The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy



Abstract
:
1. Introduction
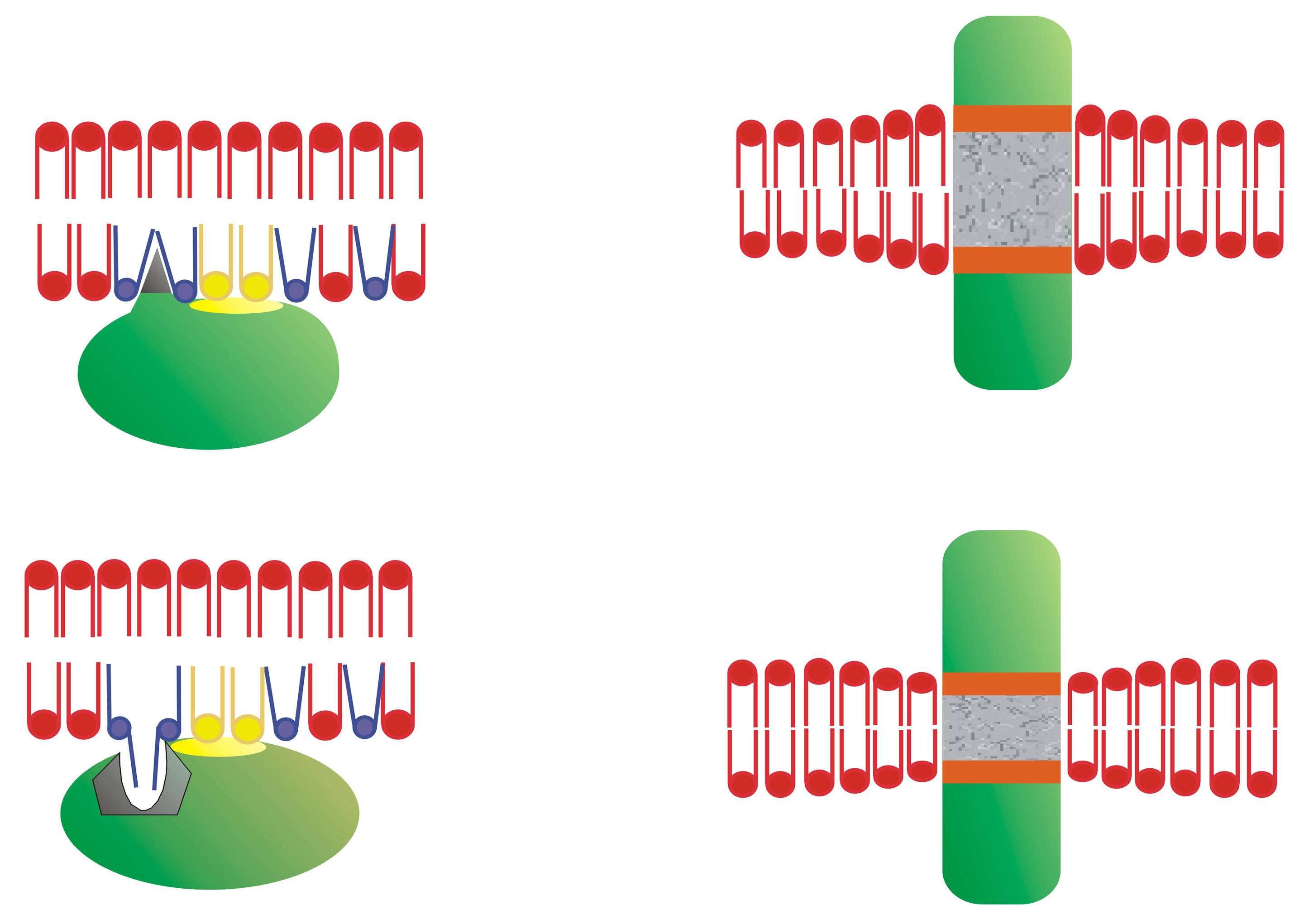
2. How Protein Structure Influences Protein–Lipid Interactions
3. How Membrane Lipid Structure Influences Protein–Lipid Interactions
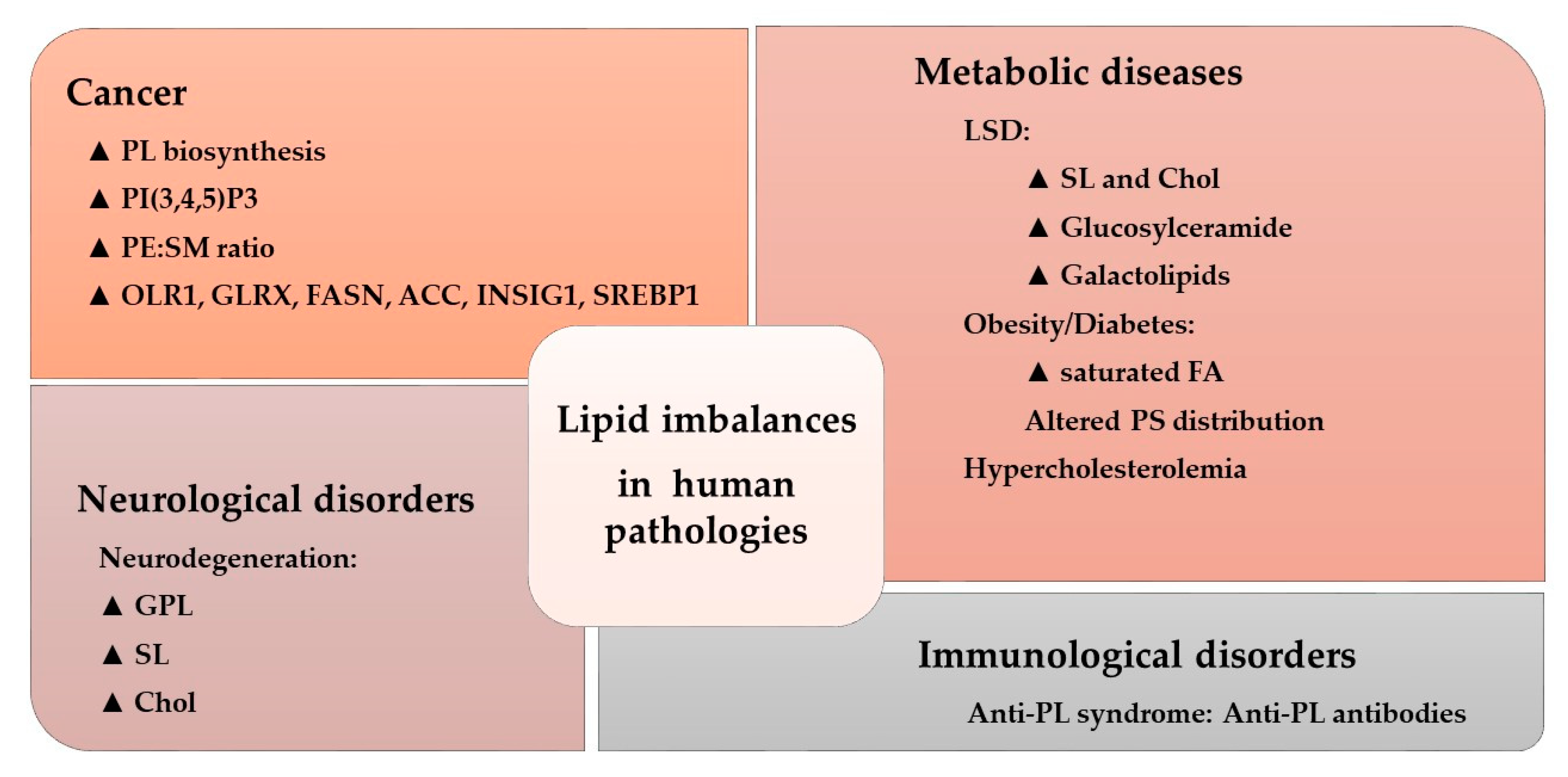
4. Altered Membrane Lipid and Amphitropic Protein Interactions in Human Diseases
5. Protein–Lipid Interactions in Cancer
6. Protein–Lipid Interactions in Neuroregeneration
7. Lipid–Protein Interactions in Diabetes
8. Protein–Lipid Interactions in Cardiovascular Diseases (CVDs)
9. Protein–Lipid Interactions in Infectious Diseases
10. Protein–Lipid Interactions and Cell Switches
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2OAA | 2-hydroxy arachidonic acid |
| 2OHOA | 2-hydroxyoleic acid |
| AA | Arachidonic acid |
| ACE | Angiotensin converting enzyme |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| BA | Benzyl alcohol |
| Chol | Cholesterol |
| CLPs | Covalent-lipid proteins |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| cPLA2 | Cytosolic phospholipase A2 |
| CVDs | Cardiovascular diseases |
| DAG | Diacylglycerol |
| DHA | Docosahexaenoic acid |
| EGF | Endothelial growth factor |
| eNOS | Nitric oxide synthase |
| ER | Endoplasmic reticulum |
| FABPs | Fatty acid binding proteins |
| FFAs | Free fatty acids |
| FTase | Farnesyl transferase |
| GCMs | Chol-rich microdomains |
| GFP | Green fluorescent protein |
| GFR | Growth factor receptor |
| GGTase | Geranylgeranyl transferase |
| GPCRs | G-protein coupled receptors |
| GPR40 | G-protein coupled receptor 40 |
| HDX-MS | Hydrogen–deuterium exchange mass spectrometry |
| HII | Non-lamellar-prone |
| HMG CoA | 3-hydroxy-3-methylglutaryl coenzyme A |
| HRP | Horseradish peroxidase |
| Hsp | Heat shock protein |
| HSR | Heat shock response |
| ICMT | Isoprenyl carboxyl methyltransferase |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| LBPs | Lipid-binding proteins |
| Ld | Liquid disordered |
| LDL-Chol | Low-density lipoprotein Chol |
| Lo | Liquid ordered |
| LOX | Lipoxygenase |
| LPA | Lysophosphatidic acid |
| LPC | lysoPC |
| LPE | lysoPE |
| LPL | Lipoprotein lipase |
| LPS | LysoPC |
| MBP | Myelin basic protein |
| MDA | Malondialdehyde |
| MFS | Major facilitator superfamily |
| MLT | Membrane-lipid therapy, melitherapy |
| MS | Multiple sclerosis |
| MS | Mass spectrometry |
| MUFAs | Monounsaturated fatty acids |
| NO | Nitric oxide |
| NPD1 | Neuroprotetin D1 |
| NSPCs | Neural Stem/Progenitor Cells |
| NVLT | Non-vesicular lipid transport |
| OA | Oleic acid |
| OSBP | Oxysterol binding protein |
| PC | Phosphatidylcholine |
| PD | Parkinson’s disease |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PE | Phosphatidylethanolamine |
| PH | Pleckstrin homology |
| PI3K | PI3 Kinase |
| PKC | Protein kinase C |
| PLA2 | Phospholipase A2 |
| PLCβ | Phospholipase Cβ |
| PPAR | Peroxisome proliferator-activated receptor |
| PS | Phosphatidylserine |
| PtdIns | Phosphoinositides |
| PTEN | Phosphatase and tensin homologue |
| RCE1 | Ras converting enzyme 1 |
| RTKs | Receptor tyrosine kinases |
| SAMs | Sterile alpha motifs |
| S–D | Sprague–Dawley |
| SFA | Saturated fatty acid |
| SHRs | Spontaneously hypertensive rats |
| SLC | Solute carrier |
| SM | Sphingomyelin |
| StAR | Steroidogenic acute regulatory protein |
| T2DM | Type-2 diabetes mellitus |
| TFEB | Transcription factor EB |
| TMS | Transmembrane segments |
| VEGF | Vascular endothelial growth factor |
References
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Gomeza, J.; Joly, C.; Kuhn, R.; Knöpfel, T.; Bockaert, J.; Pin, J.-P. The Second Intracellular Loop of Metabotropic Glutamate Receptor 1 Cooperates with the Other Intracellular Domains to Control Coupling to G-proteins. J. Biol. Chem. 1996, 271, 2199–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudermann, T.; Kalkbrenner, F.; Schultz, G. Diversity and Selectivity of Receptor-G Protein Interaction. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 429–459. [Google Scholar] [CrossRef]
