Differentiation and Suppresses Hepatic Stellate Cell Activation: Blockade of Smad3/Smad4 Signalling. Basic Clin. Pharmacol. Toxicol. 2017, 120, 560–570. [Google Scholar] [CrossRef]
- Liang, M.; Wang, J.; Chu, H.; Zhu, X.; He, H.; Liu, Q.; Qiu, J.; Zhou, X.; Guan, M.; Xue, Y.; et al. Interleukin-22 inhibits bleomycin-induced pulmonary fibrosis. Mediators Inflamm. 2013, 2013, 209179. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Liu, S.; Liu, Y.; Guo, C.; Li, H.; Li, W.; Jin, X.; Zhang, K.; Zhao, P.; Wei, L.; et al. Up-regulation of interleukin-22 mediates liver fibrosis via activating hepatic stellate cells in patients with hepatitis C. Clin. Immunol. Orlando Fla 2015, 158, 77–87. [Google Scholar] [CrossRef]
- Galati, D.; De Martino, M.; Trotta, A.; Rea, G.; Bruzzese, D.; Cicchitto, G.; Stanziola, A.A.; Napolitano, M.; Sanduzzi, A.; Bocchino, M. Peripheral depletion of NK cells and imbalance of the Treg/Th17 axis in idiopathic pulmonary fibrosis patients. Cytokine 2014, 66, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Ye, Q.; Qiu, M.; Hao, Y.; Han, J.; Zeng, H. Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 170. [Google Scholar] [CrossRef] [Green Version]
- Boveda-Ruiz, D.; D’Alessandro-Gabazza, C.N.; Toda, M.; Takagi, T.; Naito, M.; Matsushima, Y.; Matsumoto, T.; Kobayashi, T.; Gil-Bernabe, P.; Chelakkot-Govindalayathil, A.-L.; et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology 2013, 218, 245–254. [Google Scholar] [CrossRef]
- Moyé, S.; Bormann, T.; Maus, R.; Sparwasser, T.; Sandrock, I.; Prinz, I.; Warnecke, G.; Welte, T.; Gauldie, J.; Kolb, M.; et al. Regulatory T Cells Limit Pneumococcus-Induced Exacerbation of Lung Fibrosis in Mice. J. Immunol. 2020, 204, 2429–2438. [Google Scholar] [CrossRef]
- Peng, X.; Moore, M.W.; Peng, H.; Sun, H.; Gan, Y.; Homer, R.J.; Herzog, E.L. CD4+CD25+FoxP3+ Regulatory Tregs inhibit fibrocyte recruitment and fibrosis via suppression of FGF-9 production in the TGF-β1 exposed murine lung. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.C.; Ryan, K.; Ahmed, N.; Plitas, G.; Chaudhry, U.I.; Kingham, T.P.; Naheed, S.; Nguyen, C.; Somasundar, P.; Espat, N.J.; et al. Obstructive jaundice expands intrahepatic regulatory T cells which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J. Immunol. 2011, 187, 1150–1156. [Google Scholar] [CrossRef] [Green Version]
- Kalekar, L.A.; Cohen, J.N.; Prevel, N.; Sandoval, P.M.; Mathur, A.N.; Moreau, J.M.; Lowe, M.M.; Nosbaum, A.; Wolters, P.J.; Haemel, A.; et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Do Valle Duraes, F.; Lafont, A.; Beibel, M.; Martin, K.; Darribat, K.; Cuttat, R.; Waldt, A.; Naumann, U.; Wieczorek, G.; Gaulis, S.; et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Chen, G.; Dong, Z.; Liu, H.; Liu, Y.; Duan, S.; Liu, Y.; Liu, F.; Chen, H. mTOR Signaling Regulates Protective Activity of Transferred CD4+Foxp3+ T Cells in Repair of Acute Kidney Injury. J. Immunol. 2016, 197, 3917–3926. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Qiu, J.; Bostick, J.W.; Ueda, A.; Schjerven, H.; Li, S.; Jobin, C.; Chen, Z.E.; Zhou, L. Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell Rep. 2017, 21, 2277–2290. [Google Scholar] [CrossRef] [Green Version]
