Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway

Abstract
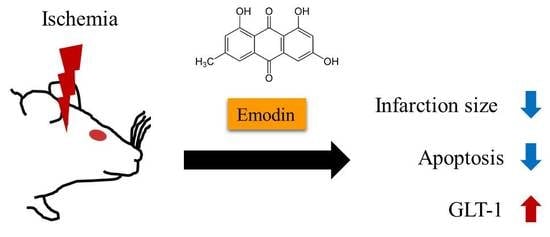
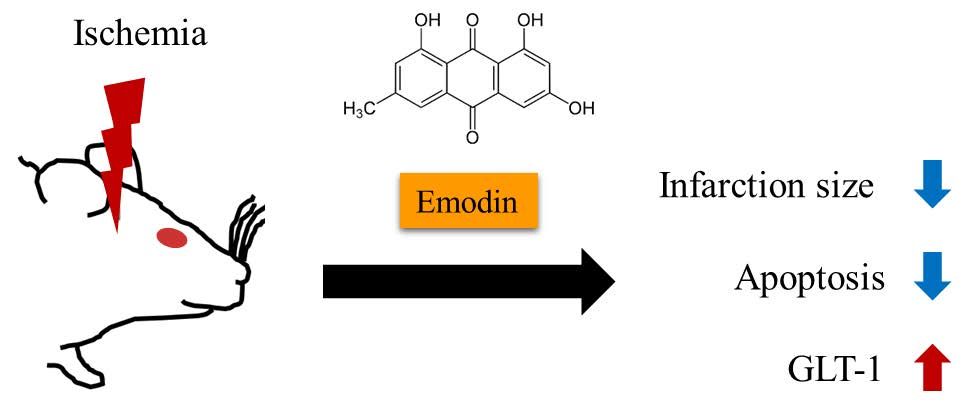
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
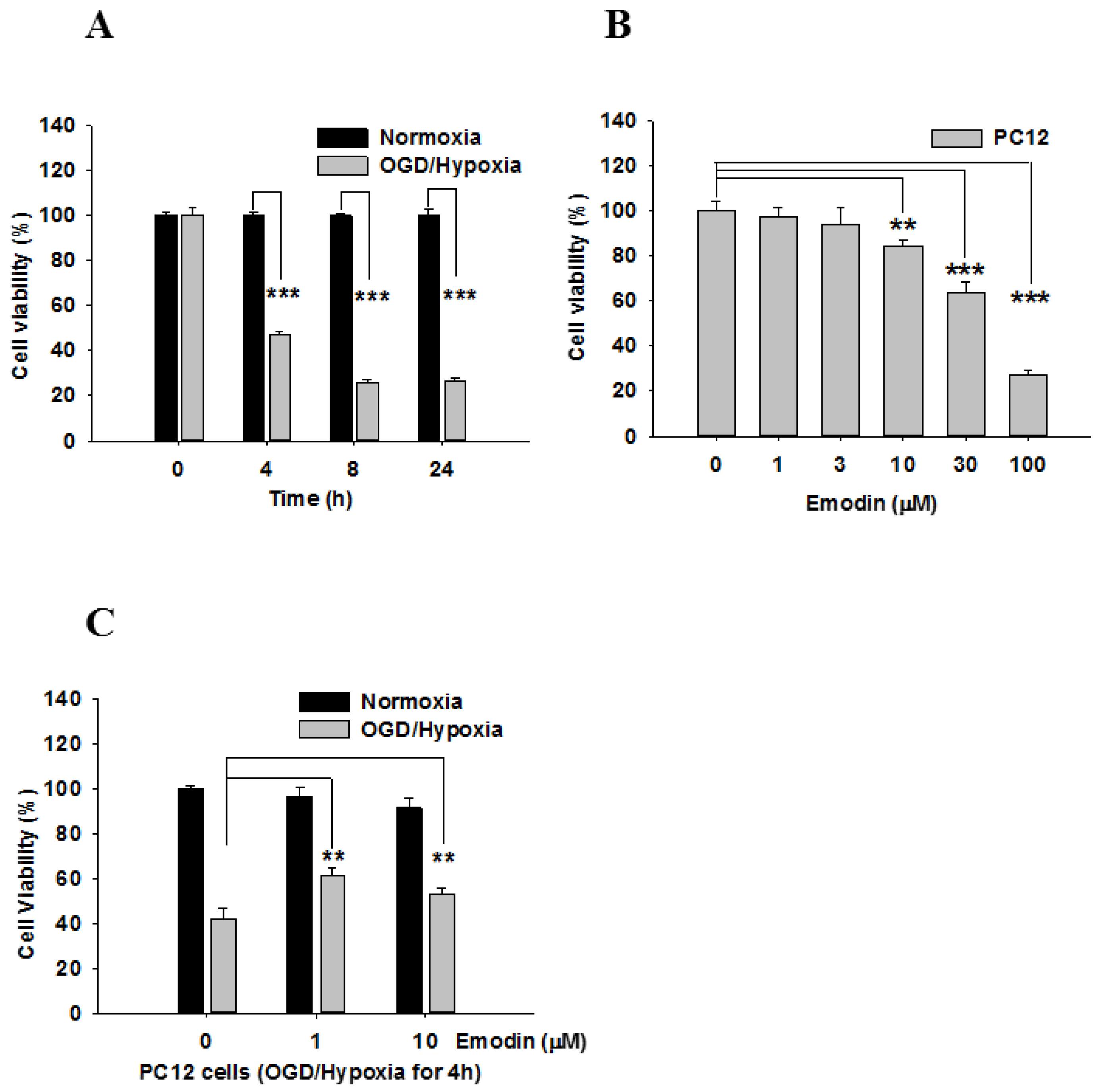
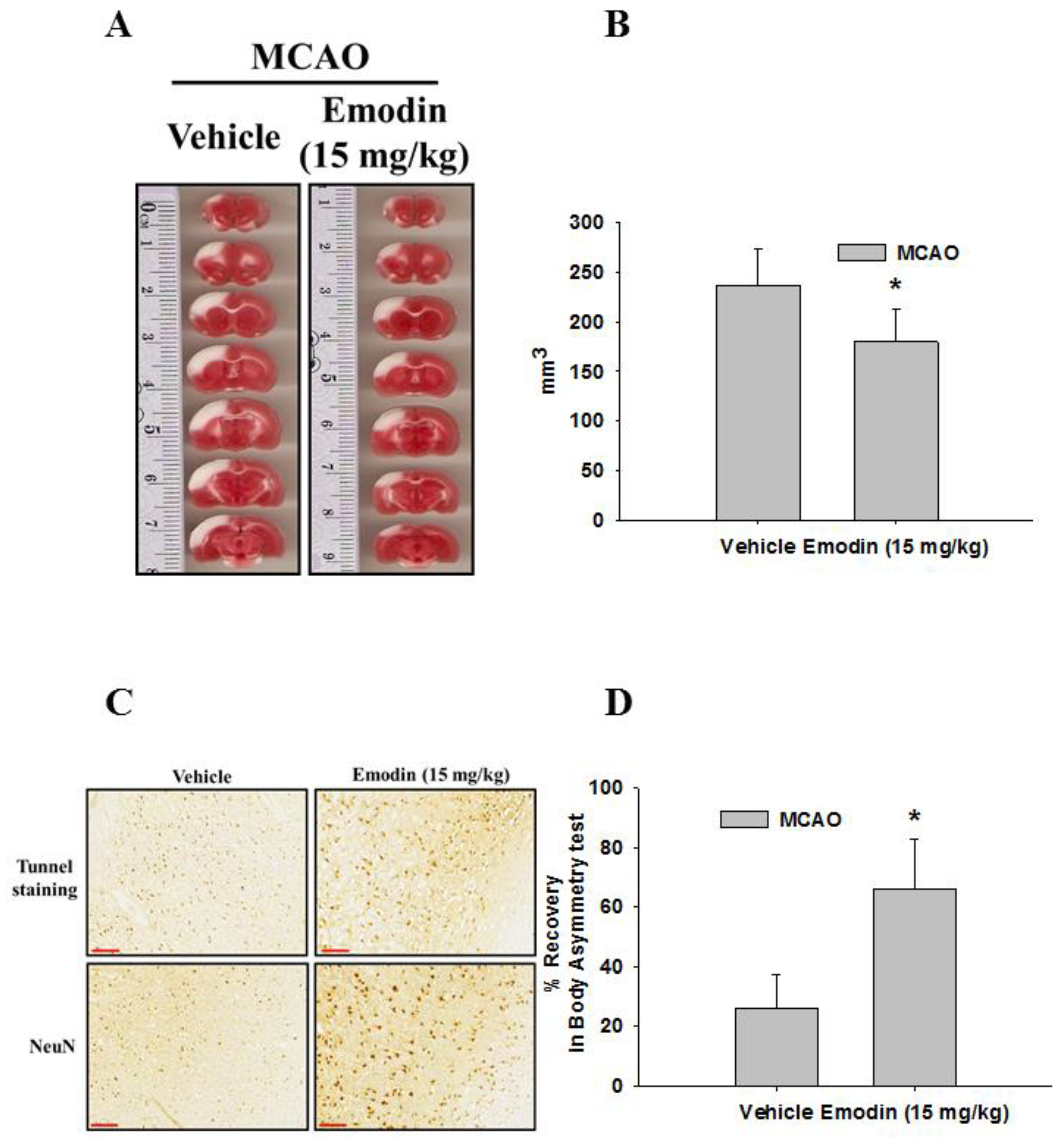
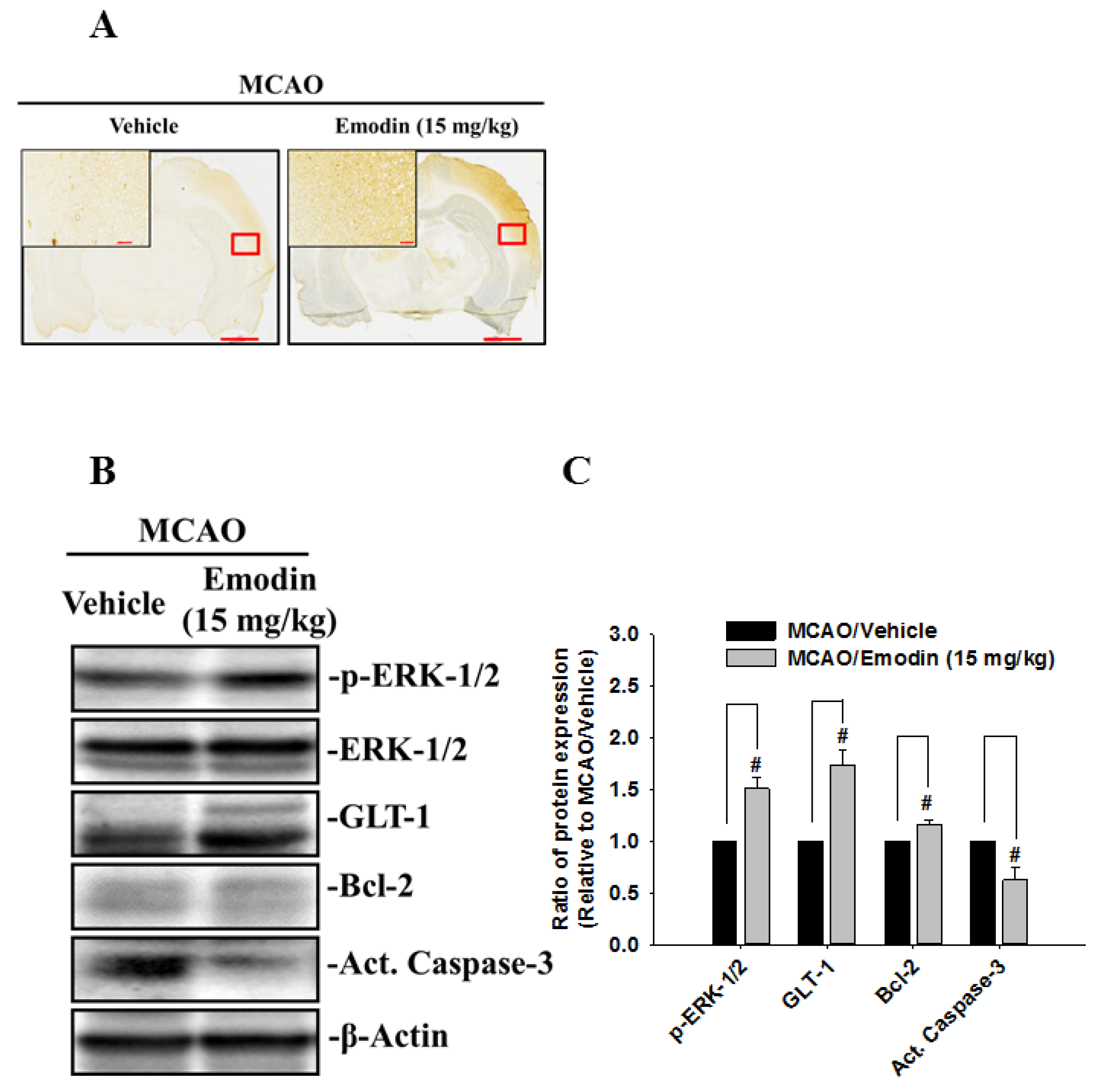
2. Results
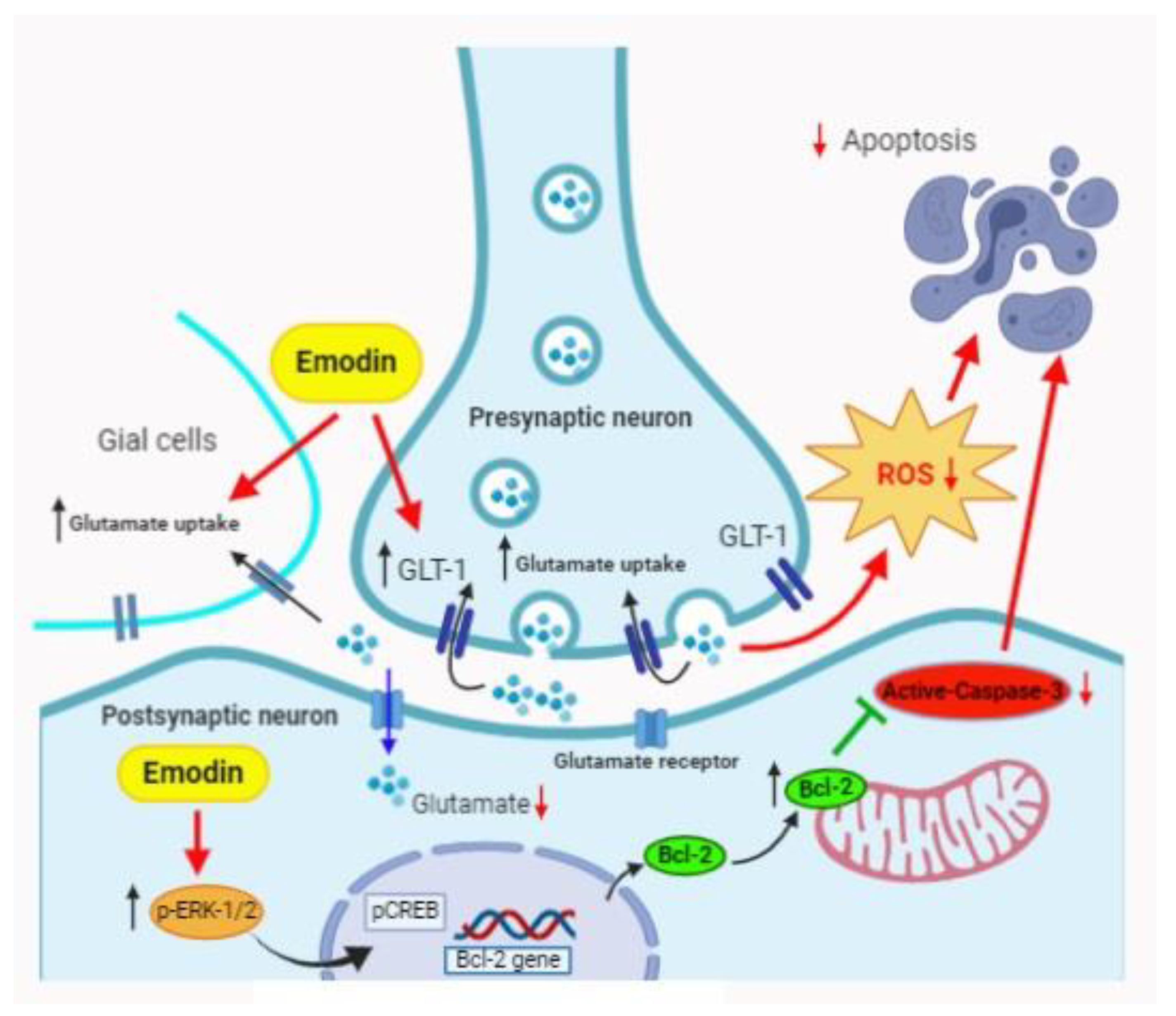
3. Discussion
4. Material and Methods
4.1. MCAO
4.2. Brain Tissue Staining
4.3. Immunohistochemical Staining and Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Staining
4.4. Body Asymmetry
4.5. Cell Culture and Oxygen-Glucose Deprivation (OGD)-Hypoxia
4.6. Viability Assay
4.7. ROS Production
4.8. Western Blotting
4.9. Glutamate Release Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slomka, A.; Switonska, M.; Sinkiewicz, W.; Zekanowska, E. Haemostatic factors do not account for worse outcomes from ischaemic stroke in patients with higher C-reactive protein concentrations. Ann. Clin. Biochem. 2017, 54, 378–385. [Google Scholar] [CrossRef]
- Repici, M.; Mariani, J.; Borsello, T. Neuronal death and neuroprotection: A review. Methods Mol. Biol. 2007, 399, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakka, V.P.; Gusain, A.; Mehta, S.L.; Raghubir, R. Molecular mechanisms of apoptosis in cerebral ischemia: Multiple neuroprotective opportunities. Mol. Neurobiol. 2008, 37, 7–38. [Google Scholar] [CrossRef] [PubMed]