- Chen, X.; Dai, J.-C.; Greenfield, E.M. Termination of immediate-early gene expression after stimulation by parathyroid hormone or isoproterenol. Am. J. Physiol. Physiol. 2002, 283, C1432–C1440. [Google Scholar] [CrossRef] [Green Version]
- Kleppisch, T.; Voigt, V.; Allmann, R.; Offermanns, S. G(alpha)q-deficient mice lack metabotropic glutamate receptor-dependent long-term depression but show normal long-term potentiation in the hippocampal CA1 region. J. Neurosci. 2001, 21, 4943–4948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayoub, M.A.; Al-Senaidy, A.; Pin, J.-P. Receptor-G Protein Interaction Studied by Bioluminescence Resonance Energy Transfer: Lessons from Protease-Activated Receptor 1. Front. Endocrinol. 2012, 3, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, R.; Tarnawski, A. Signal transduction cascades triggered by EGF receptor activation: Relevance to gastric injury repair and ulcer healing. Dig. Dis. Sci. 1998, 43, 14S–22S. [Google Scholar]
- Escriba, P.V.; Sastre, M.; Garcia-Sevilla, J.A. Disruption of cellular signaling pathways by daunomycin through destabilization of nonlamellar membrane structures. Proc. Natl. Acad. Sci. USA 1995, 92, 7595–7599. [Google Scholar] [CrossRef] [Green Version]
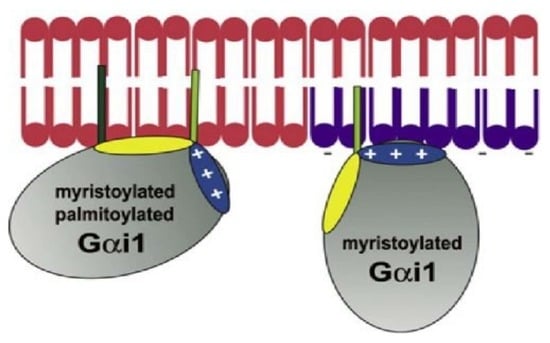
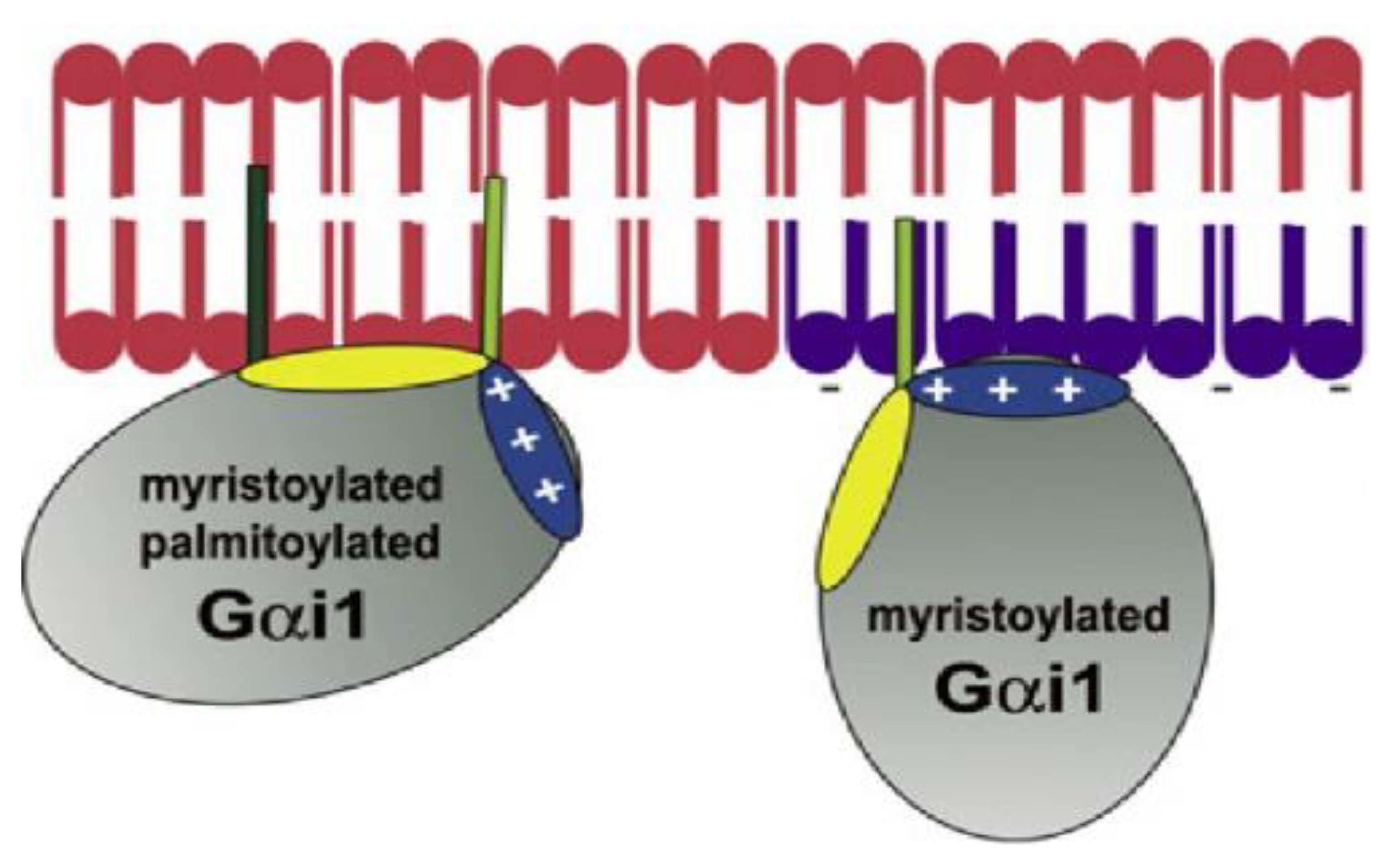
- Álvarez, R.; López, D.J.; Casas, J.; Lladó, V.; Higuera, M.; Nagy, T.; Barceló, M.; Busquets, X.; Escribá, P.V. G protein-membrane interactions I: Gαi1 myristoyl and palmitoyl modifications in protein-lipid interactions and its implications in membrane microdomain localization. Biochim. Biophys. Acta 2015, 1851, 1511–1520. [Google Scholar] [CrossRef]
- Seifert, R.; Wenzel-Seifert, K. Constitutive activity of G-protein-coupled receptors: Cause of disease and common property of wild-type receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 2002, 366, 381–416. [Google Scholar] [CrossRef]
- Alemany, R.; Terés, S.; Baamonde, C.; Benet, M.; Vögler, O.; Escribá, P.V. 2-Hydroxyoleic Acid. Hypertension 2004, 43, 249–254. [Google Scholar] [CrossRef]
- Escriba, P.V.; Ferrer-Montiel, A.V.; Ferragut, J.A.; Gonzalez-Ros, J.M. Role of membrane lipids in the interaction of daunomycin with plasma membranes from tumor cells: Implications in drug-resistance phenomena. Biochemistry 1990, 29, 7275–7282. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Martin, M.L.; de Almeida, R.F.M.; Noguera-Salva, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Luth, A.; Kleuser, B.; Halver, J.E.; Escriba, P.V. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.; Vögler, O.; Casas, J.; Barceló, F.; Alemany, R.; Prades, J.; Nagy, T.; Baamonde, C.; Kasprzyk, P.G.; Terés, S.; et al. Membrane structure modulation, protein kinase C alpha activation, and anticancer activity of minerval. Mol. Pharmacol. 2005, 67, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.; Price, S.L.; Fiol-Deroque, M.A.; Marcilla-Etxenike, A.; Ahyayauch, H.; Barceló-Coblijn, G.; Terés, S.; Katsouri, L.; Ordinas, M.; López, D.J.; et al. Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1838, 1680–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teres, S.; Barcelo-Coblijn, G.; Benet, M.; Alvarez, R.; Bressani, R.; Halver, J.E.; Escriba, P.V. Oleic acid content is responsible for the reduction in blood pressure induced by olive oil. Proc. Natl. Acad. Sci. USA 2008, 105, 13811–13816. [Google Scholar] [CrossRef] [Green Version]
- Lopez, D.H.; Fiol-deRoque, M.A.; Noguera-Salvà, M.A.; Terés, S.; Campana, F.; Piotto, S.; Castro, J.A.; Mohaibes, R.J.; Escribá, P.V.; Busquets, X. 2-hydroxy arachidonic acid: A new non-steroidal anti-inflammatory drug. PLoS ONE 2013, 8, e72052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvius, J.R. Cholesterol modulation of lipid intermixing in phospholipid and glycosphingolipid mixtures. Evaluation using fluorescent lipid probes and brominated lipid quenchers. Biochemistry 1992, 31, 3398–3408. [Google Scholar] [CrossRef] [PubMed]
