Pharmacological Inhibition of Cyclin-Dependent Kinases Triggers Anti-Fibrotic Effects in Hepatic Stellate Cells In Vitro

 , and
, and
Abstract
:1. Introduction
2. Results
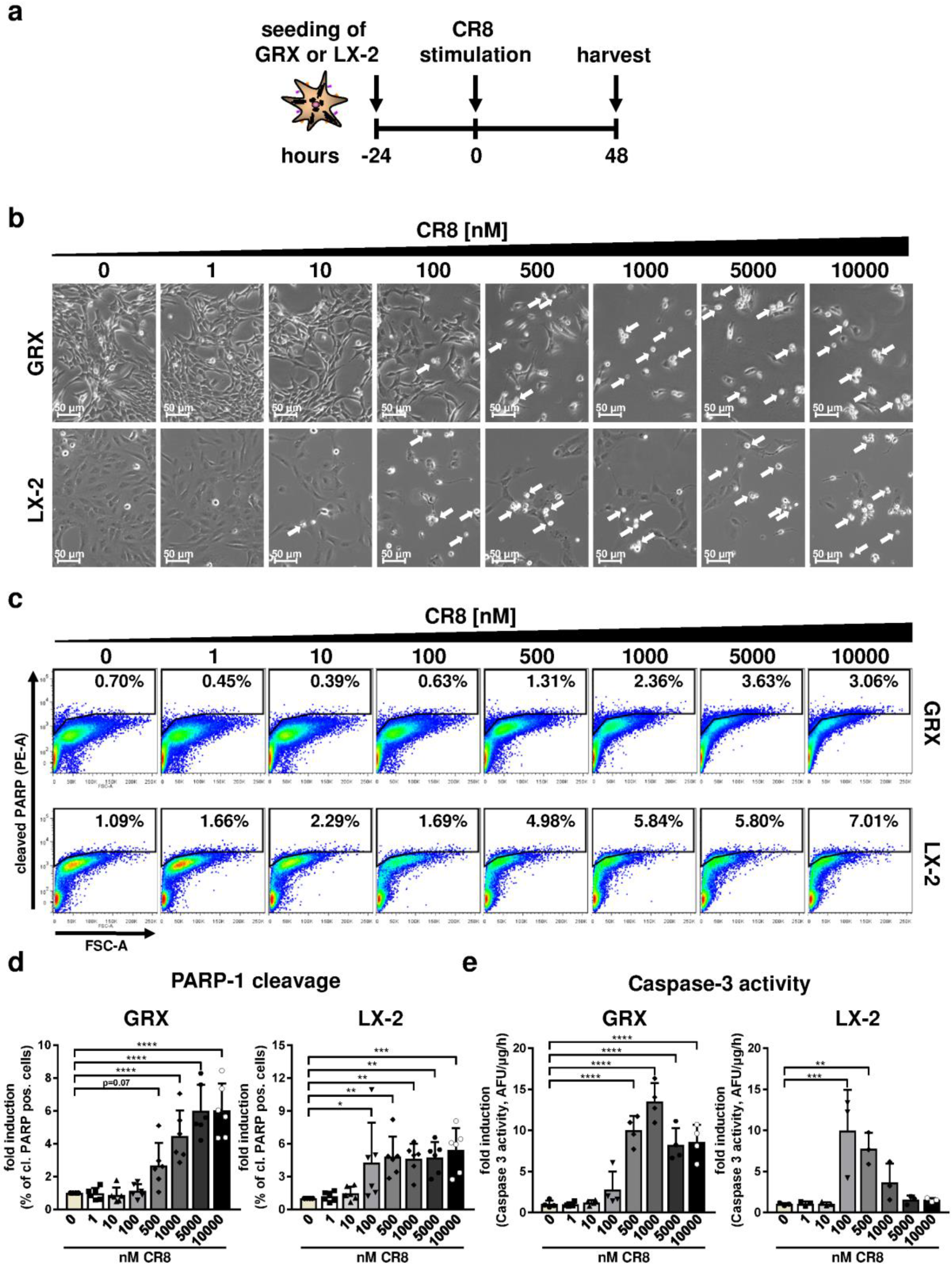
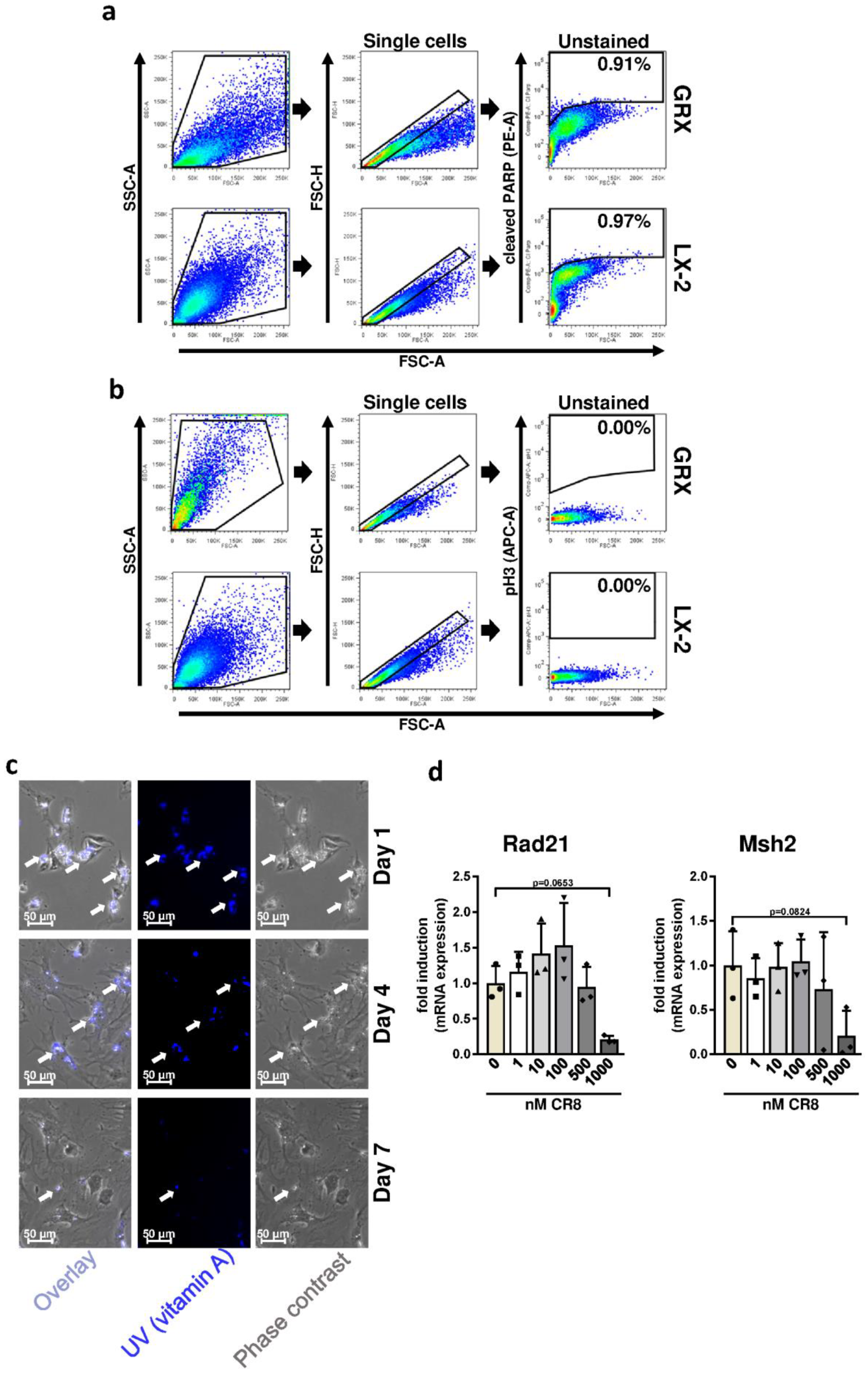
2.1. CR8 Induces Apoptosis in Immortalized Murine and Human HSC Lines
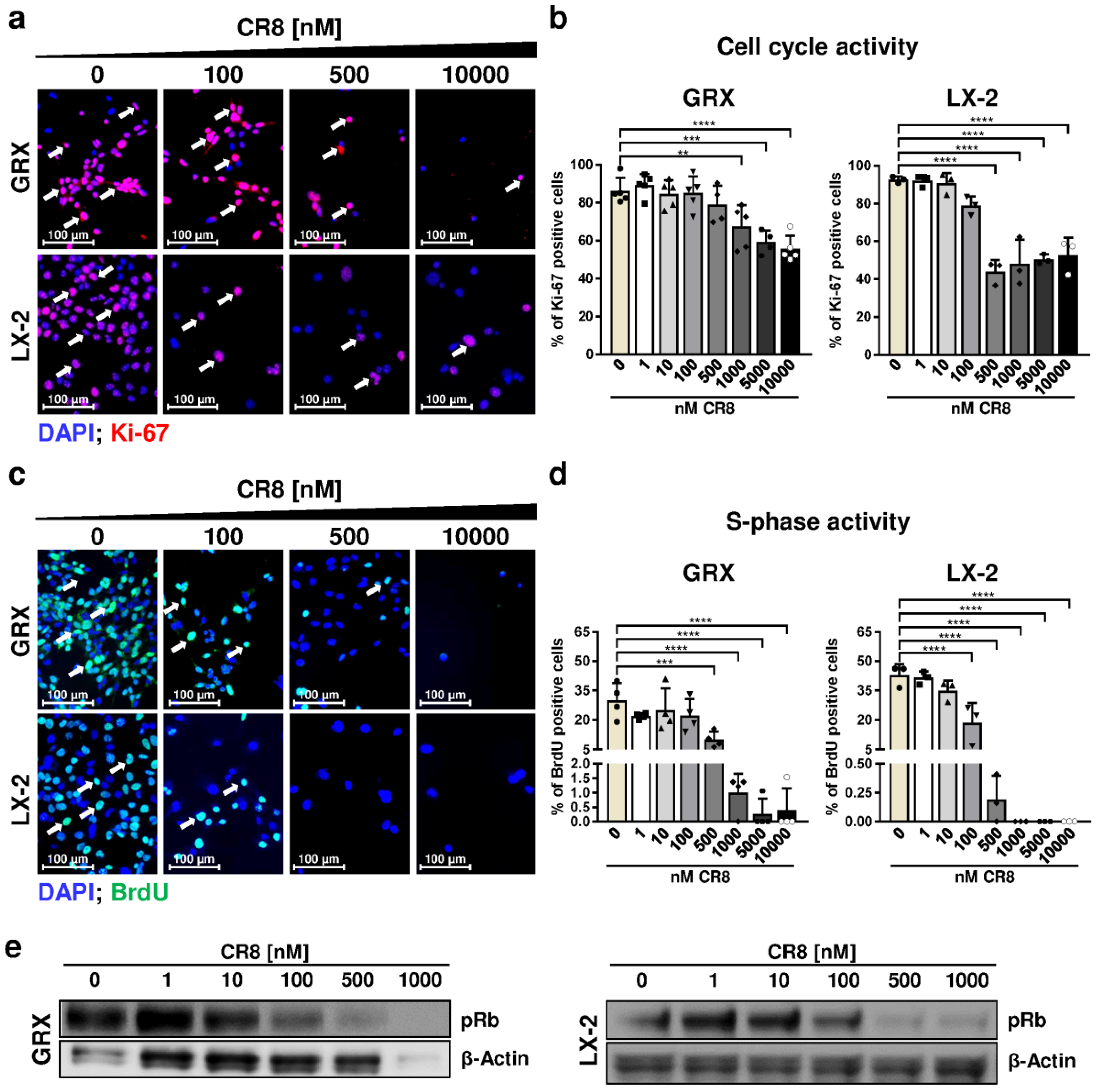
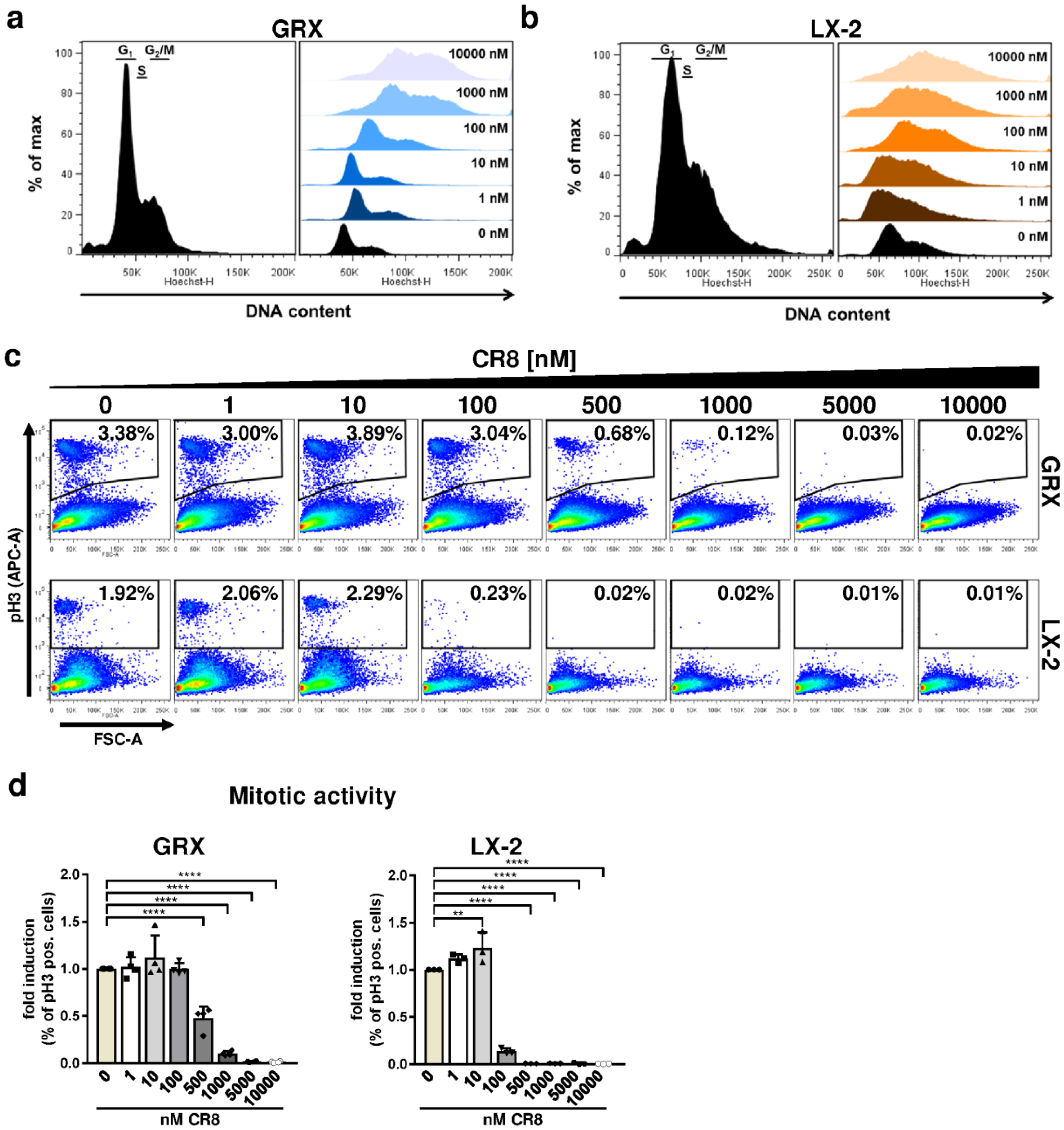
2.2. Pharmacological Inhibition of Cdks Limits Cell Cycle Activity and Triggers G2 Arrest in Murine and Human HSC Cell Lines
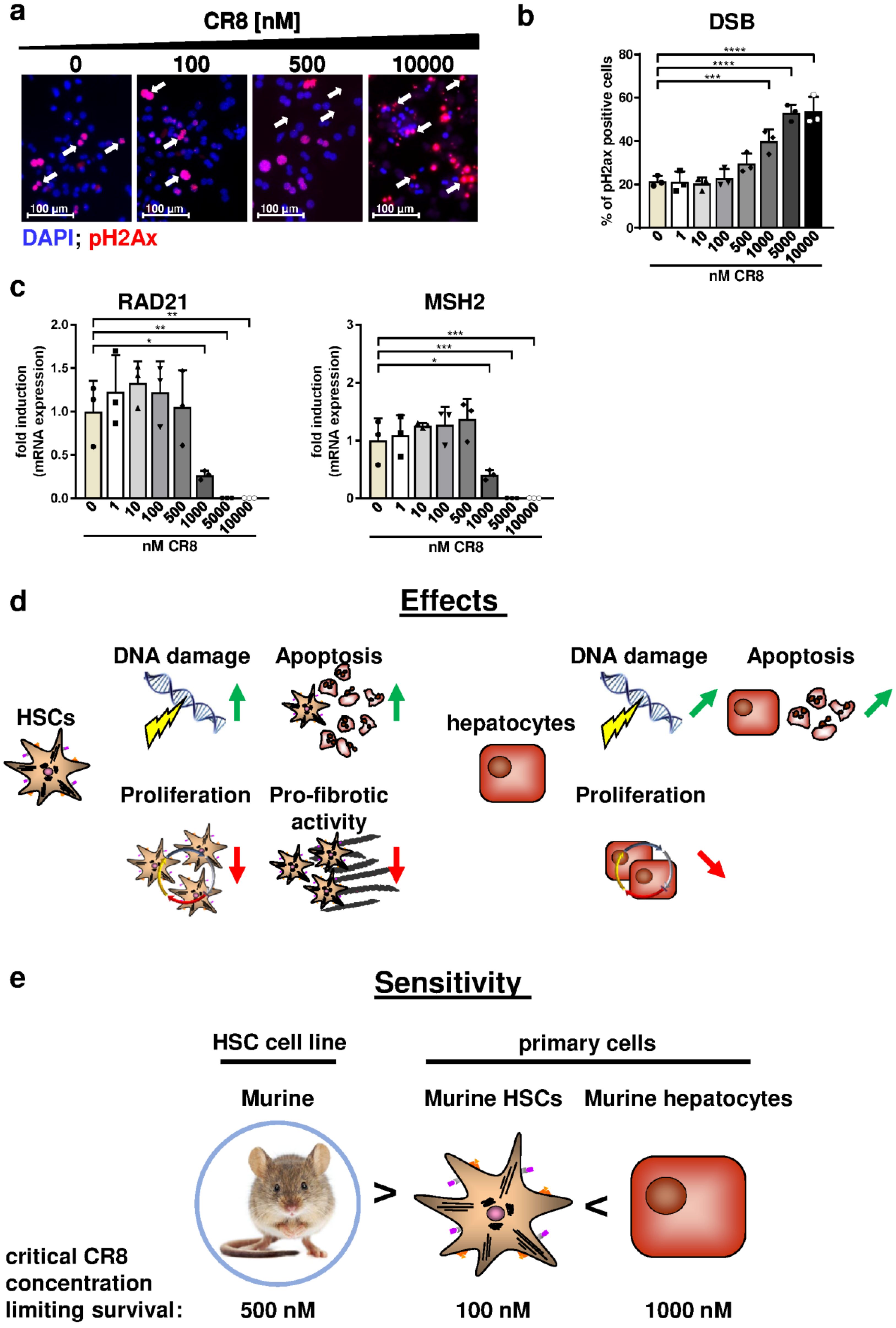
2.3. CR8 Dose-Dependently Induces DNA Double-Strand Breaks in Murine and Human HSC Lines
2.4. CR8-Treatment Reduces the Pro-Fibrotic Properties of HSC Lines In Vitro
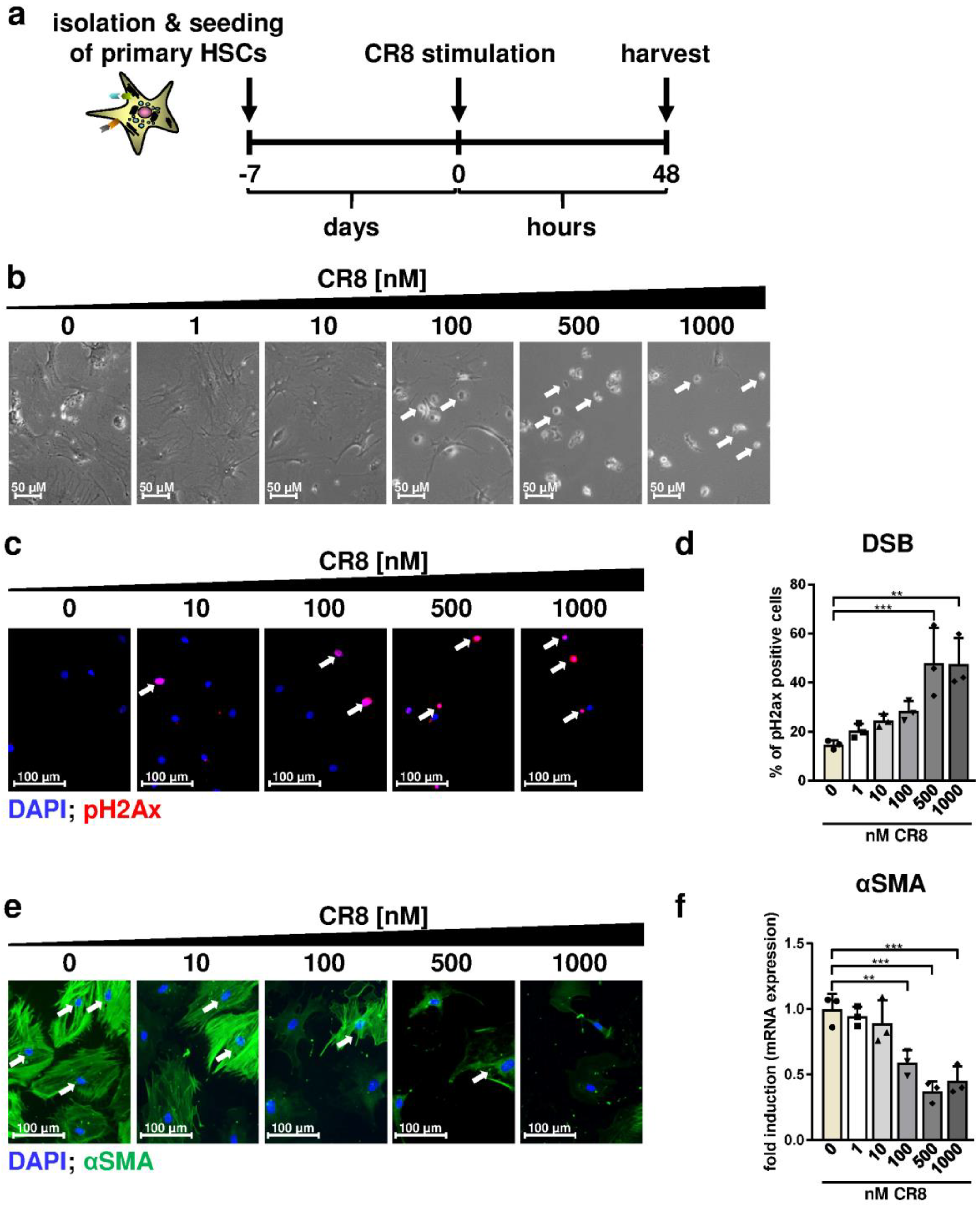
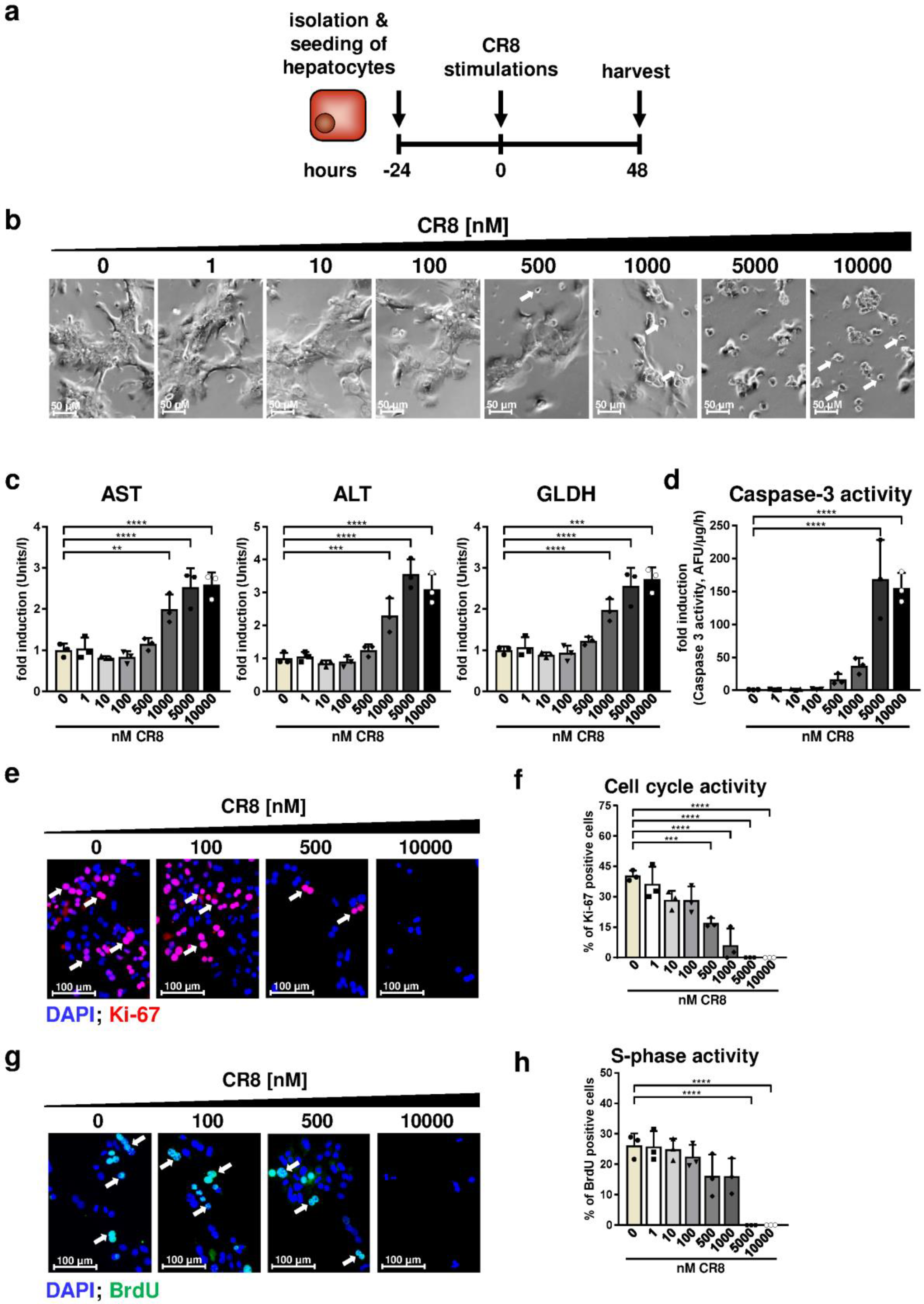
2.5. CR8-Mediates Anti-Fibrotic Effects and Triggers DNA Damage in Primary Murine HSCs
2.6. Primary Hepatocytes Are Less Sensitive to CR8 Compared to HSCs with Regard to Cell Survival and DNA Damage Induction
3. Discussion
4. Materials and Methods
4.1. Cell Culture Procedures
4.2. Measurement of Aminotransferase and Glutamate Dehydrogenase Activity
4.3. Quantitative Real-Time PCR (qPCR)
4.4. BrdU Incorporation Assay
4.5. Histology and Immunoblot Analysis
4.6. Determination of Caspase-3 Activity
4.7. Fluorescence Activated Cell Sorting (FACS)
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| αSMA | Alpha-smooth-muscle actin |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| ATP | Adenosine triphosphate |
| BrdU | 5-bromo-2′-deoxyuridine |
| CCl4 | Carbon tetrachloride |
| Ccn | Cyclin |