- Lv, Q.; Wang, K.; Qiao, S.; Yang, L.; Xin, Y.; Dai, Y.; Wei, Z. Norisoboldine, a natural AhR agonist, promotes Treg differentiation and attenuates colitis via targeting glycolysis and subsequent NAD + /SIRT1/SUV39H1/H3K9me3 signaling pathway. Cell Death Dis. 2018, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Takei, H.; Yasuoka, H.; Yoshimoto, K.; Takeuchi, T. Aryl hydrocarbon receptor signals attenuate lung fibrosis in the bleomycin-induced mouse model for pulmonary fibrosis through increase of regulatory T cells. Arthritis Res. Ther. 2020, 22, 20. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Xu, W.; Xiong, S. Adoptive Transfer of Regulatory T Cells Protects against Coxsackievirus B3-Induced Cardiac Fibrosis. PLoS ONE 2013, 8, e74955. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Gregorini, M.; Corradetti, V.; Rocca, C.; Pattonieri, E.F.; Valsania, T.; Milanesi, S.; Serpieri, N.; Bedino, G.; Esposito, P.; Libetta, C.; et al. Mesenchymal Stromal Cells Prevent Renal Fibrosis in a Rat Model of Unilateral Ureteral Obstruction by Suppressing the Renin-Angiotensin System via HuR. PLoS ONE 2016, 11, e0148542. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Li, J.; Guan, H.; Cai, W.; Bai, X.; Fang, X.; Hu, X.; Wang, Y.; Wang, H.; Zheng, Z.; et al. Anti-Fibrotic Actions of Interleukin-10 against Hypertrophic Scarring by Activation of PI3K/AKT and STAT3 Signaling Pathways in Scar-Forming Fibroblasts. PLoS ONE 2014, 9, e98228. [Google Scholar] [CrossRef]
- Birjandi, S.Z.; Palchevskiy, V.; Xue, Y.Y.; Nunez, S.; Kern, R.; Weigt, S.S.; Lynch, J.P.; Chatila, T.A.; Belperio, J.A. CD4+CD25hiFoxp3+ Cells Exacerbate Bleomycin-Induced Pulmonary Fibrosis. Am. J. Pathol. 2016, 186, 2008–2020. [Google Scholar] [CrossRef] [Green Version]
- Langhans, B.; Krämer, B.; Louis, M.; Nischalke, H.D.; Hüneburg, R.; Staratschek-Jox, A.; Odenthal, M.; Manekeller, S.; Schepke, M.; Kalff, J.; et al. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J. Hepatol. 2013, 59, 229–235. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Dawson, N.A.J.; Huang, Q.; Dunne, J.V.; Levings, M.K.; Broady, R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J. Allergy Clin. Immunol. 2015, 135, 946–955. [Google Scholar] [CrossRef]
- Taylor, D.K.; Mittereder, N.; Kuta, E.; Delaney, T.; Burwell, T.; Dacosta, K.; Zhao, W.; Cheng, L.I.; Brown, C.; Boutrin, A.; et al. T follicular helper-like cells contribute to skin fibrosis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Asai, Y.; Chiba, H.; Nishikiori, H.; Kamekura, R.; Yabe, H.; Kondo, S.; Miyajima, S.; Shigehara, K.; Ichimiya, S.; Takahashi, H. Aberrant populations of circulating T follicular helper cells and regulatory B cells underlying idiopathic pulmonary fibrosis. Respir. Res. 2019, 20. [Google Scholar] [CrossRef]