- Mabrey, S.; Mateo, P.L.; Sturtevant, J.M. High-sensitivity scanning calorimetric study of mixtures of cholesterol with dimyristoyl- and dipalmitoylphosphatidylcholines. Biochemistry 1978, 17, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V. Membrane-lipid therapy: A new approach in molecular medicine. Trends Mol. Med. 2006, 12, 34–43. [Google Scholar] [CrossRef]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goñi, F.M. “Rafts”: A nickname for putative transient nanodomains. Chem. Phys. Lipids 2019, 218, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Vögler, O.; Casas, J.; Capó, D.; Nagy, T.; Borchert, G.; Martorell, G.; Escribá, P.V. The Gbetagamma dimer drives the interaction of heterotrimeric Gi proteins with nonlamellar membrane structures. J. Biol. Chem. 2004, 279, 36540–36545. [Google Scholar] [CrossRef] [Green Version]
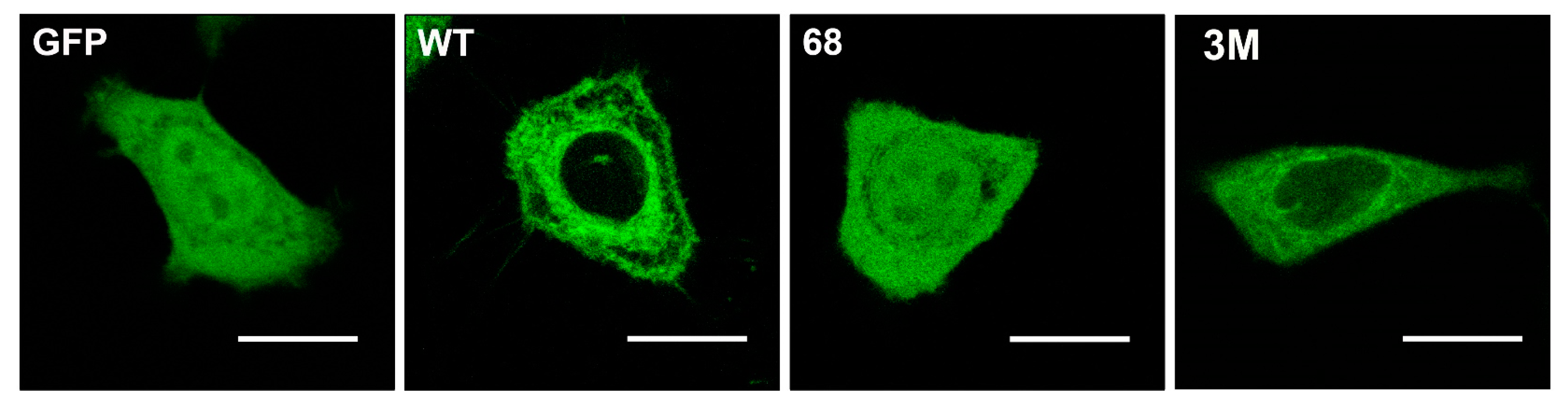
- Noguera-Salvà, M.A.; Guardiola-Serrano, F.; Martin, M.L.; Marcilla-Etxenike, A.; Bergo, M.O.; Busquets, X.; Escribá, P.V. Role of the C-terminal basic amino acids and the lipid anchor of the Gγ2 protein in membrane interactions and cell localization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V.; Sánchez-Dominguez, J.M.; Alemany, R.; Perona, J.S.; Ruiz-Gutiérrez, V. Alteration of Lipids, G Proteins, and PKC in Cell Membranes of Elderly Hypertensives. Hypertension 2003, 41, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, R.; Casas, J.; López, D.J.; Ibarguren, M.; Suari-Rivera, A.; Terés, S.; Guardiola-Serrano, F.; Lossos, A.; Busquets, X.; Kakhlon, O.; et al. Triacylglycerol mimetics regulate membrane interactions of glycogen branching enzyme: Implications for therapy. J. Lipid Res. 2017, 58, 1598–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
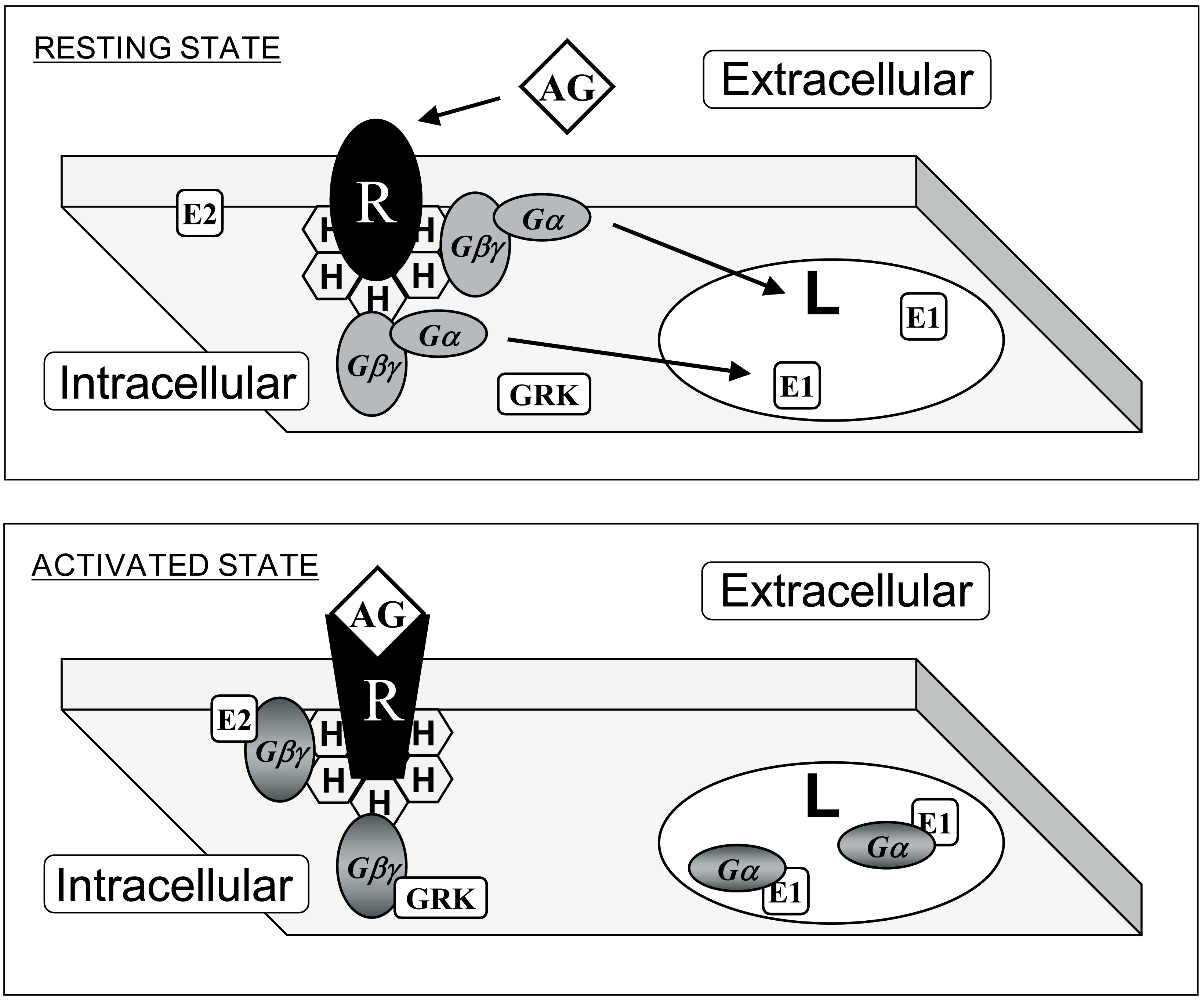
- Escribá, P.V.; Wedegaertner, P.B.; Goñi, F.M.; Vögler, O. Lipid-protein interactions in GPCR-associated signaling. Biochim. Biophys. Acta 2007, 1768, 836–852. [Google Scholar] [CrossRef] [Green Version]
- Mystek, P.; Rysiewicz, B.; Gregrowicz, J.; Dziedzicka-Wasylewska, M.; Polit, A. Gγ and Gα Identity Dictate a G-Protein Heterotrimer Plasma Membrane Targeting. Cells 2019, 8, 1246. [Google Scholar] [CrossRef] [Green Version]
- Casas, J.; Ibarguren, M.; Álvarez, R.; Terés, S.; Lladó, V.; Piotto, S.P.; Concilio, S.; Busquets, X.; López, D.J.; Escribá, P.V. G protein-membrane interactions II: Effect of G protein-linked lipids on membrane structure and G protein-membrane interactions. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1526–1535. [Google Scholar] [CrossRef]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.M.; Syed, S.A.; Zaki, O.; Bottanelli, F.; Zheng, H.; Hacke, M.; Xi, Z.; Rivera-Molina, F.; Graham, M.; Rebane, A.A.; et al. S-Palmitoylation Sorts Membrane Cargo for Anterograde Transport in the Golgi. Dev. Cell 2018, 47, 479–493.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, I.; Lingwood, D.; Grzybek, M.; Coskun, U.; Simons, K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 22050–22054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponimaskin, E.; Dumuis, A.; Gaven, F.; Barthet, G.; Heine, M.; Glebov, K.; Richter, D.W.; Oppermann, M. Palmitoylation of the 5-hydroxytryptamine4a receptor regulates receptor phosphorylation, desensitization, and beta-arrestin-mediated endocytosis. Mol. Pharmacol. 2005, 67, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.E.; Middleton, P.; Hepler, J.R.; Taussig, R.; Gilman, A.G.; Mumby, S.M. Lipid modifications of G proteins: Alpha subunits are palmitoylated. Proc. Natl. Acad. Sci. USA 1993, 90, 3675–3679. [Google Scholar] [CrossRef] [Green Version]
- Wedegaertner, P.B. Lipid modifications and membrane targeting of G alpha. Biol. Signals Recept. 1998, 7, 125–135. [Google Scholar] [CrossRef]
- Wedegaertner, P. Reversible Palmitoylation in G-Protein Signaling. In Handbook of Cell Signaling; Elsevier: Amsterdam, The Netherlands, 2003; pp. 651–656. [Google Scholar]
- Escribá, P.V.; Ozaita, A.; Ribas, C.; Miralles, A.; Fodor, E.; Farkas, T.; García-Sevilla, J.A. Role of lipid polymorphism in G protein-membrane interactions: Nonlamellar-prone phospholipids and peripheral protein binding to membranes. Proc. Natl. Acad. Sci. USA 1997, 94, 11375–11380. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, R.; Heck, M.; Henklein, P.; Henklein, P.; Kleuss, C.; Hofmann, K.P.; Ernst, O.P. Sequence of interactions in receptor-G protein coupling. J. Biol. Chem. 2004, 279, 24283–24290. [Google Scholar] [CrossRef]