- Ten, V.; Galkin, A. Mechanism of mitochondrial complex I damage in brain ischemia/reperfusion injury. A hypothesis. Mol. Cell. Neurosci. 2019, 100, 103408. [Google Scholar] [CrossRef]
- Sergeeva, S.P.; Savin, A.A.; Litvitsky, P.F.; Lyundup, A.V.; Kiseleva, E.V.; Gorbacheva, L.R.; Breslavich, I.D.; Kucenko, K.I.; Balyasin, M.V. [Apoptosis as a systemic adaptive mechanism in ischemic stroke]. Zhurnal Nevrol. Psikhiatrii Im. S.S. Korsakova 2018, 118, 38–45. [Google Scholar] [CrossRef]
- Sakai, S.; Shichita, T. Inflammation and neural repair after ischemic brain injury. Neurochem. Int. 2019, 130, 104316. [Google Scholar] [CrossRef]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Dempsy, R.; Hatcher, J.F. Integration of cytokine biology and lipid metabolism in stroke. Front. Biosci. 2008, 13, 1250–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun-Guang, W.; Jun-Qing, Y.; Bei-Zhong, L.; Dan-Ting, J.; Chong, W.; Liang, Z.; Dan, Z.; Yan, W. Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur. J. Pharmacol. 2010, 627, 33–41. [Google Scholar] [CrossRef]
- Cha, T.L.; Chuang, M.J.; Tang, S.H.; Wu, S.T.; Sun, K.H.; Chen, T.T.; Sun, G.H.; Chang, S.Y.; Yu, C.P.; Ho, J.Y.; et al. Emodin modulates epigenetic modifications and suppresses bladder carcinoma cell growth. Mol. Carcinog. 2015, 54, 167–177. [Google Scholar] [CrossRef]