- Brodeur, T.Y.; Robidoux, T.E.; Weinstein, J.S.; Craft, J.; Swain, S.L.; Marshak-Rothstein, A. IL-21 Promotes Pulmonary Fibrosis through the Induction of Pro-fibrotic CD8+ T Cells. J. Immunol. 2015, 195, 5251–5260. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Laurence, A.; Elias, K.M.; O’Shea, J.J. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 2007, 282, 34605–34610. [Google Scholar] [CrossRef] [Green Version]
- Komura, K.; Yanaba, K.; Horikawa, M.; Ogawa, F.; Fujimoto, M.; Tedder, T.F.; Sato, S. CD19 regulates the development of bleomycin-induced pulmonary fibrosis in a mouse model. Arthritis Rheum. 2008, 58, 3574–3584. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Iwata, Y.; Komura, K.; Ogawa, F.; Hara, T.; Muroi, E.; Takenaka, M.; Shimizu, K.; Hasegawa, M.; Fujimoto, M.; et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. Am. J. Pathol. 2008, 172, 1650–1663. [Google Scholar] [CrossRef] [Green Version]
- Saito, E.; Fujimoto, M.; Hasegawa, M.; Komura, K.; Hamaguchi, Y.; Kaburagi, Y.; Nagaoka, T.; Takehara, K.; Tedder, T.F.; Sato, S. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J. Clin. Investig. 2002, 109, 1453–1462. [Google Scholar] [CrossRef]
- Asano, N.; Fujimoto, M.; Yazawa, N.; Shirasawa, S.; Hasegawa, M.; Okochi, H.; Tamaki, K.; Tedder, T.F.; Sato, S. B Lymphocyte signaling established by the CD19/CD22 loop regulates autoimmunity in the tight-skin mouse. Am. J. Pathol. 2004, 165, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Invest. 2005, 115, 3072–3082. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Kobayashi, T.; Mizumaki, K.; Kano, M.; Sawada, T.; Tennichi, M.; Okamura, A.; Hamaguchi, Y.; Iwakura, Y.; Hasegawa, M.; et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci. Adv. 2018, 4, eaas9944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, N.; Kuroki, K.; Fujimoto, M.; Murakami, Y.; Tedder, T.F.; Tokunaga, K.; Takehara, K.; Sato, S. Association of a functional CD19 polymorphism with susceptibility to systemic sclerosis. Arthritis Rheum. 2004, 50, 4002–4007. [Google Scholar] [CrossRef] [PubMed]
- François, A.; Chatelus, E.; Wachsmann, D.; Sibilia, J.; Bahram, S.; Alsaleh, G.; Gottenberg, J.-E. B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthritis Res. Ther. 2013, 15, R168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoussis, D.; Tsamandas, A.; Antonopoulos, I.; Filippopoulou, A.; Papachristou, D.J.; Papachristou, N.I.; Andonopoulos, A.P.; Liossis, S.-N. B cell depletion therapy upregulates Dkk-1 skin expression in patients with systemic sclerosis: Association with enhanced resolution of skin fibrosis. Arthritis Res. Ther. 2016, 18, 118. [Google Scholar] [CrossRef] [Green Version]
- Bosello, S.; De Santis, M.; Lama, G.; Spanò, C.; Angelucci, C.; Tolusso, B.; Sica, G.; Ferraccioli, G. B cell depletion in diffuse progressive systemic sclerosis: Safety, skin score modification and IL-6 modulation in an up to thirty-six months follow-up open-label trial. Arthritis Res. Ther. 2010, 12, R54. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Dai, W.; Li, C.; Lu, X.; Chen, Y.; Weng, D.; Chen, J. Role of IL-10-producing regulatory B cells in modulating T-helper cell immune responses during silica-induced lung inflammation and fibrosis. Sci. Rep. 2016, 6, 28911. [Google Scholar] [CrossRef]
