Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
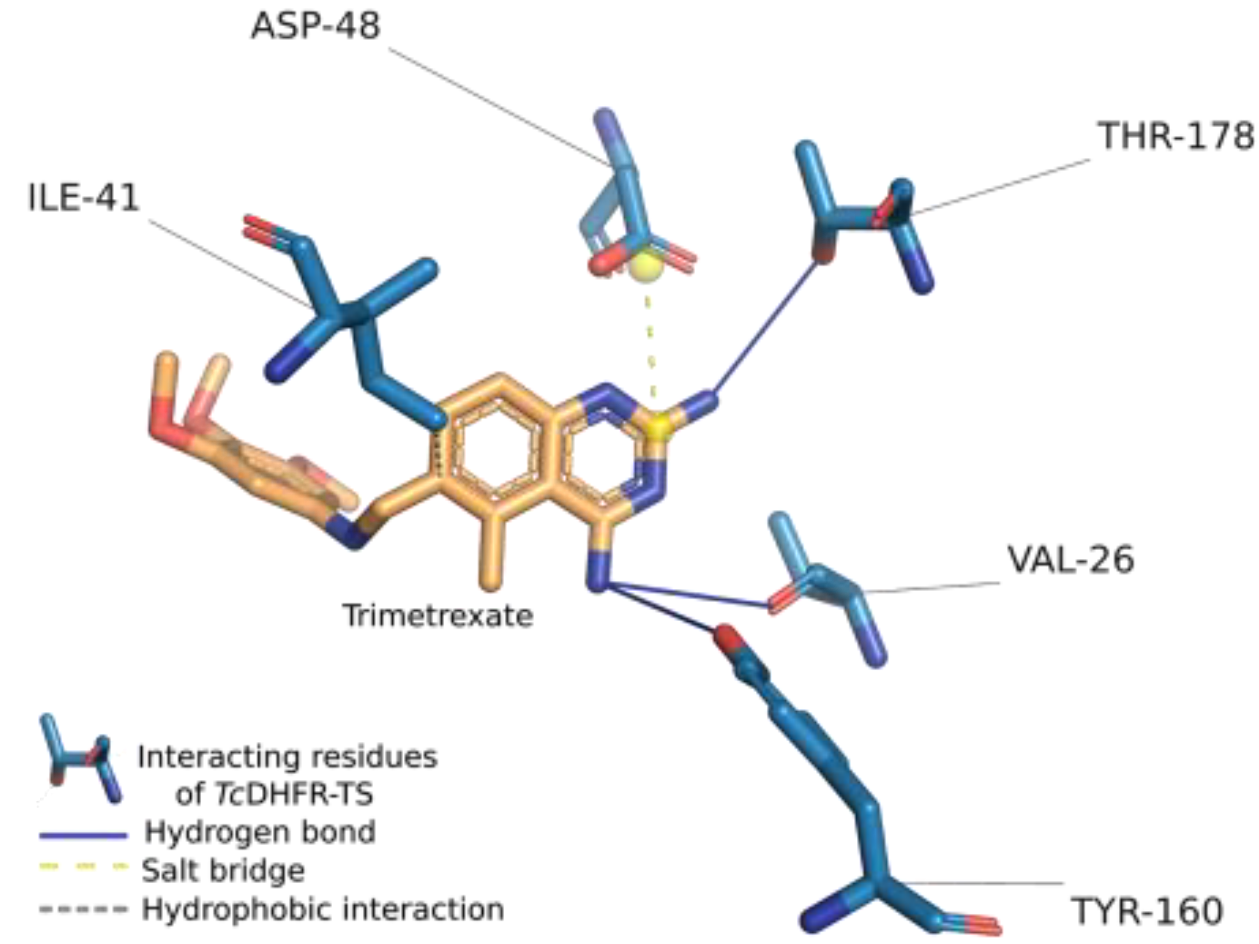
2.1. Assessment of Trimetrexate-TcDFHR-TS Complex
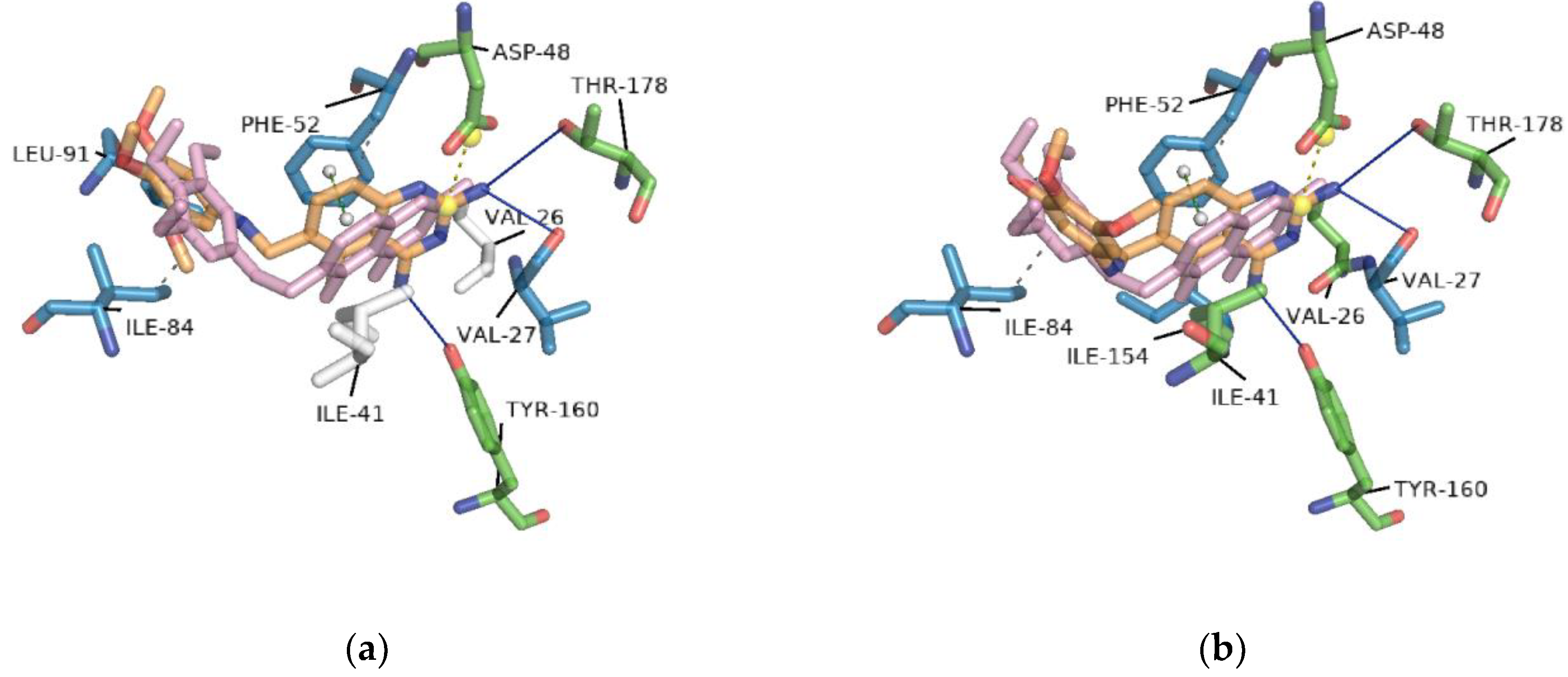
2.2. Redocking
2.3. Interaction Profile Generation
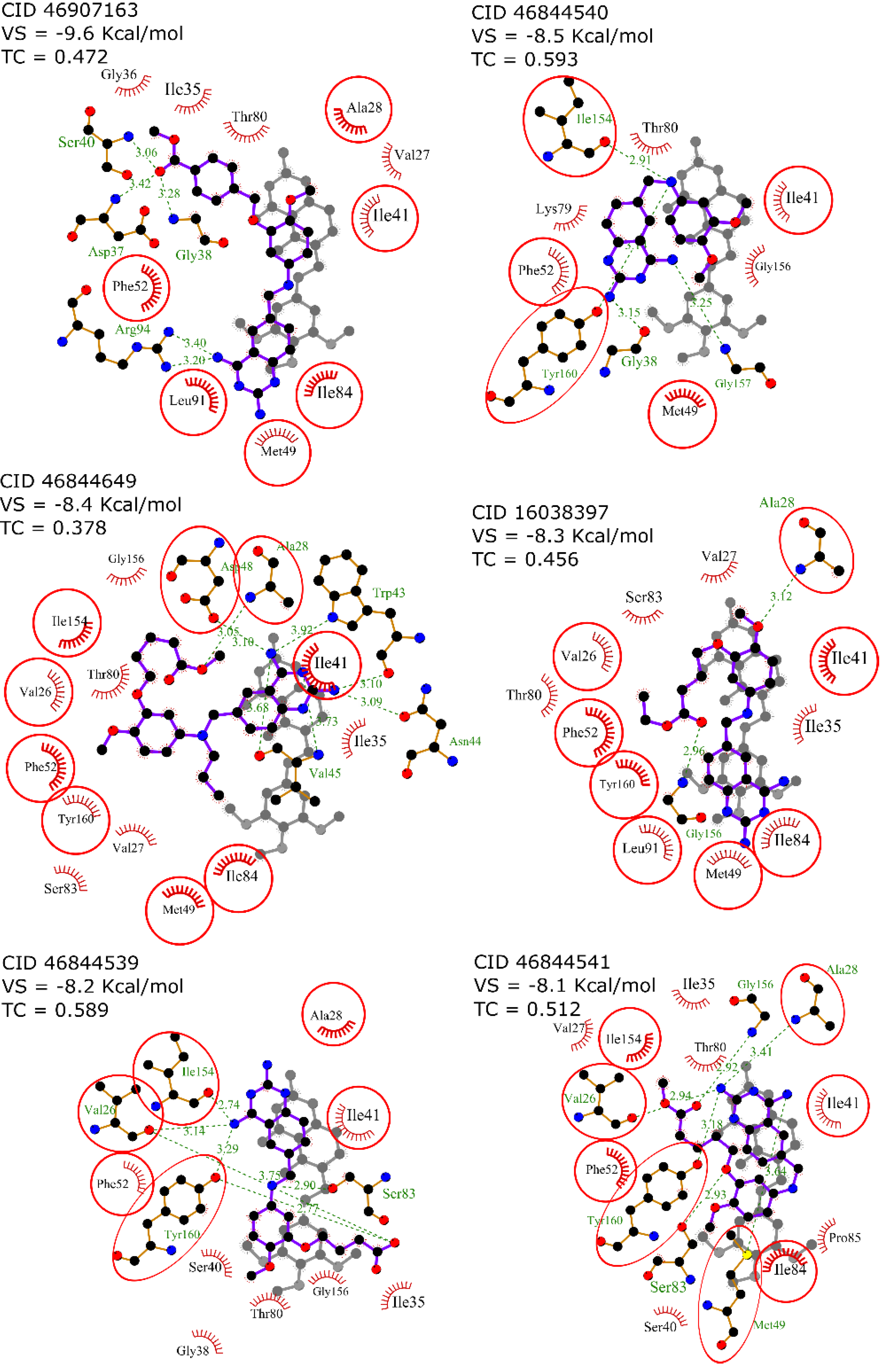
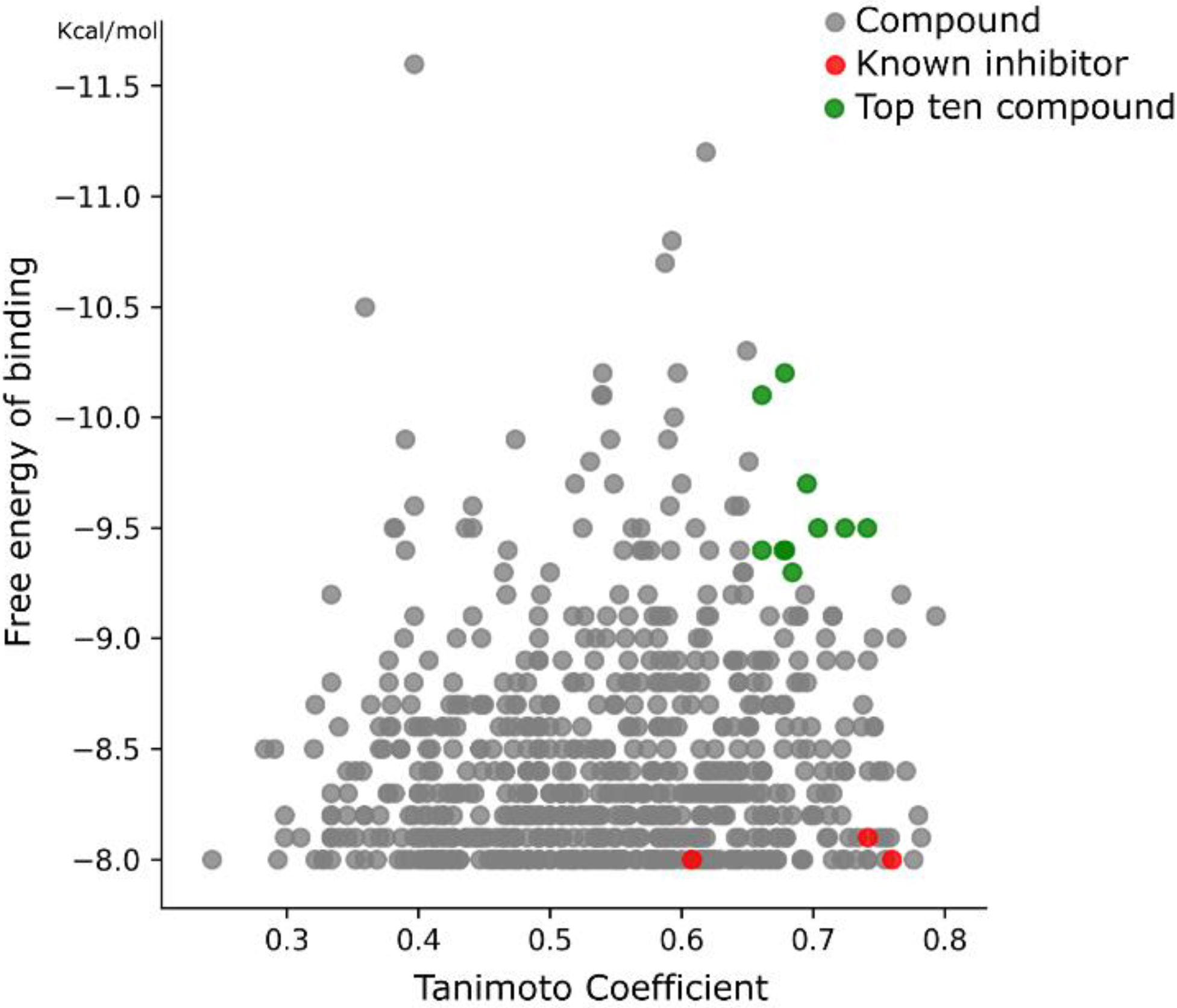
2.4. Virtual Screening
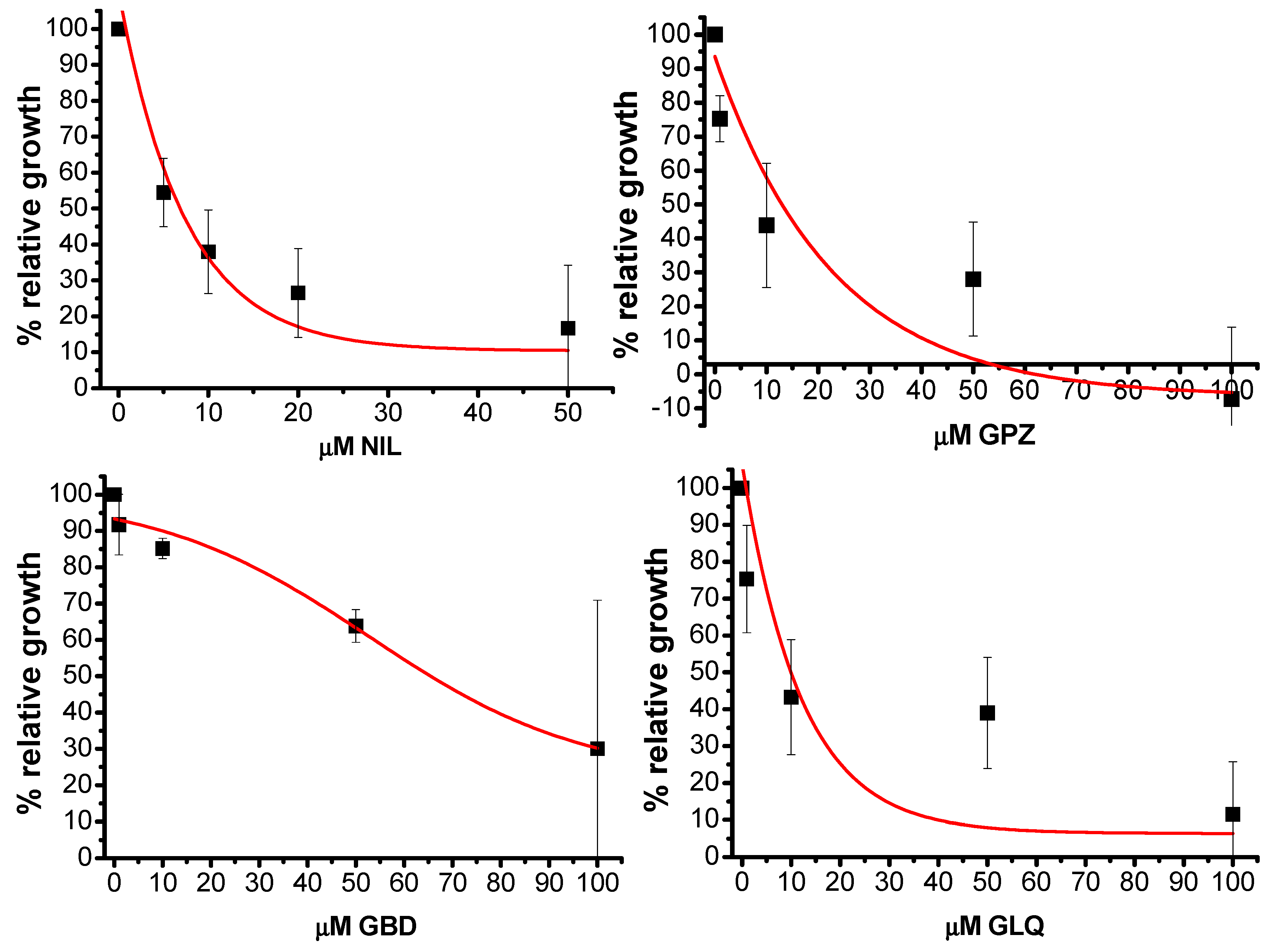
2.5. In Vitro Activity
3. Discussion
4. Materials and Methods
4.1. Protein Structure Preparation
4.2. Ligand Preparation
4.3. Molecular Docking
4.4. Interaction Profiling
4.5. Similarity Calculation
4.6. Cell Culture
4.7. Exposure to FDA-Approved Drugs
4.8. FDA-Approved Drugs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DHFR | Dihydrofolate reductase |
| TS | Thymidylate synthase |
| NIL | Nilotinib |
| GPZ | Glipizide |
| GLQ | Gliquidone |
| GBD | Glyburide |
References
- Rassi, A.; Rassi, A.; Marin-Neto, J.A. Chagas Disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Jiménez, P.; Jaimes, J.; Poveda, C.; Ramírez, J.D. A systematic review of the Trypanosoma cruzi genetic heterogeneity, host immune response and genetic factors as plausible drivers of chronic chagasic cardiomyopathy. Parasitology 2019, 141, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Scarim, C.B.; Jornada, D.H.; Chelucci, R.C.; de Almeida, L.; Dos Santos, J.L.; Chung, M.C. Current advances in drug discovery for Chagas disease. Eur. J Med. Chem. 2018, 155, 824–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meymandi, S.; Hernandez, S.; Park, S.; Sanchez, D.R.; Forsyth, C. Treatment of Chagas Disease in the United States. Curr. Treat. Options Infect. Dis. 2018, 10, 373–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junior, P.A.S.; Molina, I.; Murta, S.M.F.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Carneiro, C.M. Experimental and Clinical Treatment of Chagas Disease: A Review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef]
- Jin, G.; Wong, S.T.C. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Planer, J.D.; Hulverson, M.A.; Arif, J.A.; Ranade, R.M.; Don, R.; Buckner, F.S. Synergy testing of FDA-approved drugs identifies potent drug combinations against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2014, 8, e2977. [Google Scholar] [CrossRef] [Green Version]