- Hermans, E. Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol. Ther. 2003, 99, 25–44. [Google Scholar] [CrossRef]
- Lladó, V.; Terés, S.; Higuera, M.; Alvarez, R.; Noguera-Salva, M.A.; Halver, J.E.; Escribá, P.V.; Busquets, X. Pivotal role of dihydrofolate reductase knockdown in the anticancer activity of 2-hydroxyoleic acid. Proc. Natl. Acad. Sci. USA 2009, 106, 13754–13758. [Google Scholar] [CrossRef] [Green Version]
- Moores, S.L.; Schaber, M.D.; Mosser, S.D.; Rands, E.; O’Hara, M.B.; Garsky, V.M.; Marshall, M.S.; Pompliano, D.L.; Gibbs, J.B. Sequence dependence of protein isoprenylation. J. Biol. Chem. 1991, 266, 14603–14610. [Google Scholar]
- Baron, R.; Fourcade, E.; Lajoie-Mazenc, I.; Allal, C.; Couderc, B.; Barbaras, R.; Favre, G.; Faye, J.C.; Pradines, A. RhoB prenylation is driven by the three carboxyl-terminal amino acids of the protein: Evidenced in vivo by an anti-farnesyl cysteine antibody. Proc. Natl. Acad. Sci. USA 2000, 97, 11626–11631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, Y.; Stradley, S.J.; Gierasch, L.M.; Brown, M.S.; Goldstein, J.L. Sequence requirement for peptide recognition by rat brain p21ras protein farnesyltransferase. Proc. Natl. Acad. Sci. USA 1991, 88, 732–736. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, W.K.; Tam, A.; Fujimura-Kamada, K.; Michaelis, S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 11175–11180. [Google Scholar] [CrossRef] [Green Version]
- Boyartchuk, V.L.; Ashby, M.N.; Rine, J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 1997, 275, 1796–1800. [Google Scholar] [CrossRef]
- Clarke, S.; Vogel, J.P.; Deschenes, R.J.; Stock, J. Posttranslational modification of the Ha-ras oncogene protein: Evidence for a third class of protein carboxyl methyltransferases. Proc. Natl. Acad. Sci. USA 1988, 85, 4643–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergo, M.O.; Leung, G.K.; Ambroziak, P.; Otto, J.C.; Casey, P.J.; Young, S.G. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 2000, 275, 17605–17610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineo, C.; James, G.L.; Smart, E.J.; Anderson, R.G. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J. Biol. Chem. 1996, 271, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Barceló, F.; Prades, J.; Encinar, J.A.; Funari, S.S.; Vögler, O.; González-Ros, J.M.; Escribá, P.V. Interaction of the C-terminal region of the Ggamma protein with model membranes. Biophys. J. 2007, 93, 2530–2541. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hancock, J.F. A novel prenyl-polybasic domain code determines lipid-binding specificity of the K-Ras membrane anchor. Small GTPases 2018, 1–5. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Marenduzzo, D.; Goryachev, A.B. Lateral Dynamics of Proteins with Polybasic Domain on Anionic Membranes: A Dynamic Monte-Carlo Study. Biophys. J. 2011, 100, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deatherage, C.L.; Lu, Z.; Kroncke, B.M.; Ma, S.; Smith, J.A.; Voehler, M.W.; McFeeters, R.L.; Sanders, C.R. Structural and biochemical differences between the Notch and the amyloid precursor protein transmembrane domains. Sci. Adv. 2017, 3, e1602794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Hou, Z.; Matherly, L.H. Substituted cysteine accessibility reveals a novel transmembrane 2-3 reentrant loop and functional role for transmembrane domain 2 in the human proton-coupled folate transporter. J. Biol. Chem. 2014, 289, 25287–25295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoflack, J.; Hibert, M.F.; Trumpp-Kallmeyer, S.; Bidart, J.M. Three-dimensional models of gonado-thyrotropin hormone receptor transmembrane domain. Drug Des. Discov. 1993, 10, 157–171. [Google Scholar]
- Lou, H.; Chen, M.; Black, S.S.; Bushell, S.R.; Ceccarelli, M.; Mach, T.; Beis, K.; Low, A.S.; Bamford, V.A.; Booth, I.R.; et al. Altered antibiotic transport in OmpC mutants isolated from a series of clinical strains of multi-drug resistant E. coli. PLoS ONE 2011, 6, e25825. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Newton, A.C.; Parker, P.J.; Blumberg, P.M.; Nishizuka, Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997, 6, 477–480. [Google Scholar] [CrossRef] [Green Version]