- Ma, K.; Wang, X.; Shi, X.; Lin, X.; Xiao, F.; Ma, X.; Liu, D.; Lu, L. The expanding functional diversity of plasma cells in immunity and inflammation. Cell. Mol. Immunol. 2019, 17, 421–422. [Google Scholar] [CrossRef]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.-F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 201, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Ding, Y.; Fu, Y.; Zhou, L.; Dong, X.; Chen, S.; Wu, H.; Nai, W.; Zheng, H.; Xu, W.; et al. mTOR Overactivation in Mesenchymal cells Aggravates CCl4− Induced liver Fibrosis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, A.; Yanaba, K.; Yoshizaki, A.; Iwata, Y.; Komura, K.; Ogawa, F.; Takenaka, M.; Shimizu, K.; Asano, Y.; Hasegawa, M.; et al. Treatment with rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 2010, 62, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Vigeland, C.L.; Collins, S.L.; Chan-Li, Y.; Hughes, A.H.; Oh, M.-H.; Powell, J.D.; Horton, M.R. Deletion of mTORC1 Activity in CD4+ T Cells Is Associated with Lung Fibrosis and Increased γδ T Cells. PLoS ONE 2016, 11, e0163288. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Platé, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. J. Scleroderma Relat. Disord. 2020, 5, 40–50. [Google Scholar] [CrossRef]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Surinkaew, S.; Naud, P.; Qi, X.-Y.; Gillis, M.-A.; Shi, Y.-F.; Tardif, J.-C.; Dobrev, D.; Nattel, S. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc. Res. 2017, 113, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Mertens, P.R.; Gressner, A.M.; Dooley, S. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J. Hepatol. 2007, 46, 295–303. [Google Scholar] [CrossRef]
- Inagaki, Y.; Nemoto, T.; Kushida, M.; Sheng, Y.; Higashi, K.; Ikeda, K.; Kawada, N.; Shirasaki, F.; Takehara, K.; Sugiyama, K.; et al. Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology 2003, 38, 890–899. [Google Scholar] [CrossRef]
- Kemmner, S.; Bachmann, Q.; Steiger, S.; Lorenz, G.; Honarpisheh, M.; Foresto-Neto, O.; Wang, S.; Carbajo-Lozoya, J.; Alt, V.; Schulte, C.; et al. STAT1 regulates macrophage number and phenotype and prevents renal fibrosis after ischemia-reperfusion injury. Am. J. Physiol.-Ren. Physiol. 2018, 316, F277–F291. [Google Scholar] [CrossRef]
- Huang, F.; Wang, Q.; Guo, F.; Zhao, Y.; Ji, L.; An, T.; Song, Y.; Liu, Y.; He, Y.; Qin, G. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine 2019, 48, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef] [Green Version]
- Zehender, A.; Huang, J.; Györfi, A.-H.; Matei, A.-E.; Trinh-Minh, T.; Xu, X.; Li, Y.-N.; Chen, C.-W.; Lin, J.; Dees, C.; et al. The tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat. Commun. 2018, 9, 3259. [Google Scholar] [CrossRef]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) Trans Signaling Drives a STAT3-dependent Pathway That Leads to Hyperactive Transforming Growth Factor-β (TGF-β) Signaling Promoting SMAD3 Activation and Fibrosis via Gremlin Protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yuan, H.; Cao, W.; Wang, T.; Chen, W.; Yu, H.; Fu, Y.; Jiang, B.; Zhou, H.; Guo, H.; et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics 2019, 9, 3980–3991. [Google Scholar] [CrossRef]
