Epidermal Growth Factor (EGF) Augments the Invasive Potential of Human Glioblastoma Multiforme Cells via the Activation of Collaborative EGFR/ROS-Dependent Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
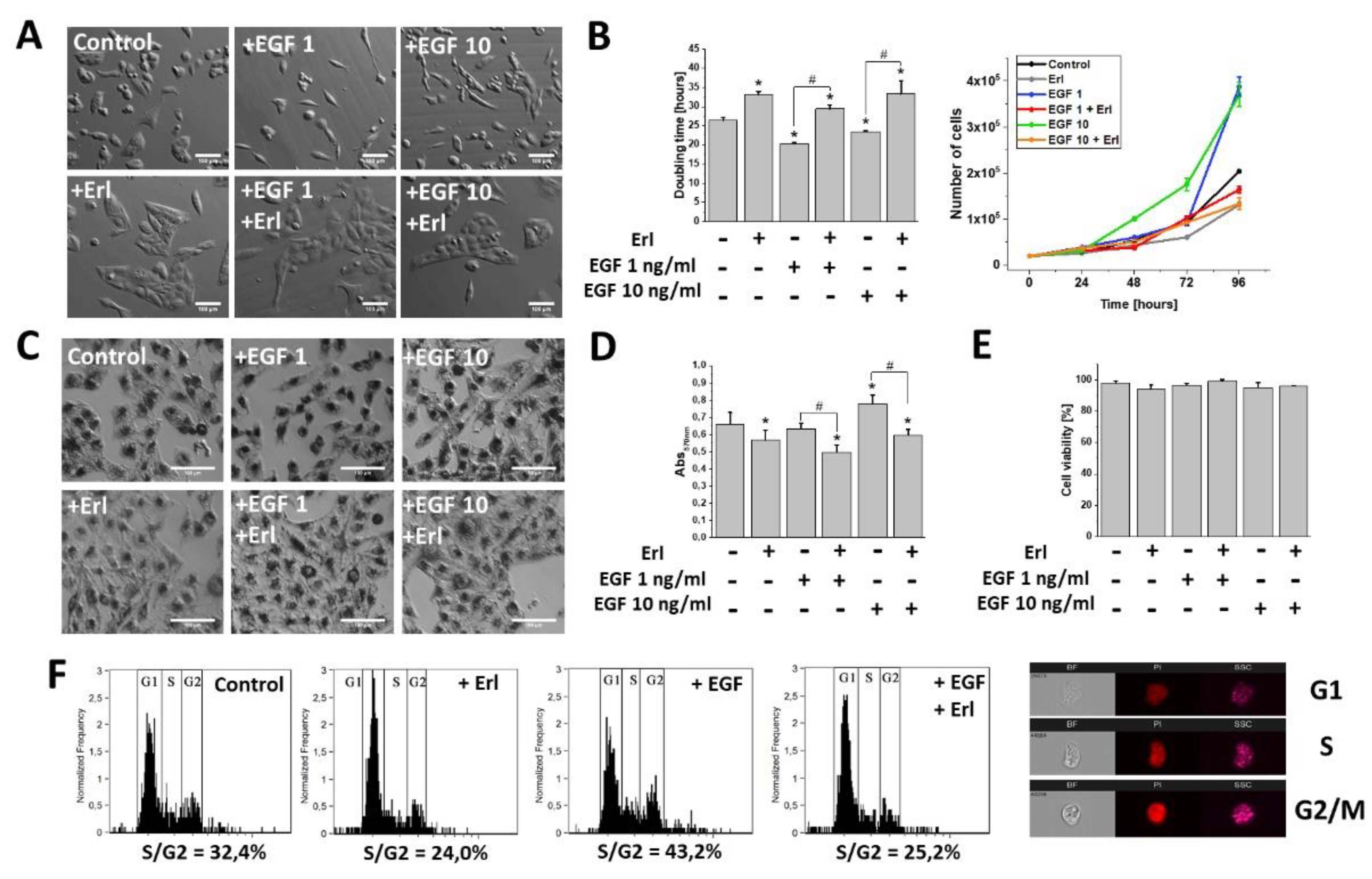
2.1. EGF Induced Proliferation, Metabolic Activity, and Morphological Changes in GBM Cells
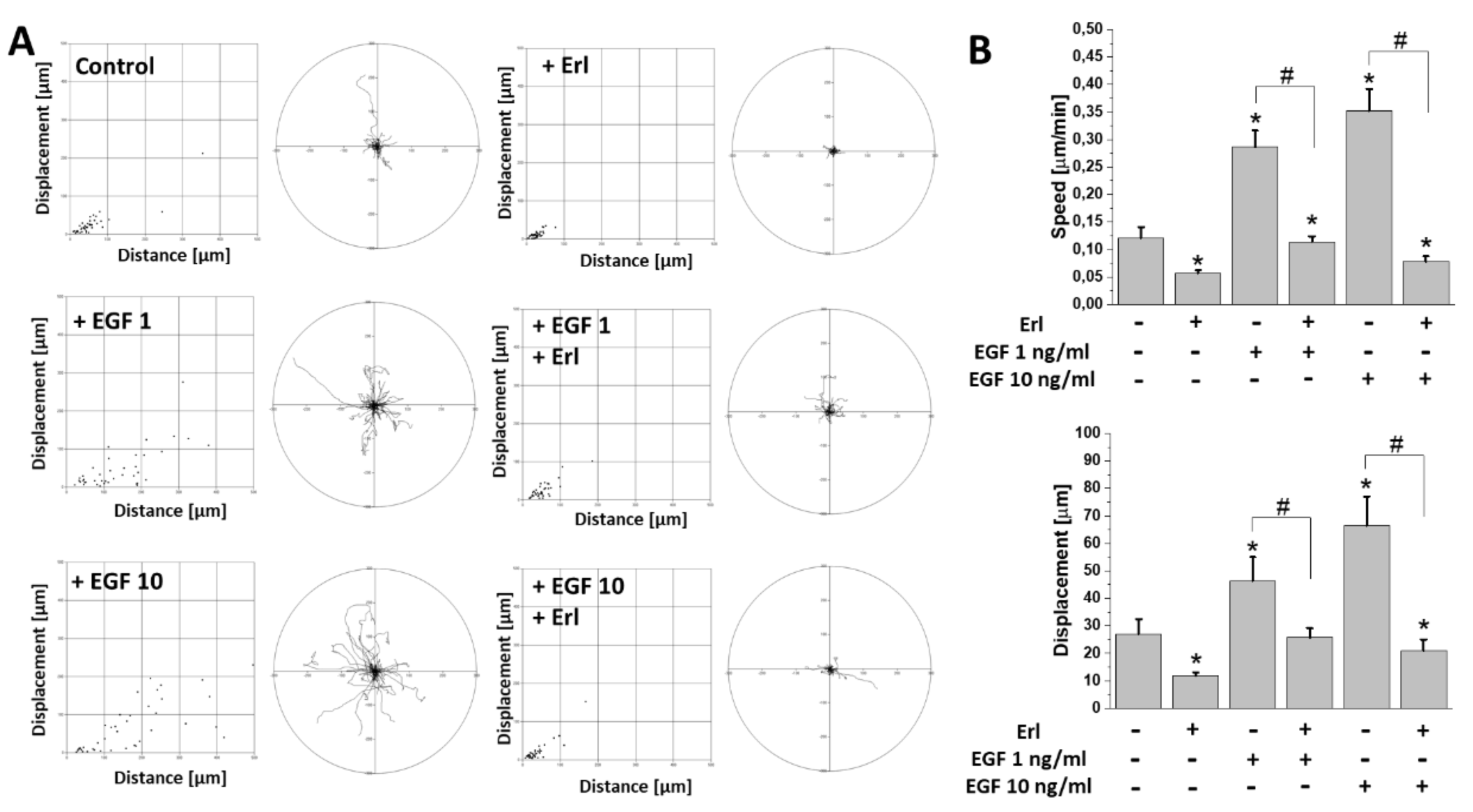
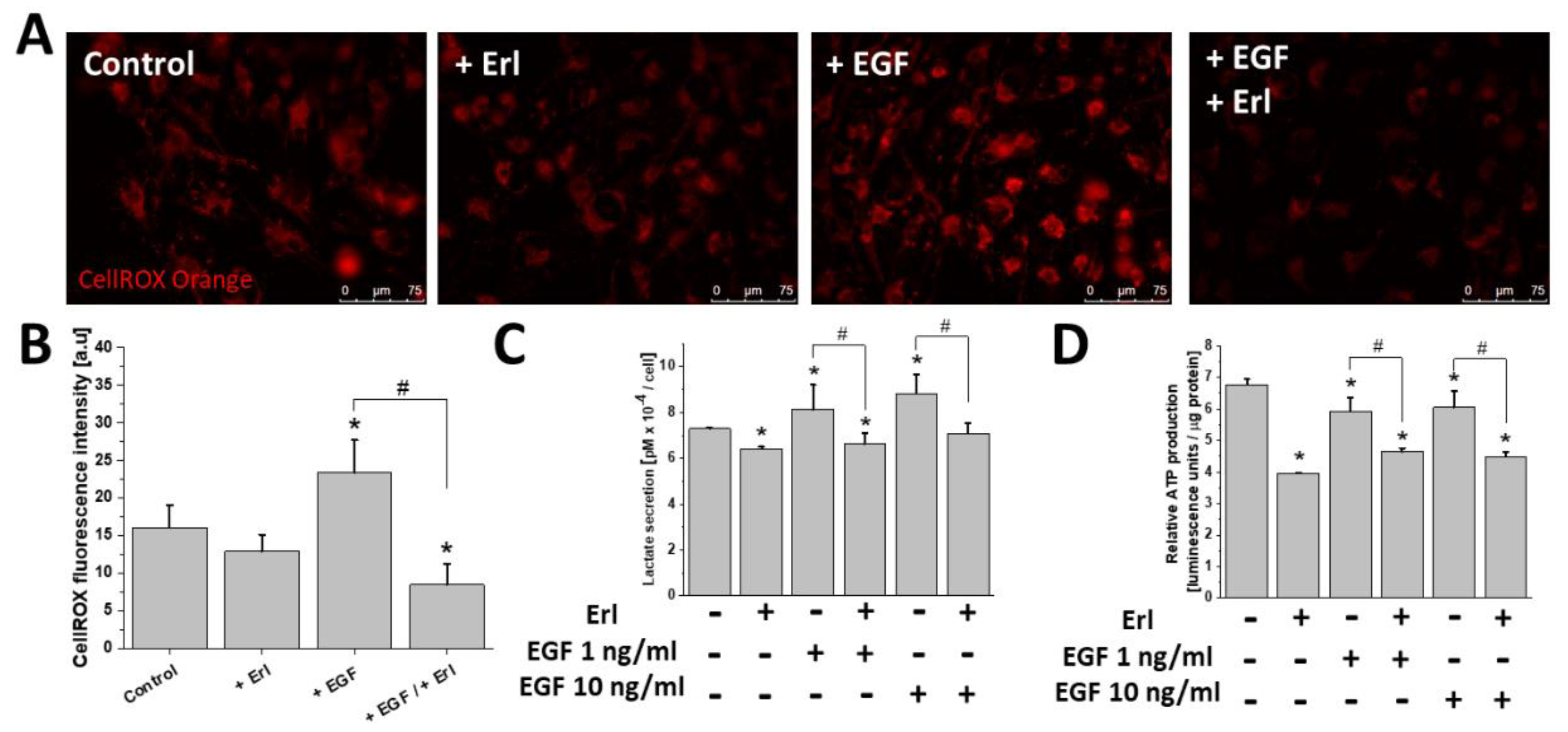
2.2. EGF Augmented T98G Cell Motility and Intracellular ATP/Lactate Production
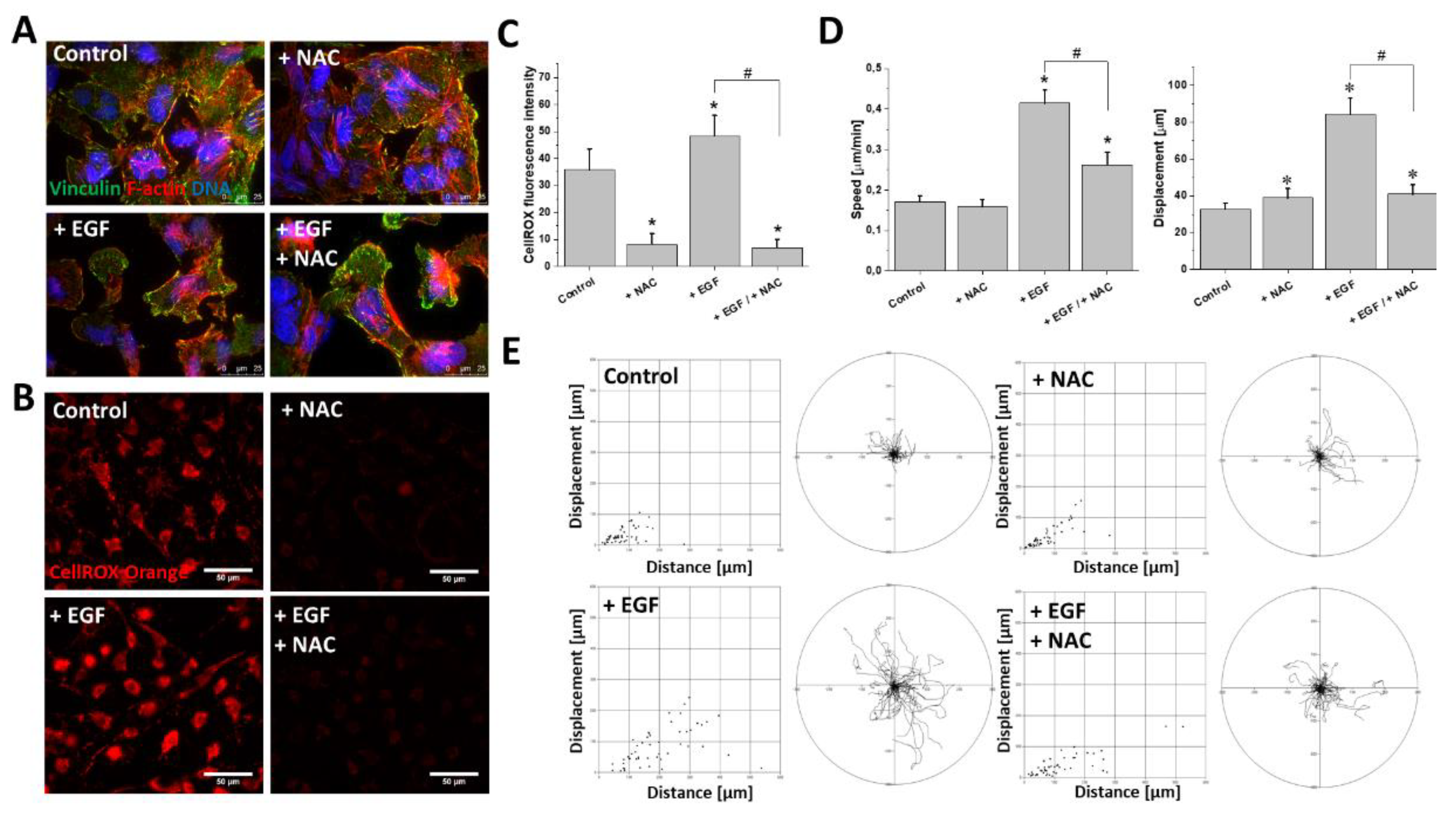
2.3. EGF Induced Cytoskeletal Rearrangements and Invasiveness of T98G Cells
2.4. ROS Upregulation Was Responsible for EGF-Induced T98G Motility
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Proliferation, Viability, and MTT Assays
4.3. Cell Migration and Transmigration
4.4. Immunocytochemistry
4.5. Immunoblotting
4.6. Mitochondrial ROS Production Detection
4.7. Image Cytometry and Cell Cycle Analysis
4.8. ATP and Lactate Production Measurements
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| Erl | Erlotinib |
| GBM | Glioblastoma multiforme |
| HRP | Horseradish Peroxidase |
| NAC | N-acetyl-L-cysteineReactive Oxygen Species |
| ROS | Reactive Oxygen Species |
| mtROS | Mitochondrial Reactive Oxygen Species |
| TIRF | Total Internal Reflection Fluorescence |
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Combs, S.E.; Edler, L.; Rausch, R.; Welzel, T.; Wick, W.; Debus, J. Generation and validation of a prognostic score to predict outcome after re-irradiation of recurrent glioma. Acta Oncol. 