- Corbalán-García, S.; Gómez-Fernández, J.C. Protein kinase C regulatory domains: The art of decoding many different signals in membranes. Biochim. Biophys. Acta 2006, 1761, 633–654. [Google Scholar] [CrossRef]
- Corbalán-García, S.; Gómez-Fernández, J.C. Classical protein kinases C are regulated by concerted interaction with lipids: The importance of phosphatidylinositol-4,5-bisphosphate. Biophys. Rev. 2014, 6, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Boije af Gennäs, G.; Talman, V.; Yli-Kauhaluoma, J.; Tuominen, R.K.; Ekokoski, E. Current status and future prospects of C1 domain ligands as drug candidates. Curr. Top. Med. Chem. 2011, 11, 1370–1392. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Watanabe, M.; Quinn, P.J.; Kato, S.; Murayama, S.; Ohki, K.; Hatta, I. Effects of diacylglycerol on the structure and phase behaviour of non-bilayer forming phospholipid. Biophys. Chem. 1999, 77, 37–48. [Google Scholar] [CrossRef]
- Hofmann, J. Protein kinase C isozymes as potential targets for anticancer therapy. Curr. Cancer Drug Targets 2004, 4, 125–146. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Alkon, D.L.; Sun, M.-K.; Nelson, T.J. PKC signaling deficits: A mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007, 28, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Churchill, E.; Budas, G.; Vallentin, A.; Koyanagi, T.; Mochly-Rosen, D. PKC isozymes in chronic cardiac disease: Possible therapeutic targets? Annu. Rev. Pharmacol. Toxicol. 2008, 48, 569–599. [Google Scholar] [CrossRef] [PubMed]
- Baier, G.; Wagner, J. PKC inhibitors: Potential in T cell-dependent immune diseases. Curr. Opin. Cell Biol. 2009, 21, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, P.M.; Kedei, N.; Lewin, N.E.; Yang, D.; Czifra, G.; Pu, Y.; Peach, M.L.; Marquez, V.E. Wealth of opportunity—The C1 domain as a target for drug development. Curr. Drug Targets 2008, 9, 641–652. [Google Scholar] [CrossRef] [Green Version]
- Kohout, S.C.; Corbalán-García, S.; Gómez-Fernández, J.C.; Falke, J.J. C2 domain of protein kinase C alpha: Elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry 2003, 42, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Verdaguer, N.; Corbalan-Garcia, S.; Ochoa, W.F.; Fita, I.; Gómez-Fernández, J.C. Ca(2+) bridges the C2 membrane-binding domain of protein kinase Calpha directly to phosphatidylserine. EMBO J. 1999, 18, 6329–6338. [Google Scholar] [CrossRef] [Green Version]
- Sutton, R.B.; Davletov, B.A.; Berghuis, A.M.; Südhof, T.C.; Sprang, S.R. Structure of the first C2 domain of synaptotagmin I: A novel Ca2+/phospholipid-binding fold. Cell 1995, 80, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Essen, L.O.; Perisic, O.; Cheung, R.; Katan, M.; Williams, R.L. Crystal structure of a mammalian phosphoinositide-specific phospholipase C delta. Nature 1996, 380, 595–602. [Google Scholar] [CrossRef]
- Perisic, O.; Fong, S.; Lynch, D.E.; Bycroft, M.; Williams, R.L. Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 1998, 273, 1596–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, W.F.; Corbalán-Garcia, S.; Eritja, R.; Rodríguez-Alfaro, J.A.; Gómez-Fernández, J.C.; Fita, I.; Verdaguer, N. Additional binding sites for anionic phospholipids and calcium ions in the crystal structures of complexes of the C2 domain of protein kinase calpha. J. Mol. Biol. 2002, 320, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Corbalan-Garcia, S.; Gómez-Fernández, J.C. Signaling through C2 domains: More than one lipid target. Biochim. Biophys. Acta 2014, 1838, 1536–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widder, K.; Harauz, G.; Hinderberger, D. Myelin basic protein (MBP) charge variants show different sphingomyelin-mediated interactions with myelin-like lipid monolayers. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183077. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.; Hossain, K.R.; Cornelius, F.; Clarke, R.J. Penetration of phospholipid membranes by poly-l-lysine depends on cholesterol and phospholipid composition. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183128. [Google Scholar] [CrossRef] [PubMed]