- Martí-Rodrigo, A.; Alegre, F.; Moragrega, Á.B.; García-García, F.; Martí-Rodrigo, P.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Apostolova, N.; Esplugues, J.V.; Blas-García, A. Rilpivirine attenuates liver fibrosis through selective STAT1-mediated apoptosis in hepatic stellate cells. Gut 2020, 69, 920–932. [Google Scholar] [CrossRef]
- Xu, S.; Mao, Y.; Wu, J.; Feng, J.; Li, J.; Wu, L.; Yu, Q.; Zhou, Y.; Zhang, J.; Chen, J.; et al. TGF-β/Smad and JAK/STAT pathways are involved in the anti-fibrotic effects of propylene glycol alginate sodium sulphate on hepatic fibrosis. J. Cell. Mol. Med. 2020, 24, 5224–5237. [Google Scholar] [CrossRef] [Green Version]
- Nishikomori, R.; Usui, T.; Wu, C.-Y.; Morinobu, A.; O’Shea, J.J.; Strober, W. Activated STAT4 has an essential role in Th1 differentiation and proliferation that is independent of its role in the maintenance of IL-12R beta 2 chain expression and signaling. J. Immunol. 2002, 169, 4388–4398. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Kawasaki, A.; Hasegawa, M.; Fujimoto, M.; Takehara, K.; Kawaguchi, Y.; Kawamoto, M.; Hara, M.; Sato, S. Association of STAT4 polymorphism with systemic sclerosis in a Japanese population. Ann. Rheum. Dis. 2009, 68, 1375–1376. [Google Scholar] [CrossRef]
- Dieudé, P.; Guedj, M.; Wipff, J.; Ruiz, B.; Hachulla, E.; Diot, E.; Granel, B.; Sibilia, J.; Tiev, K.; Mouthon, L.; et al. STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum. 2009, 60, 2472–2479. [Google Scholar] [CrossRef]
- Avouac, J.; Fürnrohr, B.G.; Tomcik, M.; Palumbo, K.; Zerr, P.; Horn, A.; Dees, C.; Akhmetshina, A.; Beyer, C.; Distler, O.; et al. Inactivation of the transcription factor STAT-4 prevents inflammation-driven fibrosis in animal models of systemic sclerosis. Arthritis Rheum. 2011, 63, 800–809. [Google Scholar] [CrossRef]
- El Sharkawy, R.; Thabet, K.; Lampertico, P.; Petta, S.; Mangia, A.; Berg, T.; Metwally, M.; Bayoumi, A.; Boonstra, A.; Brouwer, W.P.; et al. A STAT4 variant increases liver fibrosis risk in Caucasian patients with chronic hepatitis B. Aliment. Pharmacol. Ther. 2018, 48, 564–573. [Google Scholar] [CrossRef]
- Hackett, T.-L.; Shaheen, F.; Zhou, S.; Wright, J.L.; Churg, A. Fibroblast Signal Transducer and Activator of Transcription 4 Drives Cigarette Smoke–Induced Airway Fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 830–839. [Google Scholar] [CrossRef]