2013, 52, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Linde, M.E.; Brahm, C.G.; de Witt Hamer, P.C.; Reijneveld, J.C.; Bruynzeel, A.; Vandertop, W.P.; van de Ven, P.M.; Wagemakers, M.; van der Weide, H.L.; Enting, R.H.; et al. Treatment outcome of patients with recurrent glioblastoma multiforme: A retrospective multicenter analysis. J. Neurooncol. 2017, 135, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, M.; Liermann, J.; Verma, V.; Bernhardt, D.; Bougatf, N.; Paul, A.; Rieken, S.; Debus, J.; Adeberg, S. Survival and recurrence patterns of multifocal glioblastoma after radiation therapy. Cancer Manag. Res. 2018, 10, 4229–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arevalo, O.D.; Soto, C.; Rabiei, P.; Kamali, A.; Ballester, L.Y.; Esquenazi, Y.; Zhu, J.J.; Riascos, R.F. Assessment of Glioblastoma Response in the Era of Bevacizumab: Longstanding and Emergent Challenges in the Imaging Evaluation of Pseudoresponse. Front. Neurol. 2019, 10, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, L.; Coll, C.; Erthal, L.C.S.; de la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef]
- Mazaris, P.; Hong, X.; Altshuler, D.; Schultz, L.; Poisson, L.M.; Jain, R.; Mikkelsen, T.; Rosenblum, M.; Kalkanis, S. Key determinants of short-term and long-term glioblastoma survival: A 14-year retrospective study of patients from the Hermelin Brain Tumor Center at Henry Ford Hospital. Clin. Neurol. Neurosurg. 2014, 120, 103–112. [Google Scholar] [CrossRef]