| CcnE1 | Cyclin E1 |
| CcnE2 | Cyclin E2 |
| Cdk | Cyclin-dependent kinase |
| Col1A1 | Collagen type 1 alpha chain 1 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMSO | Dimethyl sulfoxide |
| DSBs | Double-strand breaks |
| ECM | Extracellular matrix |
| FACS | Fluorescence-associated cell sorting, flow cytometry |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GLDH | Glutamate dehydrogenase |
| HSCs | Hepatic Stellate Cells |
| h | Hours |
| i.e., | id est |
| MSH2 | MutS homolog 2 |
| nM | nanomol |
| PARP-1 | Poly (ADP-ribose)-polymerase 1 |
| pH3 | phospho-histone H3 |
| pH2Ax | phosphorylated histone H2Ax |
| Rb | Retinoblastoma protein |
| siRNA | Small interfering Ribonucleic acid |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Origin | Orientation | Sequence in 5′-3′Orientation |
|---|---|---|---|
| αSMA | murine | sense | TGACAGAGCCACCACTGAACC |
| antisense | TCCAGAGTCCAGCACAATACCAGT | ||
| human | sense | CAGCCAAGCACTGTC | |
| antisense | CCCACCATCACCCCCTGA | ||
| Col1A1 | murine | sense | TCTGACTGGAAGAGCGGAGAG |
| antisense | GGCACAGACGGCTGAGTAGG | ||
| human | sense | GGAATGAAGGGACACAGAGGTT | |
| antisense | AGTAGCACCATCATTTCCACGA | ||
| GAPDH | murine | sense | TGTTGAAGTCACAGGAGACAACCT |
| antisense | AACCTGCCAAGTATGATGACATCA | ||
| human | sense | TGTTGAAGTCAGAGGAGACCACCT | |
| antisense | AACCTGCCAAATATGATGACATCA | ||
| MSH2 | murine | sense | TCCATCCTCAGGTCAGCAAC |
| antisense | TGGGTGGCAAACATGCAAAA | ||
| RAD21 | murine | sense | GCCGAGATCCAGGTTTCTTC |
| antisense | ACATGGGCTTTGGTTAGCTTC |
| Product | Company |
|---|---|
| Primary antibodies (non-conjugated) | |