- Hosui, A.; Kimura, A.; Yamaji, D.; Zhu, B.; Na, R.; Hennighausen, L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-β and STAT3 activation. J. Exp. Med. 2009, 206, 819–831. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Fullard, N.; Przyborski, S.; van Laar, J.M. IL-13 mediates collagen deposition via STAT6 and microRNA-135b: A role for epigenetics. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Kaviratne, M.; Hesse, M.; Leusink, M.; Cheever, A.W.; Davies, S.J.; McKerrow, J.H.; Wakefield, L.M.; Letterio, J.J.; Wynn, T.A. IL-13 Activates a Mechanism of Tissue Fibrosis That Is Completely TGF-β Independent. J. Immunol. 2004, 173, 4020–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, S.-Y.; Wang, X.; Vijayan, S.; Tang, Y.; Kim, Y.O.; Padberg, K.; Regen, T.; Molokanova, O.; Chen, T.; Bopp, T.; et al. IL-4 Receptor Alpha Signaling through Macrophages Differentially Regulates Liver Fibrosis Progression and Reversal. EBioMedicine 2018, 29, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, A.P.; Cavassani, K.A.; Hullinger, R.; Rosada, R.S.; Fong, D.J.; Murray, L.; Hesson, D.P.; Hogaboam, C.M. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J. Allergy Clin. Immunol. 2010, 126, 712–721. [Google Scholar] [CrossRef]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients With Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open-label extension study. Lancet Respir. Med. 2019, 7, 657–664. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Gaul, S.; Geisler, L.J.; McGeough, M.D.; Johnson, C.D.; Zagorska, A.; Li, L.; Wree, A.; Barry, V.; Mikaelian, I.; Jih, L.J.; et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Devisscher, L.; Tacke, F. Targeting CCR2/5 in the treatment of nonalcoholic steatohepatitis (NASH) and fibrosis: Opportunities and challenges. Expert Opin. Investig. Drugs 2020, 29, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Sun, X.; Zhao, J.; Xia, L.; Li, J.; Xu, M.; Wang, B.; Guo, H.; Yu, C.; Gao, Y.; et al. CCL5 deficiency promotes liver repair by improving inflammation resolution and liver regeneration through M2 macrophage polarization. Cell. Mol. Immunol. 2020, 17, 753–764. [Google Scholar] [CrossRef]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C.; et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.-S.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis: Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020. [Google Scholar] [CrossRef] [Green Version]
- Thiebaut, M.; Launay, D.; Rivière, S.; Mahévas, T.; Bellakhal, S.; Hachulla, E.; Fain, O.; Mekinian, A. Efficacy and safety of rituximab in systemic sclerosis: French retrospective study and literature review. Autoimmun. Rev. 2018, 17, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Keir, G.J.; Maher, T.M.; Hansell, D.M.; Denton, C.P.; Ong, V.H.; Singh, S.; Wells, A.U.; Renzoni, E.A. Severe interstitial lung disease in connective tissue disease: Rituximab as rescue therapy. Eur. Respir. J. 2012, 40, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, M.; Kreutzer, F.P.; Kapsner, L.A.; Mitzka, S.; Just, A.; Perbellini, F.; Terracciano, C.M.; Xiao, K.; Geffers, R.; Bogdan, C.; et al. Integrative Bioinformatic Analyses of Global Transcriptome Data Decipher Novel Molecular Insights into Cardiac Anti-Fibrotic Therapies. Int. J. Mol. Sci. 2020, 21, 4727. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kumar Tekade, R.; Kalia, K. Kaempferol in ameliorating diabetes-induced fibrosis and renal damage: An in vitro and in vivo study in diabetic nephropathy mice model. Phytomedicine Int. J. Phytother. Phytopharm. 2020, 76, 153235. [Google Scholar] [CrossRef]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Zhang, J.; Wang, Y.; Yu, C.; Zhang, J.; Song, X.; Lv, C. Feifukang ameliorates pulmonary fibrosis by inhibiting JAK-STAT signaling pathway. BMC Complement. Altern. Med. 2018, 18, 234. [Google Scholar] [CrossRef]
