Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
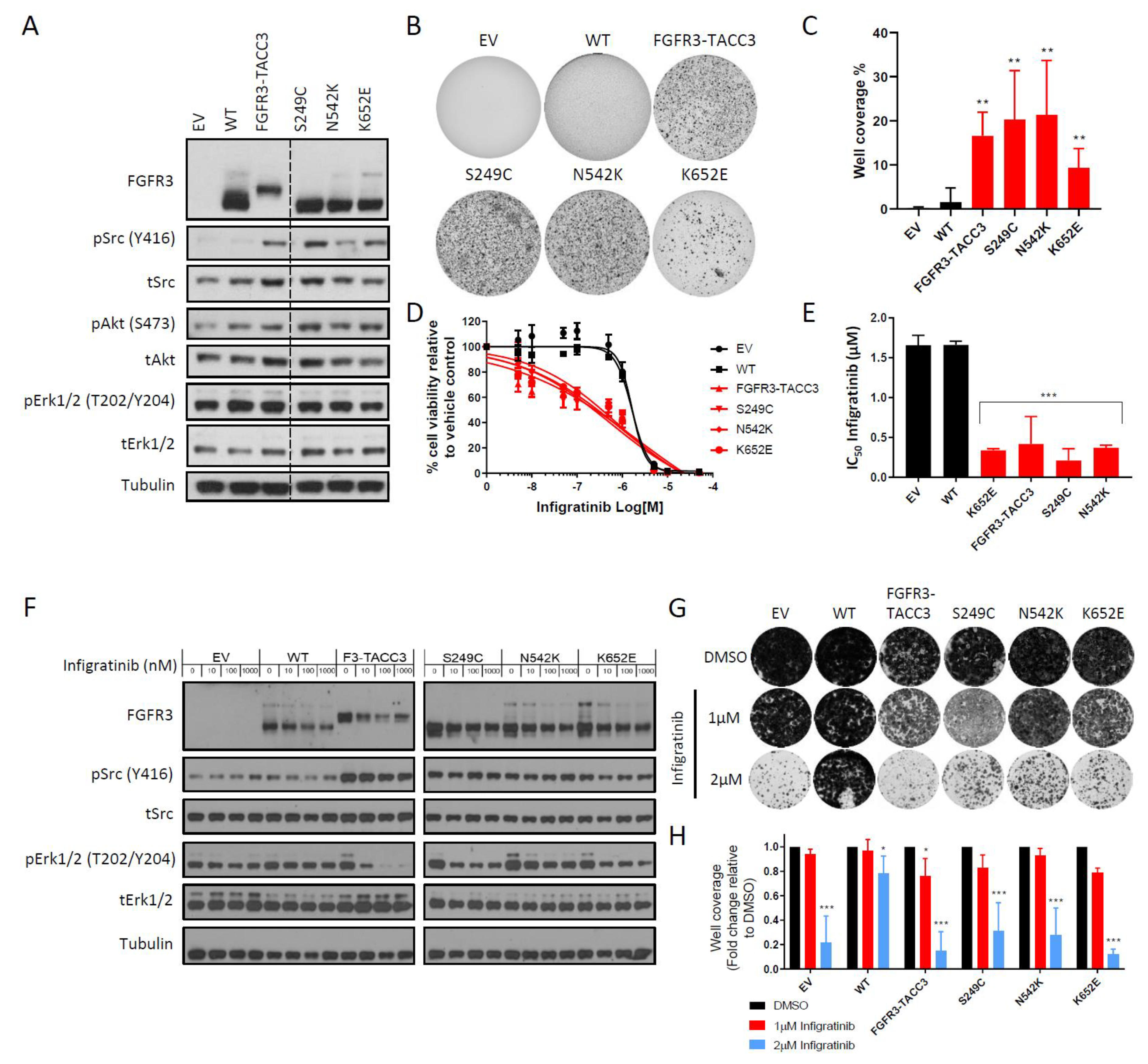
2.1. FGFR3-TACC3 Gene Fusion and Activating FGFR3 Point Mutants are Intrinsically Resistant to Infigratinib in the NIH-3T3 Cell Line Model
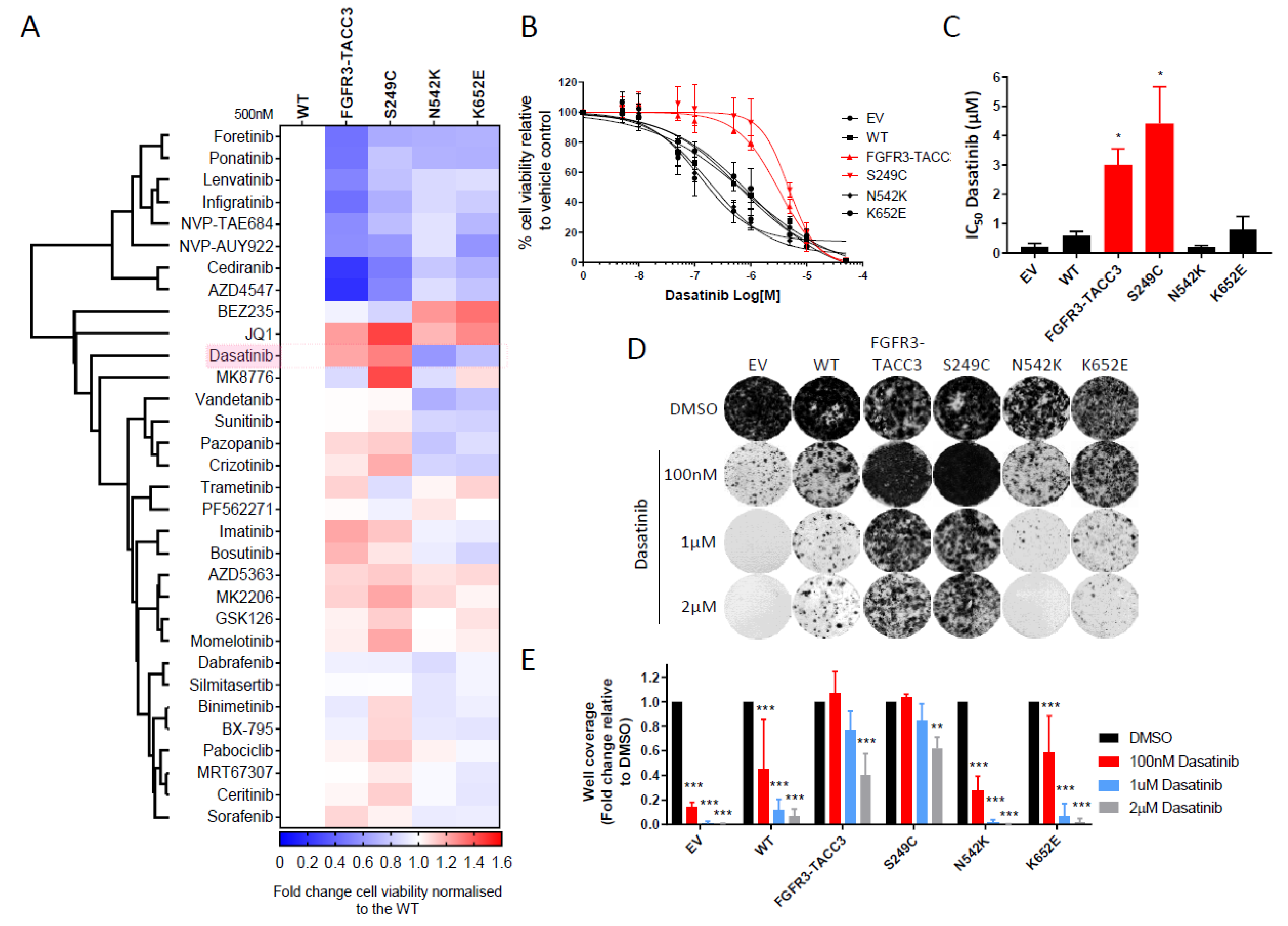
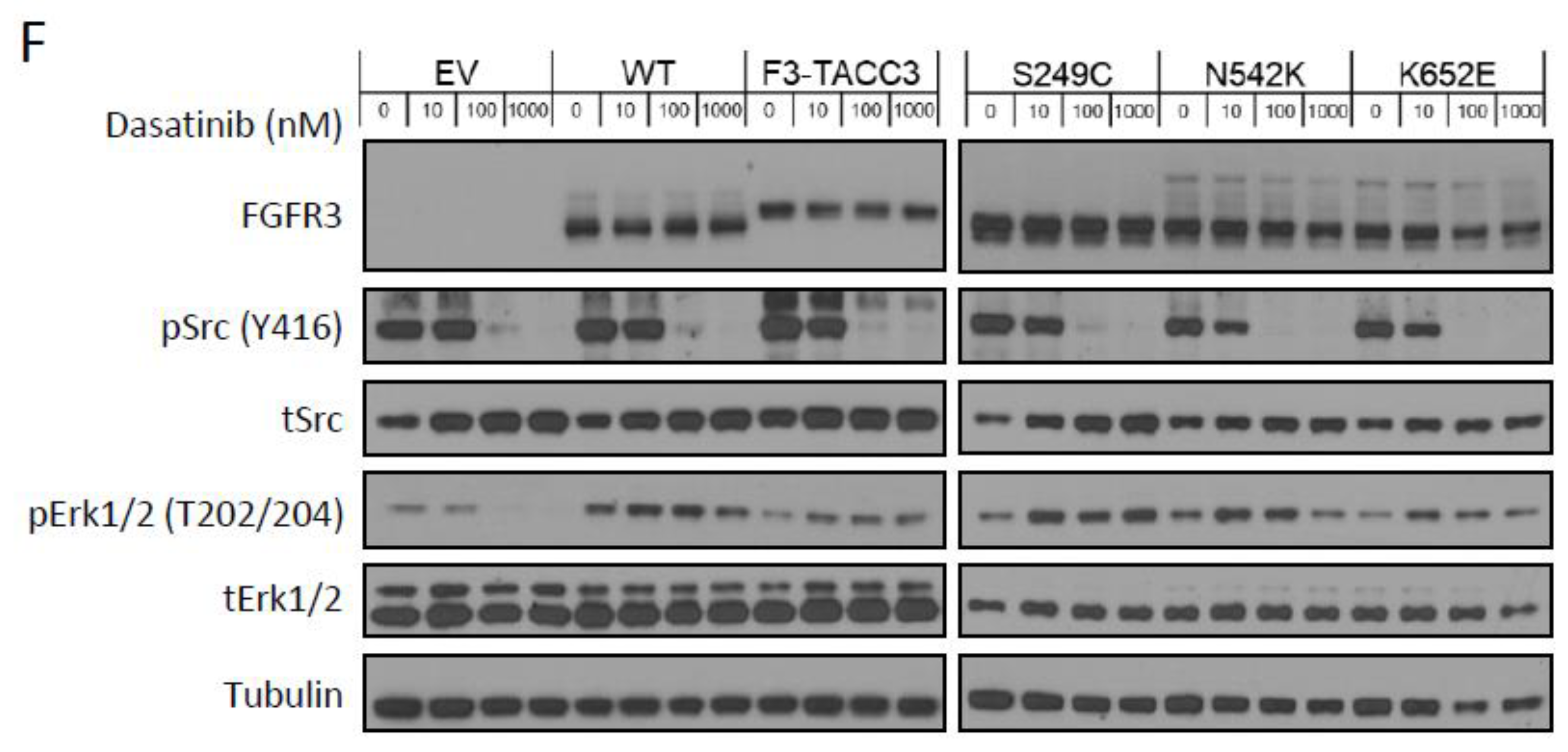
2.2. FGFR3-TACC3 and S249C Expression Confers Resistance to Dasatinib
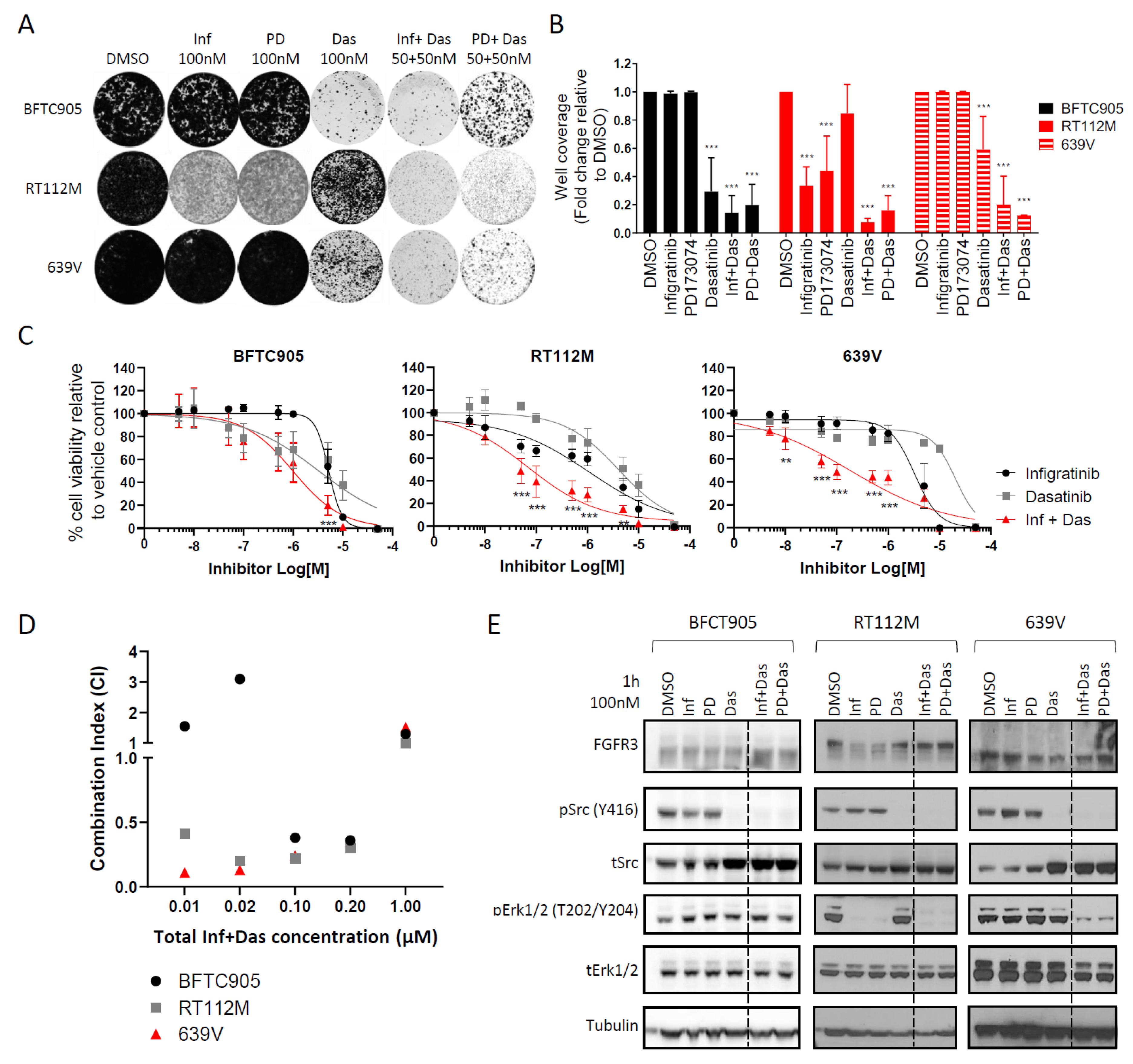
2.3. Dasatinib Sensitizes NIH-3T3 Cells with FGFR3 Molecular Alterations to Infigratinib Treatment
2.4. Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancer Cell Lines with FGFR3 Molecular Alterations
3. Discussion
4. Methods and Materials
4.1. Cell Lines
4.2. Generation of Plasmids and Stable Cell Lines
4.3. Western Blotting and Antibodies
4.4. Anchorage Independent Cell Growth in Soft Agar
4.5. Colony Formation Assay
4.6. Cell Viability Assay and Small Molecule Inhibitor Screen
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556 e25. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Hurst, C.D.; Platt, F.M.; Taylor, C.F.; Knowles, M.A. Novel tumor subgroups of urothelial carcinoma of the bladder defined by integrated genomic analysis. Clin. Cancer Res. 2012, 18, 5865–5877. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, J.; Lamy, P.; Nordentoft, I.; Algaba, F.; Hoyer, S.; Ulhoi, B.P.; Vang, S.; Reinert, T.; Hermann, G.G.; Mogensen, K.; et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 2016, 30, 27–42. [Google Scholar] [CrossRef]