| αSMA [1A4], A2547 | Sigma-Aldrich |
| ß-Actin, A2066 | Sigma-Aldrich |
| BrdU [IIB5], sc-32323 | Santa Cruz Biotechnology |
| Ki-67 [SP6], ab16667 | Abcam |
| Phospho-Histone H2A.X, Ser139 (20E3), #9718 | Cell Signaling Technology |
| Phospho-Rb, Ser807/811, #9308 | Cell Signaling Technology |
| Primary antibodies (fluorescence-labeled) | |
| PE Mouse Anti-Cleaved PARP [F21-852], Asp214 | BD Bioscience |
| Phospho-Histone H3, Ser10 (D2C8) XP, Alexa Fluor 647 Conjugate, #3458 | Cell Signaling Technology |
| Secondary antibodies (HRP-conjugated) | |
| mouse anti-rabbit IgG-HRP, sc-2357 | Santa Cruz Biotechnology |
| m-IgGκ BP-HRP, sc-516102 | Santa Cruz Biotechnology |
| Secondary antibodies (fluorescence-labeled) | |
| Alexa Fluor 488 goat anti-mouse IgG, A-11029 | Invitrogen |
| Alexa Fluor 488 goat anti-rabbit IgG, A-11008 | Invitrogen |
| Alexa Fluor 594 donkey anti-rabbit IgG, A-21207 | Invitrogen |
References
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology 2017, 65, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Bangen, J.M.; Hammerich, L.; Sonntag, R.; Baues, M.; Haas, U.; Lambertz, D.; Longerich, T.; Lammers, T.; Tacke, F.; Trautwein, C.; et al. Targeting CCl4 -induced liver fibrosis by RNA interference-mediated inhibition of cyclin E1 in mice. Hepatology 2017, 66, 1242–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Weiskirchen, R. Update on hepatic stellate cells: Pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev. Gastroenterol. Hepatol. 2012, 6, 67–80. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [Green Version]