- Tang, Q.; Wu, J.; Zheng, F.; Hann, S.S.; Chen, Y. Emodin Increases Expression of Insulin-Like Growth Factor Binding Protein 1 through Activation of MEK/ERK/AMPKalpha and Interaction of PPARgamma and Sp1 in Lung Cancer. Cell. Physiol. Biochem. 2017, 41, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, H.; Tong, H.; Ye, S.; Qiu, M.; Wang, Z.; Tan, W.; Liu, J.; Lin, S. Emodin potentiates the antitumor effects of gemcitabine in pancreatic cancer cells via inhibition of nuclear factor-kappaB. Mol. Med. Rep. 2011, 4, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, H.; Wei, W.; Ye, S.; Liao, W.; Gong, J.; Jiang, Z.; Wang, L.; Lin, S. Antiproliferative and antimetastatic effects of emodin on human pancreatic cancer. Oncol. Rep. 2011, 26, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.M.; Hsu, Y.A.; Tsai, Y.; Shieh, F.K.; Huang, S.H.; Wan, L.; Tsai, F.J. Emodin inhibits the growth of hepatoma cells: Finding the common anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem. Biophys. Res. Commun. 2010, 392, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wei, W.; Guo, Y.; Liu, A.; Tong, H.; Wang, Z.; Tan, W.; Liu, J.; Lin, S. Enhanced effect of gemcitabine by emodin against pancreatic cancer in vivo via cytochrome C-regulated apoptosis. Oncol. Rep. 2011, 25, 1253–1261. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.W.; Hasuo, H.; Takeya, M.; Akasu, T. Effects of emodin on synaptic transmission in rat hippocampal CA1 pyramidal neurons in vitro. Neuropharmacology 2005, 49, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, D.; Ma, H.; Liu, J. Neuroprotective effects of emodin-8-O-beta-D-glucoside in vivo and in vitro. Eur. J. Pharmacol. 2007, 577, 58–63. [Google Scholar] [CrossRef]
