Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells

 , , and
, , and 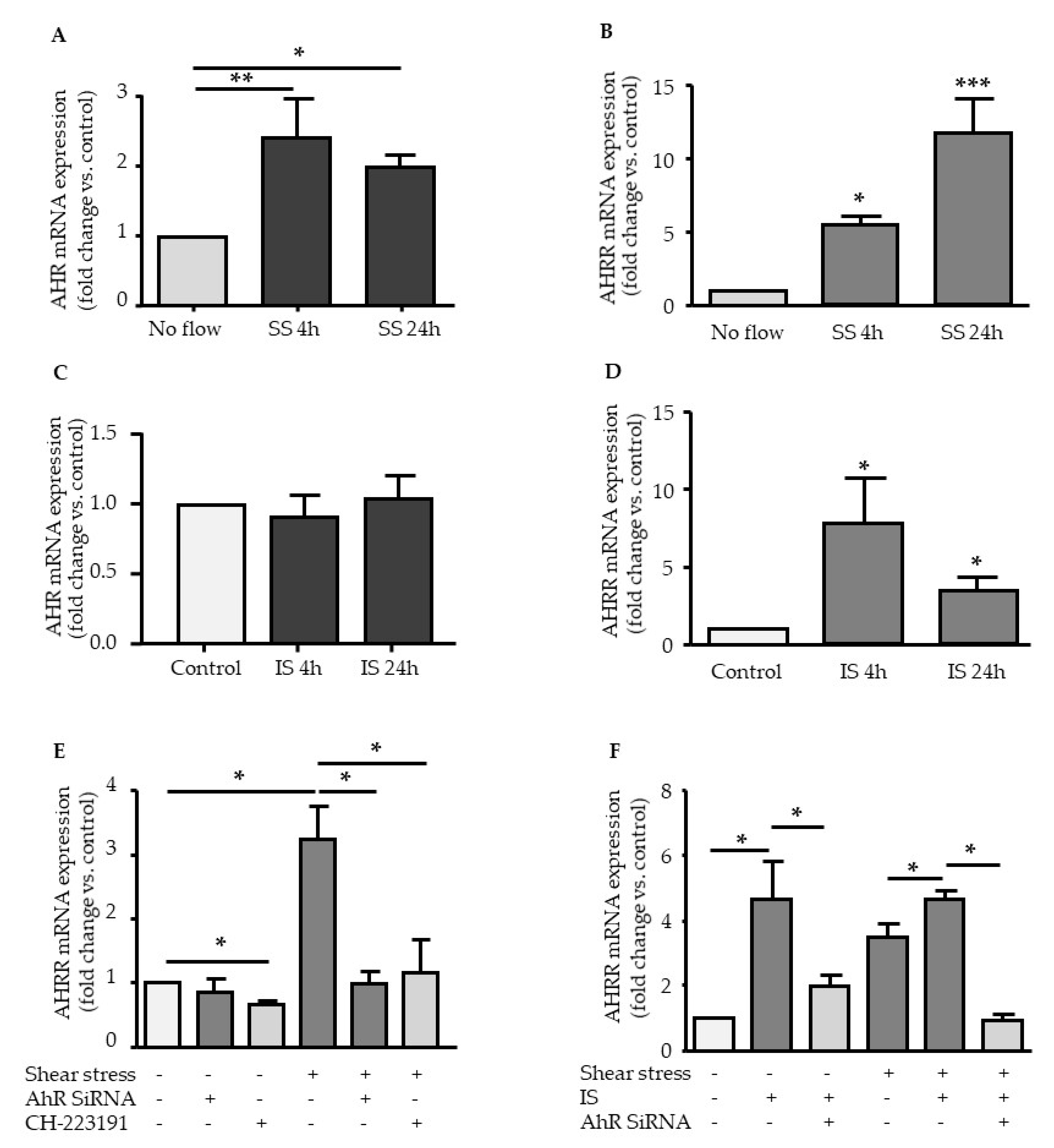{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Shear Stress and IS on AHR and AHRR Expression
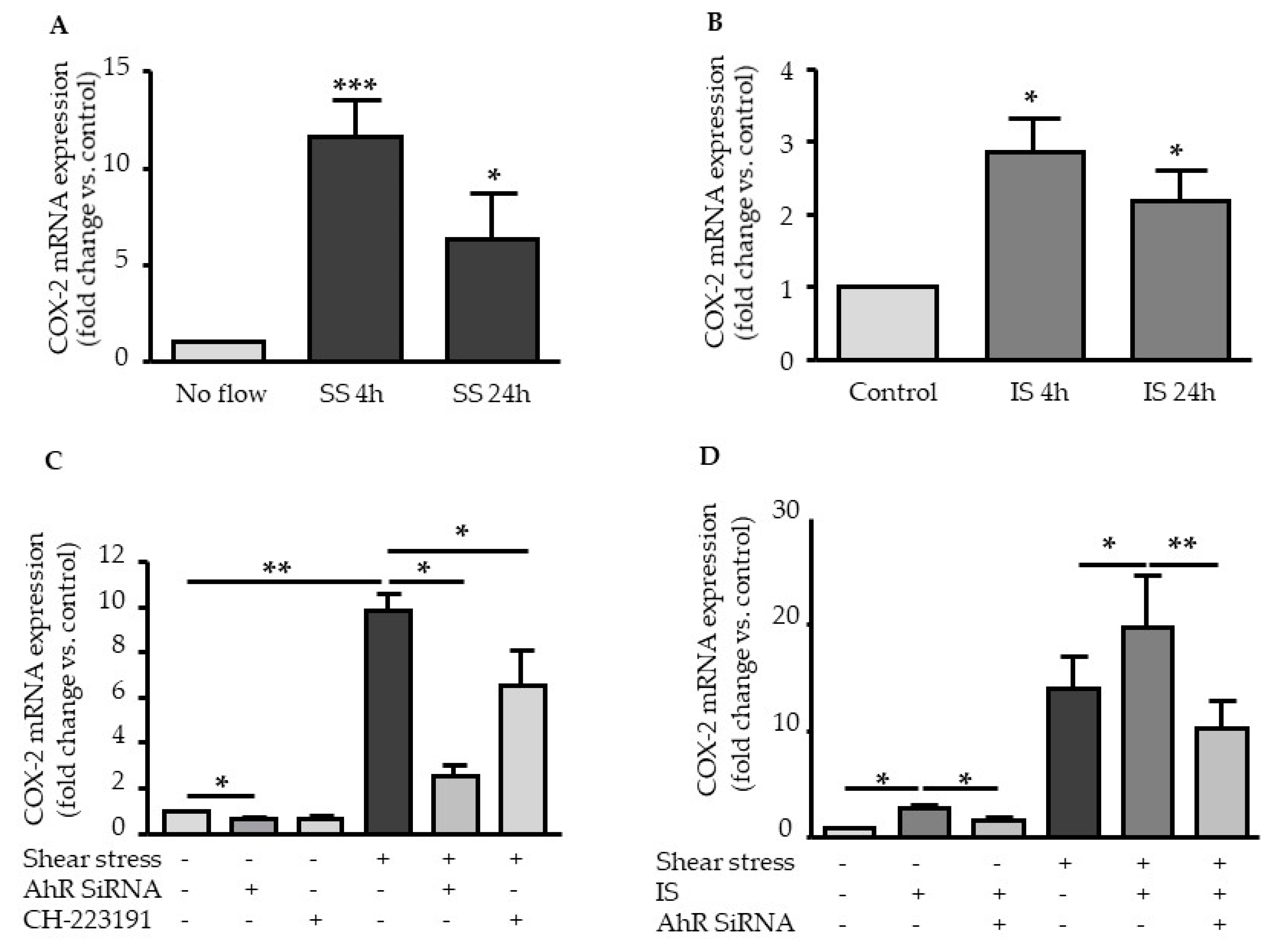
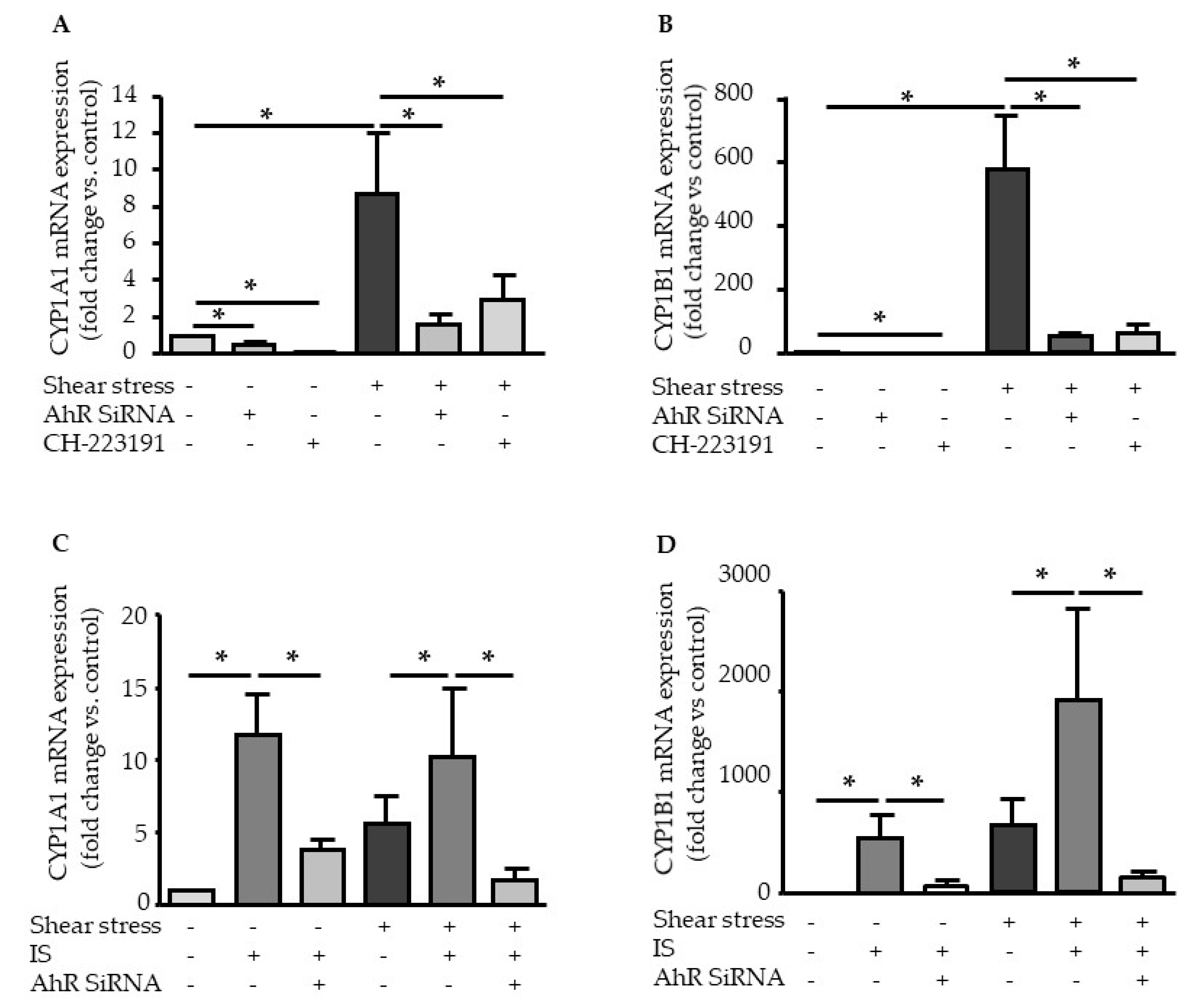
2.2. Shear Stress and IS Have AHR-Dependent Additive Effects on Upregulation of COX2, CYP1A1, and CYP1B1
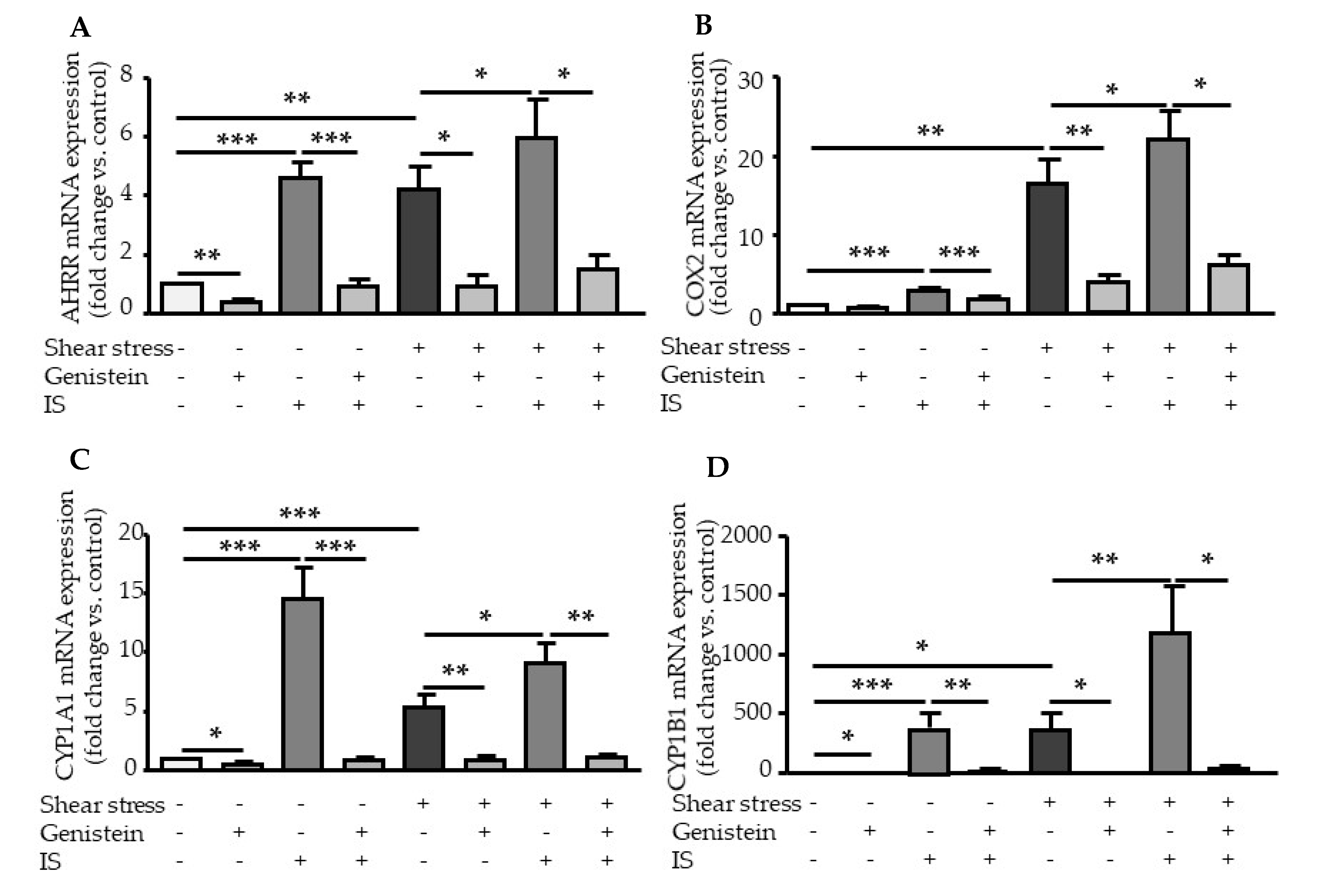
2.3. Genistein Suppressed Shear Stress- and IS-Mediated AHR Target Genes Activation
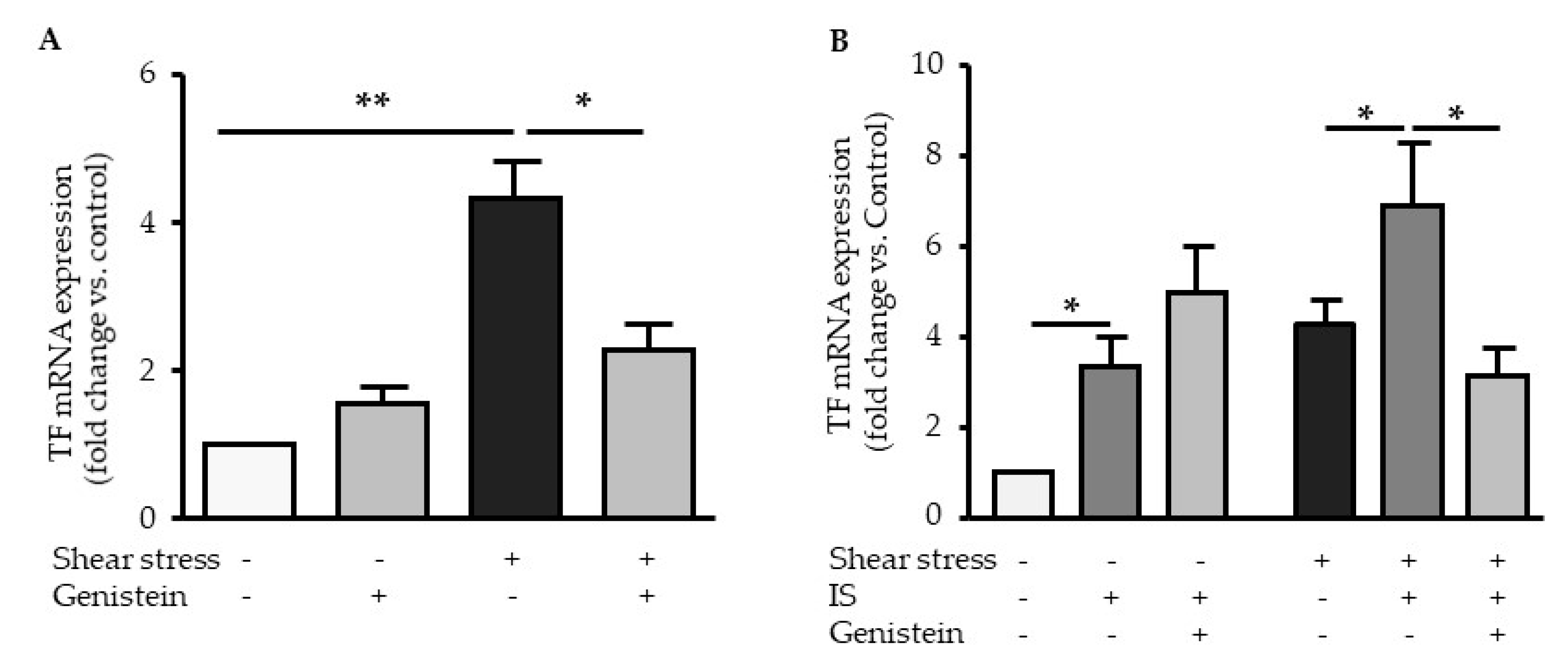
2.4. TF Upregulation by Shear Stress and IS is Dependent of AHR Activation
2.5. Genistein Suppressed Shear Stress-Mediated but not Indoxyl Sulfate-Mediated TF Expression
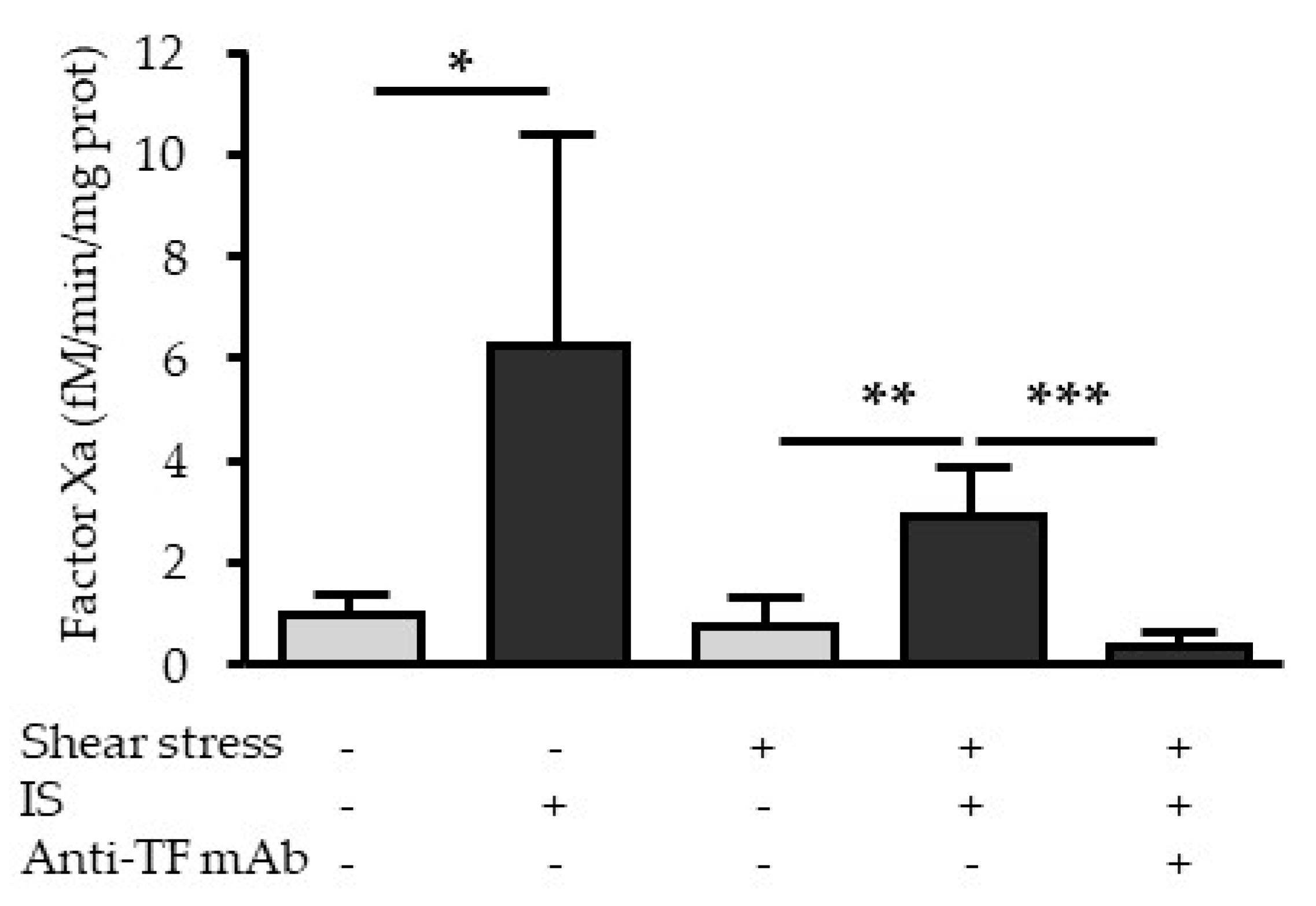
2.6. Indoxyl Sulfate Increases the Procoagulant Activity of TF under Fluid Shear Stress
3. Discussion
4. Materials and Methods