Figure 1.
Roles of immune cells in the pathogenesis of fibrosis. During inflammatory responses, many immune cell populations with diverse functions are activated to produce multiple cytokines that either activate (red arrow) or suppress (blue arrow) the differentiation, proliferation, and collagen production of myofibroblasts, promoting or suppressing the development of fibrosis in various diseases.
Figure 1.
Roles of immune cells in the pathogenesis of fibrosis. During inflammatory responses, many immune cell populations with diverse functions are activated to produce multiple cytokines that either activate (red arrow) or suppress (blue arrow) the differentiation, proliferation, and collagen production of myofibroblasts, promoting or suppressing the development of fibrosis in various diseases.


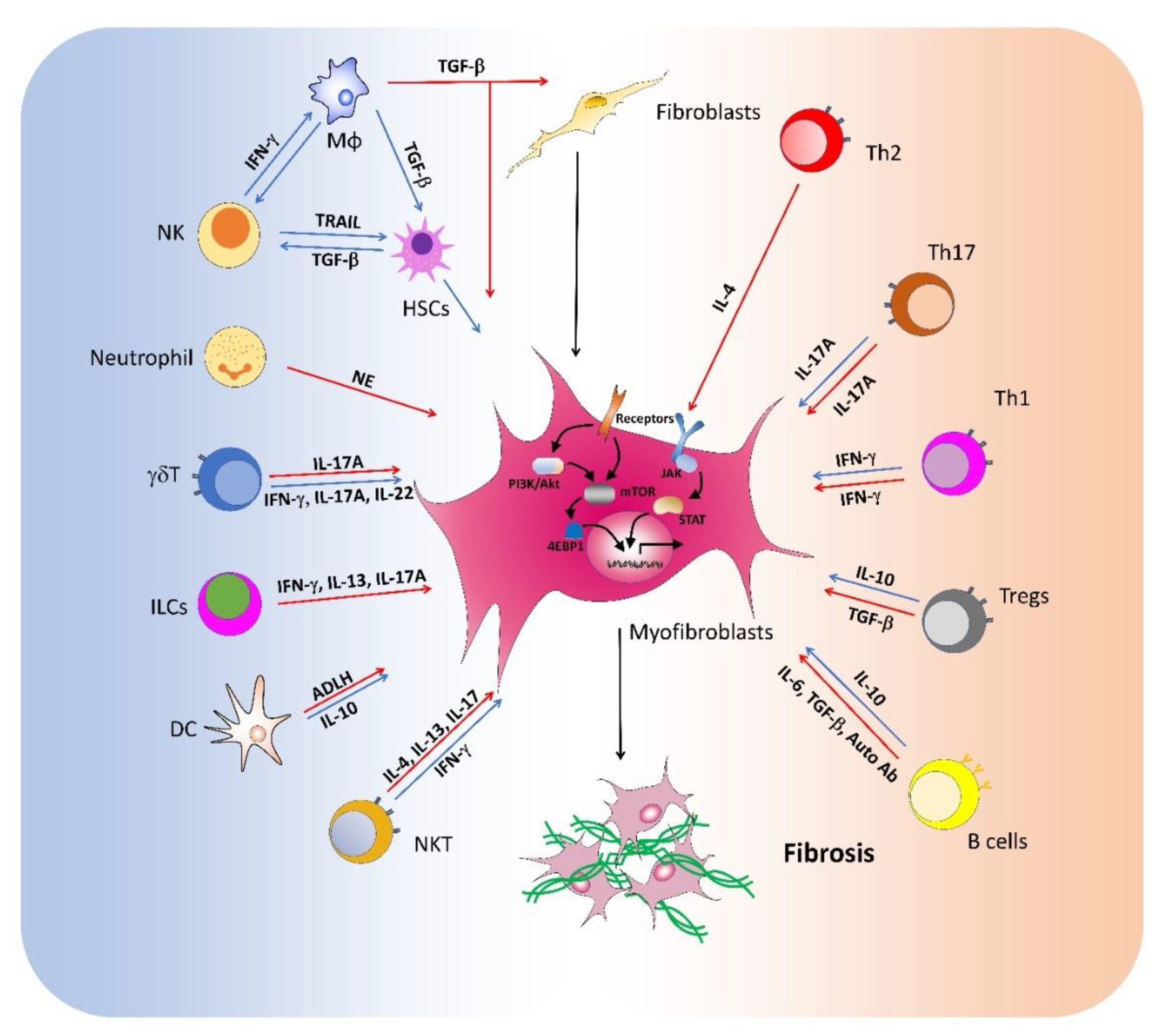{kind=link}
Table 1.
The roles of immune cells in the development fibrosis.
| Cell Subsets | Roles in Fibrosis | Diseases/Models | References |
|---|---|---|---|
| Macrophages | Promote liver fibrosis | NFκB-induced or CCl4-induced liver fibrosis BDL-induced liver fibrosis | [8,9,10,11] |
| M2 macrophages promote renal fibrosis | UUO-induced renal fibrosis | [16] | |
| Kupffer cells promote fibrosis | CCl4-induced liver fibrosis | [4,21] | |
| Kupffer cells contribute to liver fibrosis resolution | Thioacetamide-induced liver fibrosis | [22] | |
| Segregated nucleus-containing atypical monocytes or alveolar macrophages promote lung fibrosis | IL-10 or bleomycin-induced pulmonary fibrosis | [17,18,19,20] | |
| M1 cells express MMP12 to degrade ECM | CCl4-induced liver fibrosis | [15] | |
| Neutrophils | Promote lung fibrosis | Experimental hypersensitivity pneumonitis Bleomycin-induced pulmonary fibrosis | [30,31] |
| NE promote lung fibrosis | Asbestos-induced lung fibrosis | [32,33] | |
| NK cells | Suppress liver fibrosis | CCl4-induced and HBV associated liver fibrosis | [35,36,37,38] |
| Suppress cardiac fibrosis | Autoimmune myocarditis | [39] | |
| Promote liver fibrosis | Low-dose rotavirus infection | [40] | |
| Promote renal fibrosis | Injury-induced renal fibrosis | [41] | |
| ILC1 | Promote adipose fibrosis | High-fat diet-fed obese mice | [48] |
| ILC2 | Promote lung fibrosis | Bleomycin-induced pulmonary fibrosis | [45,47] |
| ILC3 | Promote liver fibrosis | CCl4-induced liver fibrosis | [44,49] |
| γδT cells | Suppress liver fibrosis | CCl4-induced liver fibrosis | [50] |
| Suppress lung fibrosis | B. subtilis-induced pulmonary fibrosis Bleomycin-induced pulmonary fibrosis | [51,52,53] | |
| Promote liver fibrosis | S. japonicum -induced liver fibrosis | [54] | |