- Nevzorova, Y.A.; Bangen, J.M.; Hu, W.; Haas, U.; Weiskirchen, R.; Gassler, N.; Huss, S.; Tacke, F.; Sicinski, P.; Trautwein, C.; et al. Cyclin E1 controls proliferation of hepatic stellate cells and is essential for liver fibrogenesis in mice. Hepatology 2012, 56, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Schulze-Gahmen, U.; Brandsen, J.; de Azevedo Junior, W.F. Structural basis for chemical inhibition of CDK2. Prog. Cell Cycle Res. 1996, 2, 137–145. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.M.; Gianella-Borradori, A. Recent progress in the discovery and development of cyclin-dependent kinase inhibitors. Expert Opin. Investig. Drugs 2005, 14, 457–477. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.; Limzerwala, J.F.; Sturmlechner, I.; Hurley, E.; Zhang, C.; Jeganathan, K.B.; Nelson, G.; Bronk, S.; Fierro Velasco, R.O.; van Deursen, E.J.; et al. Ccne1 Overexpression Causes Chromosome Instability in Liver Cells and Liver Tumor Development in Mice. Gastroenterology 2019, 157, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Azevedo, W.F.; Leclerc, S.; Meijer, L.; Havlicek, L.; Strnad, M.; Kim, S.H. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 1997, 243, 518–526. [Google Scholar] [CrossRef]
- Delehouze, C.; Godl, K.; Loaec, N.; Bruyere, C.; Desban, N.; Oumata, N.; Galons, H.; Roumeliotis, T.I.; Giannopoulou, E.G.; Grenet, J.; et al. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 2014, 33, 5675–5687. [Google Scholar] [CrossRef] [Green Version]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plenat, F. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved PARP in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. J. Histochem. Cytochem. 2009, 57, 289–300. [Google Scholar] [CrossRef]