- Li, X.; Chu, S.; Liu, Y.; Chen, N. Neuroprotective Effects of Anthraquinones from Rhubarb in Central Nervous System Diseases. Evid. Based Complement Altern. Med. 2019, 2019, 3790728. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Jin, H.; Sun, Q.R.; Xu, J.H.; Hu, H.T. Neuroprotective effects of emodin in rat cortical neurons against beta-amyloid-induced neurotoxicity. Brain Res. 2010, 1347, 149–160. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, Q.; Hu, Z.; Tang, X. Potential Neuroprotective Treatment of Stroke: Targeting Excitotoxicity, Oxidative Stress, and Inflammation. Front. Neurosci. 2019, 13, 1036. [Google Scholar] [CrossRef]
- Rao, V.L.; Dogan, A.; Todd, K.G.; Bowen, K.K.; Kim, B.T.; Rothstein, J.D.; Dempsey, R.J. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci. 2001, 21, 1876–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, F.; Shioda, N.; Moriguchi, S.; Qin, Z.H.; Fukunaga, K. Downregulation of glutamate transporters is associated with elevation in extracellular glutamate concentration following rat microsphere embolism. Neurosci. Lett. 2008, 430, 275–280. [Google Scholar] [CrossRef]
- Harvey, B.K.; Airavaara, M.; Hinzman, J.; Wires, E.M.; Chiocco, M.J.; Howard, D.B.; Shen, H.; Gerhardt, G.; Hoffer, B.J.; Wang, Y. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 2011, 6, e22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.F.; Morales, M.; Chou, J.; Chen, H.L.; Hoffer, B.; Wang, Y. Bone morphogenetic proteins are involved in fetal kidney tissue transplantation-induced neuroprotection in stroke rats. Neuropharmacology 2002, 43, 418–426. [Google Scholar] [CrossRef]