- Simões-Silva, M.R.; De Araújo, J.S.; Oliveira, G.M.; Demarque, K.C.; Peres, R.B.; D’Almeida-Melo, I.; Batista, D.G.J.; Da Silva, C.F.; Cardoso-Santos, C.; Da Silva, P.B.; et al. Drug repurposing strategy against Trypanosoma cruzi infection: In vitro and in vivo assessment of the activity of metronidazole in mono- and combined therapy. Biochem. Pharmacol. 2017, 145, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Lima, C.F.; Peres, R.B.; Silva, P.B.; Batista, M.M.; Aiub, C.A.F.; Felzenszwalb, I.; Soeiro, M.N.C. Repurposing Strategy of Atorvastatin against Trypanosoma cruzi: In Vitro Monotherapy and Combined Therapy with Benznidazole Exhibit Synergistic Trypanocidal Activity. Antimicrob. Agents Chemother. 2018, 62, e00979. [Google Scholar] [CrossRef] [Green Version]
- Reigada, C.; Valera-Vera, E.A.; Sayé, M.; Errasti, A.E.; Avila, C.C.; Miranda, M.R.; Pereira, C.A. Trypanocidal Effect of Isotretinoin through the Inhibition of Polyamine and Amino Acid Transporters in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2017, 11, e0005472. [Google Scholar] [CrossRef] [PubMed]
- Custodio, L. Developments on treatment of Chagas disease—From discovery to current times. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2576–2586. [Google Scholar] [CrossRef]
- de Godoy, A.S.; Fernandes, R.S.; Aguiar, A.C.; Bueno, R.V.; Mesquita, N.C.M.R.; Guido, R.V.C.; Oliva, G. Structural and mechanistic insight from antiviral and antiparasitic enzyme drug targets for tropical infectious diseases. Curr. Opin. Struct. Biol. 2019, 59, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.M.; Tarleton, R.L. Potential new clinical therapies for Chagas disease. Expert Rev. Clin. Pharmacol. 2014, 7, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Haupt, V.J.; Uvalle, J.E.A.; Salentin, S.; Daminelli, S.; Leonhardt, F.; Konc, J.; Schroeder, M. Computational Drug Repositioning by Target Hopping: A Use Case in Chagas Disease. Curr. Pharm. Des. 2016, 22, 3124–3134. [Google Scholar] [CrossRef] [PubMed]