- Liedtke, C.; Groger, N.; Manns, M.P.; Trautwein, C. Interferon-alpha enhances TRAIL-mediated apoptosis by up-regulating caspase-8 transcription in human hepatoma cells. J. Hepatol. 2006, 44, 342–349. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Birkenbihl, R.P.; Subramani, S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. 1992, 20, 6605–6611. [Google Scholar] [CrossRef] [Green Version]
- de Wind, N.; Dekker, M.; Berns, A.; Radman, M.; te Riele, H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995, 82, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Baunbaek, D.; Delehouze, C.; Loaec, N.; Hole, A.J.; Baumli, S.; Endicott, J.A.; Douc-Rasy, S.; Benard, J.; Oumata, N.; et al. CDK Inhibitors Roscovitine and CR8 Trigger Mcl-1 Down-Regulation and Apoptotic Cell Death in Neuroblastoma Cells. Genes Cancer 2010, 1, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukanov, N.O.; Moreno, S.E.; Natoli, T.A.; Rogers, K.A.; Smith, L.A.; Ledbetter, S.R.; Oumata, N.; Galons, H.; Meijer, L.; Ibraghimov-Beskrovnaya, O. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD. Cell Cycle 2012, 11, 4040–4046. [Google Scholar] [CrossRef] [Green Version]
- Cosimo, E.; McCaig, A.M.; Carter-Brzezinski, L.J.; Wheadon, H.; Leach, M.T.; Le Ster, K.; Berthou, C.; Durieu, E.; Oumata, N.; Galons, H.; et al. Inhibition of NF-kappaB-mediated signaling by the cyclin-dependent kinase inhibitor CR8 overcomes prosurvival stimuli to induce apoptosis in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2013, 19, 2393–2405. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, S.V.; Stoica, B.A.; Loane, D.J.; Luo, T.; Faden, A.I. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Kharebava, G.; Piao, C.; Stoica, B.A.; Dinizo, M.; Sabirzhanov, B.; Hanscom, M.; Guanciale, K.; Faden, A.I. Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS ONE 2012, 7, e42129. [Google Scholar] [CrossRef] [Green Version]
- Odajima, J.; Saini, S.; Jung, P.; Ndassa-Colday, Y.; Ficaro, S.; Geng, Y.; Marco, E.; Michowski, W.; Wang, Y.E.; DeCaprio, J.A.; et al. Proteomic Landscape of Tissue-Specific Cyclin E Functions in Vivo. PLoS Genet. 2016, 12, e1006429. [Google Scholar] [CrossRef]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Nevzorova, Y.A.; Haas, U.; Moro, N.; Sicinski, P.; Geng, Y.; Barbacid, M.; Trautwein, C.; Liedtke, C. Concurrent deletion of cyclin E1 and cyclin-dependent kinase 2 in hepatocytes inhibits DNA replication and liver regeneration in mice. Hepatology 2014, 59, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 2006, 5, 2555–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Kamradt, M.; Mulcahy, M.; Byun, Y.; Xu, H.; McKay, M.J.; Cryns, V.L. Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J. Biol. Chem. 2002, 277, 16775–16781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Latham, V.M., Jr.; Zhang, X.; Shapiro, G.I. Combined depletion of cell cycle and transcriptional cyclin-dependent kinase activities induces apoptosis in cancer cells. Cancer Res. 2006, 66, 9270–9280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, T.; Ohuchi, T.; Sumida, S.; Matsumoto, K.; Toyoshima, K. Phosphorylation