- Karki, P.; Webb, A.; Zerguine, A.; Choi, J.; Son, D.S.; Lee, E. Mechanism of raloxifene-induced upregulation of glutamate transporters in rat primary astrocytes. Glia 2014, 62, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Shen, X.; Xu, Y.; Yuan, J.; Zhao, D.; Hu, W. Emodin prevents hypoxic-ischemic neuronal injury: Involvement of the activin A pathway. Neural Regen. Res. 2013, 8, 1360–1367. [Google Scholar] [CrossRef]
- Lu, J.S.; Liu, J.X.; Zhang, W.Y.; Liang, S.W.; Wang, D.; Fang, J. [Preventive effects of emodin on cerebral ischemia injury and expression of the inflammatory factors in rats with cerebral ischemia]. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J. Chin. Mater. Med. 2005, 30, 1939–1943. [Google Scholar]
- Shrimali, D.; Shanmugam, M.K.; Kumar, A.P.; Zhang, J.; Tan, B.K.; Ahn, K.S.; Sethi, G. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013, 341, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, L.; Wang, M.; Xu, L.; Yu, L.; Fang, T.; Wu, M. Emodin-induced microglial apoptosis is associated with TRB3 induction. Immunopharmacol. Immunotoxicol. 2011, 33, 594–602. [Google Scholar] [CrossRef]