- Bellera, C.L.; Balcazar, D.E.; Vanrell, M.C.; Casassa, A.F.; Palestro, P.H.; Gavernet, L.; Labriola, C.A.; Gálvez, J.; Bruno-Blanch, L.E.; Romano, P.S.; et al. Computer-guided drug repurposing: Identification of trypanocidal activity of clofazimine, benidipine and saquinavir. Eur. J. Med. Chem. 2015, 93, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Bellera, C.L.; Balcazar, D.E.; Alberca, L.; Labriola, C.A.; Talevi, A.; Carrillo, C. Application of computer-aided drug repurposing in the search of new cruzipain inhibitors: Discovery of amiodarone and bromocriptine inhibitory effects. J. Chem. Inf. Model 2013, 53, 2402–2408. [Google Scholar] [CrossRef]
- Castilho, V.V.S.; Gonçalves, K.C.S.; Rebello, K.M.; Baptista, L.P.R.; Sangenito, L.S.; Santos, H.L.C.; Branquinha, M.H.; Santos, A.L.S.; Menna-Barreto, R.F.S.; Guimarães, A.C.; et al. Docking simulation between HIV peptidase inhibitors and Trypanosoma cruzi aspartyl peptidase. BMC Res. Notes 2018, 11, 825. [Google Scholar] [CrossRef] [Green Version]
- Rashmi, M.; Swati, D. In silico drug re-purposing against African sleeping sickness using GlcNAc-PI de-N-acetylase as an experimental target. Comput. Biol. Chem. 2015, 59, 87–94. [Google Scholar] [CrossRef]
- Ferreira, D.D.; Mesquita, J.T.; da Costa Silva, T.A.; Romanelli, M.M.; Batista, D.G.J.; da Silva, C.F.; da Gama, A.N.S.; Neves, B.J.; Melo-Filho, C.C.; Soeiro, M.N.C.; et al. Efficacy of sertraline against Trypanosoma cruzi: An in vitro and in silico study. J. Venom. Anim. Toxins. Incl. Trop. Dis. 2018, 24, 30. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Vázquez, K.; Paulino, M.; Salas, C.O.; Zarate-Ramos, J.J.; Vera, B.; Rivera, G. Trypanothione Reductase: A Target for the Development of Anti- Trypanosoma cruzi Drugs. Mini Rev. Med. Chem. 2017, 17, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Inaoka, D.K.; Iida, M.; Hashimoto, S.; Tabuchi, T.; Kuranaga, T.; Balogun, E.O.; Honma, T.; Tanaka, A.; Harada, S.; Nara, T.; et al. Design and synthesis of potent substrate-based inhibitors of the Trypanosoma cruzi dihydroorotate dehydrogenase. Bioorg. Med. Chem. 2017, 25, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Maluf, F.V.; Andricopulo, A.D.; Oliva, G.; Guido, R.V. A pharmacophore-based virtual screening approach for the discovery of Trypanosoma cruzi GAPDH inhibitors. Future Med. Chem. 2013, 5, 2019–2035. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.F.; Prokopczyk, I.M.; Zottis, A.; Oliva, G.; Andricopulo, A.D.; Trevisan, M.T.S.; Vilegas, W.; Silva, M.G.V.; Montanari, C.A. Discovery of novel Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase inhibitors. Bioorg. Med. Chem. 2009, 17, 2476–2482. [Google Scholar] [CrossRef]
- Aguilera, E.; Varela, J.; Birriel, E.; Serna, E.; Torres, S.; Yaluff, G.; de Bilbao, N.V.; Aguirre-López, B.; Cabrera, N.; Mazariegos, S.D.; et al. Potent and Selective Inhibitors of Trypanosoma cruzi Triosephosphate Isomerase with Concomitant Inhibition of Cruzipain: Inhibition of Parasite Growth through Multitarget Activity. ChemMedChem 2016, 11, 1328–1338. [Google Scholar] [CrossRef]
- Cortés-Figueroa, A.A.; Pérez-Torres, A.; Salaiza, N.; Cabrera, N.; Escalona-Montaño, A.; Rondán, A.; Aguirre-García, M.; Gómez-Puyou, A.; Pérez-Montfort, R.; Becker, I. A monoclonal antibody that inhibits Trypanosoma cruzi growth in vitro and its reaction with intracellular triosephosphate isomerase. Parasitol. Res. 2008, 102, 635–643. [Google Scholar] [CrossRef]
- Perez, B.C.; Padilla, A.M.; Xu, D.; Tarleton, R.L.; Basombrio, M.A. Knockout of the dhfr-ts gene in Trypanosoma cruzi generates attenuated parasites able to confer protection against a virulent challenge. PLoS Negl. Trop. Dis. 2011, 5, e1418. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, M.; Sallam, H.A.; Shaban, S.S.; Abdel-Wahab, S.S.; Amr, A.E.E.; Azab, M.E.; Nossier, E.S.; Al-Omar, M.A. Design, Synthesis, and Molecular Docking Study of Novel Heterocycles Incorporating 1,3,4-Thiadiazole Moiety as Potential Antimicrobial and Anticancer Agents. Molecules 2019, 24, 1066. [Google Scholar] [CrossRef] [Green Version]
- Maganti, L.; Manoharan, P.; Ghoshal, N. Probing the structure of Leishmania donovani chagasi DHFR-TS: Comparative protein modeling and protein-ligand interaction studies. J. Mol. Model 2010, 16, 1539–1547. [Google Scholar] [CrossRef]