- Lindström, M.S. Expanding the scope of candidate prognostic marker IGFBP2 in glioblastoma. Biosci. Rep. 2019, 39, BSR20190770. [Google Scholar] [CrossRef] [Green Version]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Ellert-Miklaszewska, A.; Pasierbinska, M.; Poleszak, K.; Kamińska, B. Molecular interactions between tumor and its microenvironment in malignant gliomas. Postepy Biochem. 2018, 15, 64. [Google Scholar]
- Dello Russo, C.; Lisi, L.; Tentori, L.; Navarra, P.; Graziani, G.; Combs, C.K. Exploiting Microglial Functions for the Treatment of Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Chen, X.C.; Wei, X.T.; Guan, J.H.; Shu, H.; Chen, D. EGF stimulates glioblastoma metastasis by induction of matrix metalloproteinase-9 in an EGFR-dependent mechanism. Oncotarget 2017, 8, 65969–65982. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Chang, J.; Hung, W.; Yang, Y.C.; Chien, M.H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Nakada, M.; Ueda, F.; Watanabe, T.; Arakawa, Y.; Higashi, R.; Hashimoto, M.; Nitta, H.; Hayashi, Y.; Hamada, J. Prognostic paradox: Brain damage around the glioblastoma resection cavity. J. Neurooncol. 2014, 118, 187–192. [Google Scholar] [CrossRef]
- Coskun, S.; Coskun, A.; Gursan, N.; Aydin, M.D. Post-traumatic glioblastoma multiforme: A case report. Eurasian J. Med. 2011, 43, 50–53. [Google Scholar] [CrossRef]
- Tyagi, V.; Theobald, J.; Barger, J.; Bustoros, M.; Bayin, N.S.; Modrek, A.S.; Kader, M.; Anderer, E.G.; Donahue, B.; Fatterpekar, G.; et al. Traumatic brain injury and subsequent glioblastoma development: Review of the literature and case reports. Surg. Neurol. Int. 2016, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Nolte, C.; Kirchhoff, F.; Kettenmann, H. Epidermal growth factor is a motility factor for microglial cells in vitro: Evidence for EGF receptor expression. Eur. J. Neurosci. 1997, 9, 1690–1698. [Google Scholar] [CrossRef]