- Meng, G.; Liu, Y.; Lou, C.; Yang, H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-kappaB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br. J. Pharmacol. 2010, 161, 1628–1644. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Tu, X.; Lin, G.; Xia, H.; Huang, H.; Wan, J.; Cheng, Z.; Liu, M.; Chen, G.; Zhang, H.; et al. Emodin-mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci. 2007, 81, 1332–1338. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F.; Shan, Q.; Zheng, Z.H.; Liu, C.M.; Wang, Y.J. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J. Pathol. 2010, 222, 199–212. [Google Scholar] [CrossRef]
- Zhou, G.H.; Zhang, F.; Wang, X.N.; Kwon, O.J.; Kang, D.G.; Lee, H.S.; Jin, S.N.; Cho, K.W.; Wen, J.F. Emodin accentuates atrial natriuretic peptide secretion in cardiac atria. Eur. J. Pharmacol. 2014, 735, 44–51. [Google Scholar] [CrossRef]
- Ma, Y.; Xia, X.; Cheng, J.M.; Kuang, Y.Q.; Yang, T.; Yang, L.B.; Fan, K.; Gu, J.W. Emodin inhibits inducible nitric oxide synthase in a rat model of craniocerebral explosive injury. Neurochem. Res. 2014, 39, 1809–1816. [Google Scholar] [CrossRef]
- Ming, J.; Xie, J.; Xu, P.; Ge, X.; Liu, W.; Ye, J. Effects of emodin and vitamin C on growth performance, biochemical parameters and two HSP70s mRNA expression of Wuchang bream (Megalobrama amblycephala Yih) under high temperature stress. Fish Shellfish Immunol. 2012, 32, 651–661. [Google Scholar] [CrossRef]
- Lipski, J.; Wan, C.K.; Bai, J.Z.; Pi, R.; Li, D.; Donnelly, D. Neuroprotective potential of ceftriaxone in in vitro models of stroke. Neuroscience 2007, 146, 617–629. [Google Scholar] [CrossRef]