- Osorio, E.; Aguilera, C.; Naranjo, N.; Marín, M.; Muskus, C. Biochemical characterization of the bifunctional enzyme dihydrofolate reductase-thymidylate synthase from Leishmania (Viannia) and its evaluation as a drug target. Biomedica 2013, 33, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, W.; Wang, Y.; Chang, Z.; Yang, Y.; Pu, J.; Sun, T.; Kaur, S.; Sacchettini, J.C.; Jung, H.; Wong, W.L.; et al. The identification of novel Mycobacterium tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modelling. Sci. Rep. 2015, 5, 15328. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Senkovich, O.; Schormann, N.; Chattopadhyay, D. Structures of dihydrofolate reductase-thymidylate synthase of Trypanosoma cruzi in the folate-free state and in complex with two antifolate drugs, trimetrexate and methotrexate. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 704–716. [Google Scholar] [CrossRef] [PubMed]
- González-Chávez, Z.; Vázquez, C.; Mejia-Tlachi, M.; Márquez-Dueñas, C.; Manning-Cela, R.; Encalada, R.; Rodríguez-Enríquez, S.; Michels, P.A.M.; Moreno-Sánchez, R.; Saavedra, E. Gamma-glutamylcysteine synthetase and tryparedoxin 1 exert high control on the antioxidant system in Trypanosoma cruzi contributing to drug resistance and infectivity. Redox Biol. 2019, 26, 101231. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Velu, S.E.; Murugesan, S.; Senkovich, O.; Walker, K.; Chenna, B.C.; Shinkre, B.; Desai, A.; Chattopadhyay, D. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorg. Med. Chem. 2010, 18, 4056–4066. [Google Scholar] [CrossRef]
- Schormann, N.; Senkovich, O.; Walker, K.; Wright, D.L.; Anderson, A.C.; Rosowsky, A.; Ananthan, S.; Shinkre, B.; Velu, S.; Chattopadhyay, D. Structure-based approach to pharmacophore identification, in silico screening, and three-dimensional quantitative structure-activity relationship studies for inhibitors of Trypanosoma cruzi dihydrofolate reductase function. Proteins 2008, 73, 889–901. [Google Scholar] [CrossRef]