4.1. Endothelial Cell Culture
4.2. Incubation with the Uremic Toxin IS
4.3. SiRNA Knockdown of AHR
4.4. Flow System
4.5. RNA Extraction and Quantitative RT-PCR Analysis of mRNA Expression
4.6. Chromatin Immunoprecipitation (ChIP) Assay
4.7. Measurement of TF by Enzyme-Linked Immunosorbent Assay
4.8. Procoagulant Activity of TF
4.9. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIP | aryl hydrocarbon receptor interacting protein |
| AHR | aryl hydrocarbon receptor |
| AHRR | aryl hydrocarbon receptor repressor |
| ARNT | aryl hydrocarbon nuclear translocator |
| ChIP | chromatin immunoprecipitation |
| CKD | chronic kidney disease |
| COX2 | cyclooxygenase-2 |
| CYP1A1 | cytochrome P450 family 1 subfamily A member 1 |
| CYP1B1 | cytochrome P450 family 1 subfamily B member 1 |
| HSP90 | heat shock protein 90kDa |
| HUVEC | human umbilical vein endothelial cells |
| IAA | indole-3 acetic acid |
| IS | indoxyl sulfate |
| MAPK | mitogen-activated protein kinases |
| NF-κB | nuclear factor-kappa B |
| qPCR | quantitative polymerase chain reaction |
| RT | reverse transcription |
| TF | tissue factor |
| XRE | xenobiotic response element |
References
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins (Basel) 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.C.; Dinatale, B.C.; Murray, I.A.; Flaveny, C.A.; Liu, Q.; Laurenzana, E.M.; Lin, J.M.; Strom, S.C.; Omiecinski, C.J.; Amin, S.; et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins (Basel) 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Fujii-Kuriyama, Y.; Mimura, J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 2005, 338, 311–317. [Google Scholar] [CrossRef]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of constitutive and inducible AHR signaling: Complex interactions involving the AHR repressor. Biochem. Pharmacol. 