- Meisel, A. Preventive antibiotic therapy in stroke: PASSed away? Lancet 2015. [Google Scholar] [CrossRef]
- Kawahara, K.; Kosugi, T.; Tanaka, M.; Nakajima, T.; Yamada, T. Reversed operation of glutamate transporter GLT-1 is crucial to the development of preconditioning-induced ischemic tolerance of neurons in neuron/astrocyte co-cultures. Glia 2005, 49, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Lin, H.B.; Zhou, H.; Xu, J.P. [Protective effect of polydatin on a PC12 cell model of oxygen-glucose deprivation]. Nan Fang Yi Ke Da Xue Xue Bao = J. South. Med Univ. 2010, 30, 1041–1043. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, S.W.; Lai, J.H.; Wu, J.C.-C.; Tsai, Y.-R.; Chen, Y.-H.; Kang, S.-J.; Chiang, Y.-H.; Chang, C.-F.; Chen, K.-Y. Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 2899. https://doi.org/10.3390/ijms21082899
Leung SW, Lai JH, Wu JC-C, Tsai Y-R, Chen Y-H, Kang S-J, Chiang Y-H, Chang C-F, Chen K-Y. Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway. International Journal of Molecular Sciences. 2020; 21(8):2899. https://doi.org/10.3390/ijms21082899
Chicago/Turabian StyleLeung, Stephen Wan, Jing Huei Lai, John Chung-Che Wu, Yan-Rou Tsai, Yen-Hua Chen, Shuo-Jhen Kang, Yung-Hsiao Chiang, Cheng-Fu Chang, and Kai-Yun Chen. 2020. "Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway" International Journal of Molecular Sciences 21, no. 8: 2899. https://doi.org/10.3390/ijms21082899