- Adar, R.; Monsonego-Ornan, E.; David, P.; Yayon, A. Differential activation of cysteine-substitution mutants of fibroblast growth factor receptor 3 is determined by cysteine localization. J. Bone Miner. Res. 2002, 17, 860–868. [Google Scholar] [CrossRef]
- d’Avis, P.Y.; Robertson, S.C.; Meyer, A.N.; Bardwell, W.M.; Webster, M.K.; Donoghue, D.J. Constitutive activation of fibroblast growth factor receptor 3 by mutations responsible for the lethal skeletal dysplasia thanatophoric dysplasia type I. Cell Growth Differ. 1998, 9, 71–78. [Google Scholar]
- Del Piccolo, N.; Placone, J.; Hristova, K. Effect of thanatophoric dysplasia type I mutations on FGFR3 dimerization. BioPhys. J. 2015, 108, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.N.; Meyer, A.N.; Siari, A.; Campos, A.R.; Motamedchaboki, K.; Donoghue, D.J. Oncogenic Gene Fusion FGFR3-TACC3 Is Regulated by Tyrosine Phosphorylation. Mol. Cancer Res. 2016, 14, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.; et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Invest 2013, 123, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Bellus, G.A.; McIntosh, I.; Smith, E.A.; Aylsworth, A.S.; Kaitila, I.; Horton, W.A.; Greenhaw, G.A.; Hecht, J.T.; Francomano, C.A. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat. Genet. 1995, 10, 357–359. [Google Scholar] [CrossRef]