2009, 77, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Nishimura, N.; Vogel, C.F.; Tohyama, C.; Matsumura, F. TCDD-induced cyclooxygenase-2 expression is mediated by the nongenomic pathway in mouse MMDD1 macula densa cells and kidneys. Biochem. Pharmacol. 2010, 79, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Degner, S.C.; Kemp, M.Q.; Hockings, J.K.; Romagnolo, D.F. Cyclooxygenase-2 promoter activation by the aromatic hydrocarbon receptor in breast cancer mcf-7 cells: repressive effects of conjugated linoleic acid. Nutr. Cancer 2007, 59, 248–257. [Google Scholar] [CrossRef]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2018, 93, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.E.; Sakurai, Y.; Weiss, D.; Vega, J.D.; Taylor, W.R.; Jo, H.; Eskin, S.G.; Marcus, C.B.; McIntire, L.V. Expression of CYP1A1 and CYP1B1 in human endothelial cells: regulation by fluid shear stress. Cardiovasc. Res. 2009, 81, 669–677. [Google Scholar] [CrossRef]
- Han, Z.; Miwa, Y.; Obikane, H.; Mitsumata, M.; Takahashi-Yanaga, F.; Morimoto, S.; Sasaguri, T. Aryl hydrocarbon receptor mediates laminar fluid shear stress-induced CYP1A1 activation and cell cycle arrest in vascular endothelial cells. Cardiovasc. Res. 2008, 77, 809–818. [Google Scholar] [CrossRef]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: how Kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Bock, K.W. Human AHR functions in vascular tissue: Pro- and anti-inflammatory responses of AHR agonists in atherosclerosis. Biochem. Pharmacol. 2019, 159, 116–120. [Google Scholar] [CrossRef]
- Kasai, S.; Kikuchi, H. The inhibitory mechanisms of the tyrosine kinase inhibitors herbimycin a, genistein, and tyrphostin B48 with regard to the function of the aryl hydrocarbon receptor in Caco-2 cells. Biosci. Biotechnol. Biochem. 2010, 74, 36–43. [Google Scholar] [CrossRef]