- Sánchez-del-Campo, L.; Sáez-Ayala, M.; Chazarra, S.; Cabezas-Herrera, J.; Rodríguez-López, J.N. Binding of natural and synthetic polyphenols to human dihydrofolate reductase. Int. J. Mol. Sci. 2009, 10, 5398–5410. [Google Scholar] [CrossRef] [Green Version]
- Harel, D.; Schepmann, D.; Prinz, H.; Brun, R.; Schmidt, T.J.; Wünsch, B. Natural product derived antiprotozoal agents: Synthesis, biological evaluation, and structure-activity relationships of novel chromene and chromane derivatives. J. Med. Chem. 2013, 56, 7442–7448. [Google Scholar] [CrossRef]
- Parkkila, S.; Innocenti, A.; Kallio, H.; Hilvo, M.; Scozzafava, A.; Supuran, C.T. The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian alpha-carbonic anhydrase isoforms. Bioorg. Med. Chem. Lett. 2009, 19, 4102–4106. [Google Scholar] [CrossRef]
- Radi, M.; Evensen, L.; Dreassi, E.; Zamperini, C.; Caporicci, M.; Falchi, F.; Musumeci, F.; Schenone, S.; Lorens, J.B.; Botta, M. A combined targeted/phenotypic approach for the identification of new antiangiogenics agents active on a zebrafish model: From in silico screening to cyclodextrin formulation. Bioorg. Med. Chem. Lett. 2012, 22, 5579–5583. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L. Non-kinase targets of protein kinase inhibitors. Nat. Rev. Drug Discov. 2017, 16, 424–440. [Google Scholar] [CrossRef] [PubMed]
- De Rycker, M.; Thomas, J.; Riley, J.; Brough, S.J.; Miles, T.J.; Gray, D.W. Identification of Trypanocidal Activity for Known Clinical Compounds Using a New Trypanosoma cruzi Hit-Discovery Screening Cascade. PLoS Negl. Trop. Dis. 2016, 10, e0004584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões-Silva, M.R.; De Araújo, J.S.; Peres, R.B.; Da Silva, P.B.; Batista, M.M.; De Azevedo, L.D.; Bastos, M.; Bahia, M.T.; Boechat, N.; Soeiro, M.N.C. Repurposing strategies for Chagas disease therapy: The effect of imatinib and derivatives against Trypanosoma cruzi. Parasitology 2019, 146, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Narayanana, A.; Jones, L.H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659. [Google Scholar] [CrossRef] [Green Version]