- Patani, H.; Bunney, T.D.; Thiyagarajan, N.; Norman, R.A.; Ogg, D.; Breed, J.; Ashford, P.; Potterton, A.; Edwards, M.; Williams, S.V.; et al. Landscape of activating cancer mutations in FGFR kinases and their differential responses to inhibitors in clinical use. Oncotarget 2016, 7, 24252–24268. [Google Scholar] [CrossRef] [Green Version]
- di Martino, E.; L’Hote, C.G.; Kennedy, W.; Tomlinson, D.C.; Knowles, M.A. Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type and mutation-specific manner. Oncogene 2009, 28, 4306–4316. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Bunney, T.D.; Wan, S.; Thiyagarajan, N.; Sutto, L.; Williams, S.V.; Ashford, P.; Koss, H.; Knowles, M.A.; Gervasio, F.L.; Coveney, P.V.; et al. The Effect of Mutations on Drug Sensitivity and Kinase Activity of Fibroblast Growth Factor Receptors: A Combined Experimental and Theoretical Study. EBioMedicine 2015, 2, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel RNA interference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013, 3, 1058–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Kwarcinski, F.E.; Brandvold, K.R.; Phadke, S.; Beleh, O.M.; Johnson, T.K.; Meagher, J.L.; Seeliger, M.A.; Stuckey, J.A.; Soellner, M.B. Conformation-Selective Analogues of Dasatinib Reveal Insight into Kinase Inhibitor Binding and Selectivity. ACS Chem. Biol. 2016, 11, 1296–1304. [Google Scholar] [CrossRef]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef]