- Estrada, C.; Villalobo, A. Epidermal Growth Factor Receptor in the Adult Brain. In The Cell Cycle in the Central Nervous System. Contemporary Neuroscience; Janigro, D., Ed.; Humana Press: Totowa, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Sun, D.; Bullock, M.R.; Altememi, N.; Zhou, Z.; Hagood, S.; Rolfe, A.; McGinn, M.J.; Hamm, R.; Colello, R.J. The effect of epidermal growth factor in the injured brain after trauma in rats. J. Neurotrauma 2010, 27, 923–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT Regulation by Autophagy: A New Perspective in Glioblastoma Biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, A.; Noda, K.; Hirose, I.; Ishida, S. TGF-β-SNAIL axis induces Müller glial-mesenchymal transition in the pathogenesis of idiopathic epiretinal membrane. Sci. Rep. 2019, 9, 673. [Google Scholar] [CrossRef]
- Dai, P.; Nakagami, T.; Tanaka, H.; Hitomi, T.; Takamatsu, T. Cx43 mediates TGF-beta signaling through competitive Smads binding to microtubules. Mol. Biol. Cell 2007, 18, 2264–2273. [Google Scholar] [CrossRef] [Green Version]
- Ryszawy, D.; Sarna, M.; Rak, M.; Szpak, K.; Kędracka-Krok, S.; Michalik, M.; Siedlar, M.; Zuba-Surma, E.; Burda, K.; Korohoda, W.; et al. Functional links between Snail-1 and Cx43 account for the recruitment of Cx43-positive cells into the invasive front of prostate cancer. Carcinogenesis 2014, 35, 1920–1930. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.R.; DeGennaro, M.; Lehmann, R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012, 22, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthuramalingam, K.; Cho, M.; Kim, Y. Role of NAPDH oxidase and its therapeutic intervention in TGF-β-mediated EMT progression: An in vitro analysis on HeLa cervical cancer cells. Appl. Biol. Chem. 2020, 63, 4. [Google Scholar] [CrossRef] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 10, 37349–37366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR)signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addington, C.P.; Roussas, A.; Dutta, D.; Stabenfeldt, S.E. Endogenous repair signaling after brain injury and complementary bioengineering approaches to enhance neural regeneration. Biomark. Insights 2015, 10, 43–60. [Google Scholar] [CrossRef]
- Clatz, O.; Sermesant, M.; Bondiau, P.Y.; Delingette, H.; Warfield, S.K.; Malandain, G.; Ayache, N. Realistic simulation of the 3-D growth of brain tumors in MR images coupling diffusion with biomechanical deformation. IEEE Trans. Med. Imaging 2005, 24, 1334–1346. [Google Scholar] [CrossRef] [Green Version]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ryszawy, D.; Pudełek, M.; Catapano, J.; Ciarach, M.; Setkowicz, Z.; Konduracka, E.; Madeja, Z.; Czyż, J. High doses of sodium ascorbate interfere with the expansion of glioblastoma multiforme cells in vitro and in vivo. Life Sci. 2019, 232, 116657. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudełek, M.; Catapano, J.; Kochanowski, P.; Mrowiec, K.; Janik-Olchawa, N.; Czyż, J.; Ryszawy, D. Therapeutic potential of monoterpene α-thujone, the main compound of Thuja occidentalis L. essential oil, against malignant glioblastoma multiforme cells in vitro. Fitoterapia 2019, 134, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Ryszawy, D.; Pudełek, M.; Kochanowski, P.; Janik-Olchawa, N.; Bogusz, J.; Rąpała, M.; Koczurkiewicz, P.; Mikołajczyk, J.; Borek, I.; Kędracka-Krok, S.; et al. High bisphenol A concentrations augment the invasiveness of tumor cells through Snail-1/Cx43/ERRγ-dependent epithelial-mesenchymal transition. Toxicol. In Vitro 2020, 62, 104676. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pudełek, M.; Król, K.; Catapano, J.; Wróbel, T.; Czyż, J.; Ryszawy, D. Epidermal Growth Factor (EGF) Augments the Invasive Potential of Human Glioblastoma Multiforme Cells via the Activation of Collaborative EGFR/ROS-Dependent Signaling. Int. J. Mol. Sci. 2020, 21, 3605. https://doi.org/10.3390/ijms21103605
Pudełek M, Król K, Catapano J, Wróbel T, Czyż J, Ryszawy D. Epidermal Growth Factor (EGF) Augments the Invasive Potential of Human Glioblastoma Multiforme Cells via the Activation of Collaborative EGFR/ROS-Dependent Signaling. International Journal of Molecular Sciences. 2020; 21(10):3605. https://doi.org/10.3390/ijms21103605
Chicago/Turabian StylePudełek, Maciej, Kamila Król, Jessica Catapano, Tomasz Wróbel, Jarosław Czyż, and Damian Ryszawy. 2020. "Epidermal Growth Factor (EGF) Augments the Invasive Potential of Human Glioblastoma Multiforme Cells via the Activation of Collaborative EGFR/ROS-Dependent Signaling" International Journal of Molecular Sciences 21, no. 10: 3605. https://doi.org/10.3390/ijms21103605