- Hussein, E.M.; Al-Rooqi, M.M.; El-Galil, S.M.A.; Ahmed, S.A. Design, synthesis, and biological evaluation of novel N4-substituted sulfonamides: Acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem. 2019, 13, 91. [Google Scholar] [CrossRef]
- Rub, A.; Shaker, K.; Kashif, M.; Arish, M.; Dukhyil, A.A.B.; Alshehri, B.M.; Alaidarous, M.A.; Banawas, S.; Amir, K. Repurposing Glyburide as Antileishmanial Agent to Fight Against Leishmaniasis. Protein Pept. Lett. 2019, 26, 371–376. [Google Scholar] [CrossRef]
- Palos, I.; Lara-Ramirez, E.E.; Lopez-Cedillo, J.C.; Garcia-Perez, C.; Kashif, M.; Bocanegra-Garcia, V.; Nogueda-Torres, B.; Rivera, G. Repositioning FDA Drugs as Potential Cruzain Inhibitors from Trypanosoma cruzi: Virtual Screening, In Vitro and In Vivo Studies. Molecules 2017, 22, 1015. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Martinez, A.; Hernández-Rodríguez, V.; Rodríguez-Durán, J.; Hejchman, E.; Benaim, G. Anti-Trypanosoma cruzi action of a new benzofuran derivative based on amiodarone structure. Exp. Parasitol. 2018, 189, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Properties | Interacting Residues Properties | Interaction Pattern | |||
|---|---|---|---|---|---|
| Hydrogen bond acceptor | 8 | Acidic | 1 | Hydrogen bonds | 4 |
| Hydrogen bond donor | 5 | Acyclic | 4 | Hydrophobic interactions | 2 |
| Rings | 4 | Aliphatic | 2 | Salt bridges | 1 |
| Rotatable bonds | 6 | Aromatic | 1 | hb 1: TYR-160 | 1 |
| Buried | 2 | hb: THR-178 | 2 | ||
| Charged | 1 | hb: VAL-26 | 1 | ||
| Cyclic | 1 | hi 1: ILE-41 | 2 | ||
| Hydrophobic | 2 | sb 1: ASP-48 | 1 | ||
| Large | 2 | ||||
| Medium | 3 | ||||
| Negative | 1 | ||||
| Neutral | 4 | ||||
| Polar | 3 | ||||
| Surface | 4 | ||||
| Conformation | RMSDÅ | Vina Score Kcal/mol | hb 1: TYR-160 | hb: THR-178 | hb: VAL-26 | hi 1: ILE-41 | sb 1: ASP-48 |
|---|---|---|---|---|---|---|---|
| Crystal | - | - | 1 | 2 | 1 | 2 | 1 |
| 1 | 3.044 | −8.5 | 1 | 1 | - | - | 1 |
| 2 | 10.810 | −8.4 | 2 | 1 | - | 1 | 1 |
| 3 | 3.768 | −8.4 | 1 | 1 | 1 | - | 1 |
| 4 | 10.762 | −8.4 | 2 | 1 | - | 1 | 1 |
| 5 | 3.605 | −8.1 | 1 | - | - | - | 1 |
| 6 | 2.451 | −8.0 | 1 | 1 | 1 | 1 | 1 |