- McCormick, S.M.; Eskin, S.G.; McIntire, L.V.; Teng, C.L.; Lu, C.M.; Russell, C.G.; Chittur, K.K. DNA microarray reveals changes in gene expression of shear stressed human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. USA 2001, 98, 8955–8960. [Google Scholar] [CrossRef] [Green Version]
- Eskin, S.G.; Turner, N.A.; McIntire, L.V. Endothelial cell cytochrome P450 1A1 and 1B1: up-regulation by shear stress. Endothelium 2004, 11, 1–10. [Google Scholar] [CrossRef]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- McMillan, B.J.; Bradfield, C.A. The aryl hydrocarbon receptor is activated by modified low-density lipoprotein. Proc. Natl. Acad. Sci. USA 2007, 104, 1412–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Degroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Zelaya, H.; Rothmeier, A.S.; Ruf, W. Tissue factor at the crossroad of coagulation and cell signaling. J. Thromb. Haemost. 2018, 16, 1941–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.M.; Hogg, P.J. Allosteric disulfide bonds in thrombosis and thrombolysis. J. Thromb. Haemost. 2006, 4, 2533–2541. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 1973, 52, 2745–2756. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2392. https://doi.org/10.3390/ijms21072392
Lano G, Laforêt M, Von Kotze C, Perrin J, Addi T, Brunet P, Poitevin S, Burtey S, Dou L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. International Journal of Molecular Sciences. 2020; 21(7):2392. https://doi.org/10.3390/ijms21072392
Chicago/Turabian StyleLano, Guillaume, Manon Laforêt, Clarissa Von Kotze, Justine Perrin, Tawfik Addi, Philippe Brunet, Stéphane Poitevin, Stéphane Burtey, and Laetitia Dou. 2020. "Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells" International Journal of Molecular Sciences 21, no. 7: 2392. https://doi.org/10.3390/ijms21072392