| Dendritic cells | Suppress lung fibrosis | Mice exposed to adenoviral gene transfer of TGF-β1 | [59] |
| IL-10-producing DCs suppress heart inflammation and fibrosis | Chronic Chagas disease cardiomyopathy model | [62] | |
| pDCs suppress lung and skin fibrosis | Bleomycin-induced SSc model | [64] | |
| CD11b+ DCs promote ocular mucosal fibrosis | Allergic eye disease model | [65] | |
| NKT cells/ MAIT cells | NKT cells promote liver fibrosis | CCl4-treated HBV-transgenic mice | [66] |
| CD1d-dependent NKT cells suppress kidney fibrosis | Diet-induced NAFLD mouse model | [67] | |
| iNKT cells suppress lung fibrosis | Bleomycin-induced pulmonary fibrosis | [69] | |
| invariant NKT cells promote liver injury and fibrosis | Autoimmune cholangitis mouse model, mice fed with high fat diet | [71,72] | |
| MAIT cells promote liver fibrosis | CCl4-induced or bile duct ligation-induced liver fibrosis | [76] | |
| Th1 cells | Suppress lung fibrosis | Bleomycin-induced pulmonary fibrosis | [79,80] |
| Promote peritoneal fibrosis | S. epidermidis-induced peritoneal fibrosis | [81] | |
| Promote cardiac fibrosis | Thoracic aortic constriction-induced cardiac fibrosis | [82] | |
| Th2 cells | Promote liver fibrosis | Bacterial infection-associated hepatic fibrosis | [84] |
| Promote airway fibrosis | House dust mite -induced allergic disorders | [85] | |
| Th17 cells | Promoted skin and lung fibrosis | Bleomycin-induced SSc model | [87] |
| Promote lung fibrosis | Bleomycin-induced pulmonary fibrosis | [86] | |
| Suppress lung fibrosis | B. subtilis-induced pulmonary fibrosis | [51] | |
| Tregs | Suppress lung fibrosis | Bleomycin-induced pulmonary fibrosis (late stage) | [98] |
| Suppress liver fibrosis | BDL-induced liver fibrosis | [101] | |
| Suppress skin fibrosis | Bleomycin-induced SSc model | [102] | |
| Suppress kidney fibrosis | Injury-induced renal fibrosis | [103] | |
| Tfh cells | Promote dermal fibrosis | GVHD-SSc model | [115] |
| Promote lung fibrosis | Bleomycin-induced pulmonary fibrosis | [117] | |
| B cells | Promote lung fibrosis | Bleomycin-induced pulmonary fibrosis | [119,120] |
| Promote skin fibrosis | TSK/+ mice | [121,122] | |
| Promote liver fibrosis | CCl4-induced liver fibrosis | [123] | |
| IL-6-producing B cells promote skin and lung fibrosis | Bleomycin-induced SSc model | [124] | |
| Bregs suppress skin and lung fibrosis | Bleomycin-induced SSc model | [124] | |
| Bregs promote lung fibrosis | Silica-induced pulmonary fibrosis | [129] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203. https://doi.org/10.3390/ijms21155203
AMA Style
Huang E, Peng N, Xiao F, Hu D, Wang X, Lu L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. International Journal of Molecular Sciences. 2020; 21(15):5203. https://doi.org/10.3390/ijms21155203
Chicago/Turabian StyleHuang, Enyu, Na Peng, Fan Xiao, Dajun Hu, Xiaohui Wang, and Liwei Lu. 2020. "The Roles of Immune Cells in the Pathogenesis of Fibrosis" International Journal of Molecular Sciences 21, no. 15: 5203. https://doi.org/10.3390/ijms21155203
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.