| 7 | 8.232 | −7.9 | - | - | - | - | - |
| 8 | 7.916 | −7.9 | - | - | - | - | - |
| 9 | 7.592 | −7.8 | - | - | - | - | - |
| Name | Structure | Vina Score Kcal/mol | Description |
|---|---|---|---|
| Trimetrexate |  | −8.5 | DHFR-TS inhibitor |
| Nebivolol |  | −10.2 | Treatment of hypertension |
| Nilotinib |  | −10.1 | Tyrosine kinase inhibitor |
| Glipizide |  | −9.8 | Anti-diabetes drug |
| Glyburide |  | −9.7 | Anti-diabetes drug |
| Gliquidone |  | −9.5 | Anti-diabetes drug |
| Imatinib |  | −9.5 | Tyrosine kinase inhibitor |
| Dihydro-alpha-ergocryptine |  | −9.5 | Early treatment of Parkinson’s disease |
| Dihydroergocornine |  | −9.4 | Early treatment of Parkinson’s disease |
| Darifenacin |  | −9.4 | Treatment of urinary incontinence |
| Eltrombopag |  | −9.3 | Treatment of chronic immune thrombocytopenia |
| Compound | IC50 μM T. cruzi | IC50 μM HFF1 |
|---|---|---|
| NIL | 6 ± 2 | 12 ± 6 |
| GPZ | 13.4 ± 6 | 38 ± 11 |
| GLQ | 12 ± 5 | 68 ± 14 |
| GBD | 66 ± 12 | >50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juárez-Saldivar, A.; Schroeder, M.; Salentin, S.; Haupt, V.J.; Saavedra, E.; Vázquez, C.; Reyes-Espinosa, F.; Herrera-Mayorga, V.; Villalobos-Rocha, J.C.; García-Pérez, C.A.; et al. Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling. Int. J. Mol. Sci. 2020, 21, 4270. https://doi.org/10.3390/ijms21124270
Juárez-Saldivar A, Schroeder M, Salentin S, Haupt VJ, Saavedra E, Vázquez C, Reyes-Espinosa F, Herrera-Mayorga V, Villalobos-Rocha JC, García-Pérez CA, et al. Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling. International Journal of Molecular Sciences. 2020; 21(12):4270. https://doi.org/10.3390/ijms21124270
Chicago/Turabian StyleJuárez-Saldivar, Alfredo, Michael Schroeder, Sebastian Salentin, V. Joachim Haupt, Emma Saavedra, Citlali Vázquez, Francisco Reyes-Espinosa, Verónica Herrera-Mayorga, Juan Carlos Villalobos-Rocha, Carlos A. García-Pérez, and et al. 2020. "Computational Drug Repositioning for Chagas Disease Using Protein-Ligand Interaction Profiling" International Journal of Molecular Sciences 21, no. 12: 4270. https://doi.org/10.3390/ijms21124270