- Tan, A.C.; Vyse, S.; Huang, P.H. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov. Today 2017, 22, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, Y.; Akiyama, N.; Tsukaguchi, T.; Fujii, T.; Satoh, Y.; Ishii, N.; Aoki, M. Mechanism of Oncogenic Signal Activation by the Novel Fusion Kinase FGFR3-BAIAP2L1. Mol. Cancer Ther. 2015, 14, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Acquaviva, J.; He, S.; Zhang, C.; Jimenez, J.P.; Nagai, M.; Sang, J.; Sequeira, M.; Smith, D.L.; Ogawa, L.S.; Inoue, T.; et al. FGFR3 translocations in bladder cancer: Differential sensitivity to HSP90 inhibition based on drug metabolism. Mol Cancer Res. 2014, 12, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Elliott, A.Y.; Bronson, D.L.; Cervenka, J.; Stein, N.; Fraley, E.E. Properties of cell lines established from transitional cell cancers of the human urinary tract. Cancer Res. 1977, 37, 1279–1289. [Google Scholar]
- Suwaki, N.; Vanhecke, E.; Atkins, K.M.; Graf, M.; Swabey, K.; Huang, P.; Schraml, P.; Moch, H.; Cassidy, A.M.; Brewer, D.; et al. A HIF-regulated VHL-PTP1B-Src signaling axis identifies a therapeutic target in renal cell carcinoma. Sci Transl Med. 2011, 3, 85ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mikse, O.; Liao, R.G.; Li, Y.; Tan, L.; Janne, P.A.; Gray, N.S.; Wong, K.K.; Hammerman, P.S. Ligand-associated ERBB2/3 activation confers acquired resistance to FGFR inhibition in FGFR3-dependent cancer cells. Oncogene 2015, 34, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, J.M.; Yamashita, H.; Jian, W.; Lerner, S.P.; Sonpavde, G. Dasatinib is preclinically active against Src-overexpressing human transitional cell carcinoma of the urothelium with activated Src signaling. Mol. Cancer Ther. 2010, 9, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Overdevest, J.B.; Nitz, M.D.; Williams, P.D.; Owens, C.R.; Sanchez-Carbayo, M.; Frierson, H.F.; Schwartz, M.A.; Theodorescu, D. Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Res. 2011, 71, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Hahn, N.M.; Knudsen, B.S.; Daneshmand, S.; Koch, M.O.; Bihrle, R.; Foster, R.S.; Gardner, T.A.; Cheng, L.; Liu, Z.; Breen, T.; et al. Neoadjuvant dasatinib for muscle-invasive bladder cancer with tissue analysis of biologic activity. Urol. Oncol. 2016, 34, 4.e11–4.e17. [Google Scholar] [CrossRef]
- Xu, A.M.; Huang, P.H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Morris, M.; Bagui, T.; Lee, F.Y.; Jove, R.; Haura, E.B. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res. 2006, 66, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kalyankrishna, S.; Wislez, M.; Thilaganathan, N.; Saigal, B.; Wei, W.; Ma, L.; Wistuba, I.I.; Johnson, F.M.; Kurie, J.M. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol. 2007, 170, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haura, E.B.; Tanvetyanon, T.; Chiappori, A.; Williams, C.; Simon, G.; Antonia, S.; Gray, J.; Litschauer, S.; Tetteh, L.; Neuger, A.; et al. Phase I/II study of the Src inhibitor dasatinib in combination with erlotinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1387–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, A.; Nichol, D.; Kinose, F.; Abazeed, M.E.; Marusyk, A.; Haura, E.B.; Scott, J.G. Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. ReP. 2017, 7, 1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Sedlak, J.C.; Srinivas, R.; Creixell, P.; Pritchard, J.R.; Tidor, B.; Lauffenburger, D.A.; Hemann, M.T. Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution. Cell 2016, 165, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Noujaim, J.; Payne, L.S.; Judson, I.; Jones, R.L.; Huang, P.H. Phosphoproteomics in translational research: A sarcoma perspective. Ann. Onc. 2016, 27, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, D.C.; L’Hote, C.G.; Kennedy, W.; Pitt, E.; Knowles, M.A. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. 2005, 65, 10441–10449. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, N.C.; Atkinson, E.; Bunney, T.D.; Katan, M.; Huang, P.H. Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations. Int. J. Mol. Sci. 2020, 21, 3214. https://doi.org/10.3390/ijms21093214
Lima NC, Atkinson E, Bunney TD, Katan M, Huang PH. Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations. International Journal of Molecular Sciences. 2020; 21(9):3214. https://doi.org/10.3390/ijms21093214
Chicago/Turabian StyleLima, Nadia Carvalho, Eliza Atkinson, Tom D. Bunney, Matilda Katan, and Paul H. Huang. 2020. "Targeting the Src Pathway Enhances the Efficacy of Selective FGFR Inhibitors in Urothelial Cancers with FGFR3 Alterations" International Journal of Molecular Sciences 21, no. 9: 3214. https://doi.org/10.3390/ijms21093214