of the retinoblastoma protein by cdk2. Proc. Natl. Acad. Sci USA 1992, 89, 7900–7904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever-Chroneos, Z.; Angus, S.P.; Fribourg, A.F.; Wan, H.; Todorov, I.; Knudsen, K.E.; Knudsen, E.S. Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol. Cell. Biol. 2001, 21, 4032–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. J. Hepatol. 2012, 57, 692–694. [Google Scholar] [CrossRef] [Green Version]
- McClue, S.J.; Stuart, I. Metabolism of the trisubstituted purine cyclin-dependent kinase inhibitor seliciclib (R-roscovitine) in vitro and in vivo. Drug Metab. Dispos. Biol. Fate Chem. 2008, 36, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.; Gressner, A.M.; Weiskirchen, R. Immortal hepatic stellate cell lines: Useful tools to study hepatic stellate cell biology and function? J. Cell. Mol. Med. 2007, 11, 704–722. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A.; Dierssen, U.; Valli, L.; Garuti, C.; Rump, A.; Corradini, E.; Ernst, M.; Klein, C.; Trautwein, C. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology 2007, 132, 294–300. [Google Scholar] [CrossRef]
- Weiskirchen, S.; Tag, C.G.; Sauer-Lehnen, S.; Tacke, F.; Weiskirchen, R. Isolation and Culture of Primary Murine Hepatic Stellate Cells. Methods Mol. Biol. 2017, 1627, 165–191. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Tschaharganeh, D.; Gassler, N.; Geng, Y.; Weiskirchen, R.; Sicinski, P.; Trautwein, C.; Liedtke, C. Aberrant cell cycle progression and endoreplication in regenerating livers of mice that lack a single E-type cyclin. Gastroenterology 2009, 137, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonntag, R.; Giebeler, N.; Nevzorova, Y.A.; Bangen, J.M.; Fahrenkamp, D.; Lambertz, D.; Haas, U.; Hu, W.; Gassler, N.; Cubero, F.J.; et al. Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, 9282–9287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, C.; Lambertz, D.; Schnepel, N.; Trautwein, C. Molecular mechanism of Mitomycin C-dependent caspase-8 regulation: Implications for apoptosis and synergism with interferon-alpha signalling. Apoptosis Int. J. Program. Cell Death 2007, 12, 2259–2270. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hübbers, A.; Hennings, J.; Lambertz, D.; Haas, U.; Trautwein, C.; Nevzorova, Y.A.; Sonntag, R.; Liedtke, C. Pharmacological Inhibition of Cyclin-Dependent Kinases Triggers Anti-Fibrotic Effects in Hepatic Stellate Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 3267. https://doi.org/10.3390/ijms21093267
Hübbers A, Hennings J, Lambertz D, Haas U, Trautwein C, Nevzorova YA, Sonntag R, Liedtke C. Pharmacological Inhibition of Cyclin-Dependent Kinases Triggers Anti-Fibrotic Effects in Hepatic Stellate Cells In Vitro. International Journal of Molecular Sciences. 2020; 21(9):3267. https://doi.org/10.3390/ijms21093267
Chicago/Turabian StyleHübbers, Anna, Julia Hennings, Daniela Lambertz, Ute Haas, Christian Trautwein, Yulia A. Nevzorova, Roland Sonntag, and Christian Liedtke. 2020. "Pharmacological Inhibition of Cyclin-Dependent Kinases Triggers Anti-Fibrotic Effects in Hepatic Stellate Cells In Vitro" International Journal of Molecular Sciences 21, no. 9: 3267. https://doi.org/10.3390/ijms21093267