- Koprivanacz, K.; Tőke, O.; Besztercei, B.; Juhász, T.; Radnai, L.; Merő, B.; Mihály, J.; Péter, M.; Balogh, G.; Vígh, L.; et al. The SH3 domain of Caskin1 binds to lysophosphatidic acid suggesting a direct role for the lipid in intracellular signaling. Cell. Signal. 2017, 32, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kim, B.-G.; Kim, S.; Lee, Y.; Yoon, Y. Inhibitory potential of flavonoids on PtdIns(3,4,5)P3 binding with the phosphoinositide-dependent kinase 1 pleckstrin homology domain. Bioorg. Med. Chem. Lett. 2017, 27, 420–426. [Google Scholar] [CrossRef]
- Barrera, F.N.; Poveda, J.A.; González-Ros, J.M.; Neira, J.L. Binding of the C-terminal sterile alpha motif (SAM) domain of human p73 to lipid membranes. J. Biol. Chem. 2003, 278, 46878–46885. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [Green Version]
- Triton, T.R.; Yee, G. The anticancer agent adriamycin can be actively cytotoxic without entering cells. Science 1982, 217, 248–250. [Google Scholar] [CrossRef]
- Buda, C.; Dey, I.; Balogh, N.; Horvath, L.I.; Maderspach, K.; Juhasz, M.; Yeo, Y.K.; Farkas, T. Structural order of membranes and composition of phospholipids in fish brain cells during thermal acclimatization. Proc. Natl. Acad. Sci. USA 1994, 91, 8234–8238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, T.; Kitajka, K.; Fodor, E.; Csengeri, I.; Lahdes, E.; Yeo, Y.K.; Krasznai, Z.; Halver, J.E. Docosahexaenoic acid-containing phospholipid molecular species in brains of vertebrates. Proc. Natl. Acad. Sci. USA 2000, 97, 6362–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, M.W.; Gruner, S.M. Lipid polymorphism of mixtures of dioleoylphosphatidylethanolamine and saturated and monounsaturated phosphatidylcholines of various chain lengths. Biochemistry 1987, 26, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Ibarguren, M.; López, D.J.; Encinar, J.A.; González-Ros, J.M.; Busquets, X.; Escribá, P.V. Partitioning of liquid-ordered/liquid-disordered membrane microdomains induced by the fluidifying effect of 2-hydroxylated fatty acid derivatives. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2553–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canerina-Amaro, A.; Hernandez-Abad, L.G.; Ferrer, I.; Quinto-Alemany, D.; Mesa-Herrera, F.; Ferri, C.; Puertas-Avendano, R.A.; Diaz, M.; Marin, R. Lipid raft ER signalosome malfunctions in menopause and Alzheimer’s disease. Front. Biosci. 2017, 9, 111–126. [Google Scholar]
- Yerly, S.; Ding, H.; Tauzin, S.; van Echten-Deckert, G.; Borisch, B.; Hoessli, D.C. The sphingolipid-rich rafts of ALK+ lymphomas downregulate the Lyn-Cbp/PAG signalosome. Eur. J. Haematol. 2010, 85, 93–98. [Google Scholar] [CrossRef]
- Terés, S.; Lladó, V.; Higuera, M.; Barceló-Coblijn, G.; Martin, M.L.; Noguera-Salvà, M.A.; Marcilla-Etxenike, A.; García-Verdugo, J.M.; Soriano-Navarro, M.; Saus, C.; et al. 2-Hydroxyoleate, a nontoxic membrane binding anticancer drug, induces glioma cell differentiation and autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 8489–8494. [Google Scholar] [CrossRef] [Green Version]
- Hernando, S.; Requejo, C.; Herran, E.; Ruiz-Ortega, J.A.; Morera-Herreras, T.; Lafuente, J.V.; Ugedo, L.; Gainza, E.; Pedraz, J.L.; Igartua, M.; et al. Beneficial effects of n-3 polyunsaturated fatty acids administration in a partial lesion model of Parkinson’s disease: The role of glia and NRf2 regulation. Neurobiol. Dis. 2019, 121, 252–262. [Google Scholar] [CrossRef]
- Feng, L.; Liao, W.-X.; Luo, Q.; Zhang, H.-H.; Wang, W.; Zheng, J.; Chen, D.-B. Caveolin-1 orchestrates fibroblast growth factor 2 signaling control of angiogenesis in placental artery endothelial cell caveolae. J. Cell. Physiol. 2012, 227, 2480–2491. [Google Scholar] [CrossRef] [Green Version]
- Quinn, P.J.; Chapman, D. The dynamics of membrane structure. CRC Crit. Rev. Biochem. 1980, 8, 1–117. [Google Scholar] [CrossRef]
- Bozelli, J.C.; Jennings, W.; Black, S.; Hou, Y.H.; Lameire, D.; Chatha, P.; Kimura, T.; Berno, B.; Khondker, A.; Rheinstädter, M.C.; et al. Membrane curvature allosterically regulates the phosphatidylinositol cycle, controlling its rate and acyl-chain composition of its lipid intermediates. J. Biol. Chem. 2018, 293, 17780–17791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozelli, J.C.; Epand, R.M. Role of membrane shape in regulating the phosphatidylinositol cycle at contact sites. Chem. Phys. Lipids 2019, 221, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Shin, O.-H. Exocytosis and synaptic vesicle function. Compr. Physiol. 2014, 4, 149–175. [Google Scholar] [PubMed]
- Yeagle, P.L. Lipid regulation of cell membrane structure and function. FASEB J. 1989, 3, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Goñi, F.M.; Alonso, A. Structure and functional properties of diacylglycerols in membranes. Prog. Lipid Res. 1999, 38, 1–48. [Google Scholar] [CrossRef]
- Marrink, S.-J.; Mark, A.E. Molecular view of hexagonal phase formation in phospholipid membranes. Biophys. J. 2004, 87, 3894–3900. [Google Scholar] [CrossRef] [Green Version]
- Escribá, P. V Membrane-lipid therapy: A historical perspective of membrane-targeted therapies - From lipid bilayer structure to the pathophysiological regulation of cells. Biochim. Biophys. acta. Biomembr. 2017, 1859, 1493–1506. [Google Scholar] [CrossRef]
- Bretscher, M.S. Asymmetrical lipid bilayer structure for biological membranes. Nat. New Biol. 1972, 236, 11–12. [Google Scholar] [CrossRef]
- García-Segura, L.M.; Ferragut, J.A.; Ferrer-Montiel, A.V.; Escriba, P.V.; Gonzalez-Ros, J.M. Ultrastructural alterations in plasma membranes from drug-resistant P388 murine leukemia cells. Biochim. Biophys. Acta 1990, 1029, 191–195. [Google Scholar] [CrossRef]
- Bozelli, J.C.; Hou, Y.H.; Schreier, S.; Epand, R.M. Lipid asymmetry of a model mitochondrial outer membrane affects Bax-dependent permeabilization. Biochim. Biophys. Acta - Biomembr. 2020, 183241. [Google Scholar] [CrossRef]
- Grassmé, H.; Henry, B.; Ziobro, R.; Becker, K.A.; Riethmüller, J.; Gardner, A.; Seitz, A.P.; Steinmann, J.; Lang, S.; Ward, C.; et al. β1-Integrin Accumulates in Cystic Fibrosis Luminal Airway Epithelial Membranes and Decreases Sphingosine, Promoting Bacterial Infections. Cell Host Microbe 2017, 21, 707–718.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunasekara, L.; Al-Saiedy, M.; Green, F.; Pratt, R.; Bjornson, C.; Yang, A.; Michael Schoel, W.; Mitchell, I.; Brindle, M.; Montgomery, M.; et al. Pulmonary surfactant dysfunction in pediatric cystic fibrosis: Mechanisms and reversal with a lipid-sequestering drug. J. Cyst. Fibros. 2017, 16, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, S.D.; Katz, M.H.; Parker, E.M.; Laposata, M.; Urman, M.Y.; Alvarez, J.G. A membrane lipid imbalance plays a role in the phenotypic expression of cystic fibrosis in cftr−/− mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13995–14000. [Google Scholar] [CrossRef] [Green Version]
- Ojo, J.O.; Algamal, M.; Leary, P.; Abdullah, L.; Mouzon, B.; Evans, J.E.; Mullan, M.; Crawford, F. Disruption in Brain Phospholipid Content in a Humanized Tau Transgenic Model Following Repetitive Mild Traumatic Brain Injury. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-P. The mechanism of protein kinase C activation. Trends Neurosci. 1989, 12, 425–432. [Google Scholar] [CrossRef]
- Oancea, E.; Meyer, T. Protein Kinase C as a Molecular Machine for Decoding Calcium and Diacylglycerol Signals. Cell 1998, 95, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef]
- Gilio, K.; Harper, M.T.; Cosemans, J.M.E.M.; Konopatskaya, O.; Munnix, I.C.A.; Prinzen, L.; Leitges, M.; Liu, Q.; Molkentin, J.D.; Heemskerk, J.W.M.; et al. Functional Divergence of Platelet Protein Kinase C (PKC) Isoforms in Thrombus Formation on Collagen. J. Biol. Chem. 2010, 285, 23410–23419. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Subbulakshmi, V.; Fields, A.P.; Murray, N.R.; Cathcart, M.K. Protein Kinase Cα Regulates Human Monocyte O·−2 Production and Low Density Lipoprotein Lipid Oxidation. J. Biol. Chem. 1999, 274, 3764–3771. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.-H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of PKC and PKC. Proc. Natl. Acad. Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosicek, M.; Hecimovic, S. Phospholipids and Alzheimer’s Disease: Alterations, Mechanisms and Potential Biomarkers. Int. J. Mol. Sci. 2013, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.K.; Nelson, T.J.; Verma, V.A.; Wender, P.A.; Alkon, D.L. A cellular model of Alzheimer’s disease therapeutic efficacy: PKC activation reverses Aβ-induced biomarker abnormality on cultured fibroblasts. Neurobiol. Dis. 2009, 34, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busquets, X.; Escriba, P.V.; Sastre, M.; García-Sevilla, J.A. Loss of protein kinase C-alpha beta in brain of heroin addicts and morphine-dependent rats. J. Neurochem. 1995, 64, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattan, W.E.; Chen, W.; Ma, X.; Lan, T.H.; van der Hoeven, D.; van der Hoeven, R.; Hancock, J.F. Targeting plasma membrane phosphatidylserine content to inhibit oncogenic KRAS function. Life Sci. Alliance 2019, 2, e201900431. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Swarthout, J.T.; Lobo, S.; Farh, L.; Croke, M.R.; Greentree, W.K.; Deschenes, R.J.; Linder, M.E. DHHC9 and GCP16 Constitute a Human Protein Fatty Acyltransferase with Specificity for H- and N-Ras. J. Biol. Chem. 2005, 280, 31141–31148. [Google Scholar] [CrossRef] [Green Version]
- Bleijlevens, B.; van Breemen, M.J.; Donker-Koopman, W.E.; de Koster, C.G.; Aerts, J.M.F.G. Detection of mutant protein in complex biological samples: Glucocerebrosidase mutations in Gaucher’s disease. Anal. Biochem. 2008, 372, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta Rev. Cancer 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Koval, A.; Katanaev, V.L. Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci. Rep. 2018, 8, 7329. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Garzón, V.; Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Tompa, M.; Kalovits, F.; Nagy, A.; Kalman, B. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. NeuroMol. Med. 2018, 20, 437–451. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, Z.; Tang, Y.; Chen, X.; Zhou, J. Identification of potential target genes in pancreatic ductal adenocarcinoma by bioinformatics analysis. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.; Kir, D.; Banerjee, S.; Saluja, A. Control of Apoptosis in Treatment and Biology of Pancreatic Cancer. J. Cell. Biochem. 2016, 117, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Sun, Y.; Niu, J.; Jarugumilli, G.K.; Wu, X. Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Suwala, A.K.; Hanaford, A.; Kahlert, U.D.; Maciaczyk, J. Clipping the Wings of Glioblastoma: Modulation of WNT as a Novel Therapeutic Strategy. J. Neuropathol. Exp. Neurol. 2016, 75, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Doubravska, L.; Krausova, M.; Gradl, D.; Vojtechova, M.; Tumova, L.; Lukas, J.; Valenta, T.; Pospichalova, V.; Fafilek, B.; Plachy, J.; et al. Fatty acid modification of Wnt1 and Wnt3a at serine is prerequisite for lipidation at cysteine and is essential for Wnt signalling. Cell. Signal. 2011, 23, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Lladó, V.; López, D.J.; Ibarguren, M.; Alonso, M.; Soriano, J.B.; Escribá, P.V.; Busquets, X. Regulation of the cancer cell membrane lipid composition by NaCHOleate. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Knobloch, M. The Role of Lipid Metabolism for Neural Stem Cell Regulation. Brain Plast. 2017, 3, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Fiol-deRoque, M.A.; Gutierrez-Lanza, R.; Terés, S.; Torres, M.; Barceló, P.; Rial, R.V.; Verkhratsky, A.; Escribá, P.V.; Busquets, X.; Rodríguez, J.J. Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology 2013, 14, 763–775. [Google Scholar] [CrossRef]
- Orth, M.; Bellosta, S. Cholesterol: Its Regulation and Role in Central Nervous System Disorders. Cholesterol 2012, 2012, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Dubreuil, V.; Arai, Y.; Wilsch-Brauninger, M.; Schwudke, D.; Saher, G.; Miyata, T.; Breier, G.; Thiele, C.; Shevchenko, A.; et al. Ablation of cholesterol biosynthesis in neural stem cells increases their VEGF expression and angiogenesis but causes neuron apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 8350–8355. [Google Scholar] [CrossRef] [Green Version]
- Driver, A.M.; Kratz, L.E.; Kelley, R.I.; Stottmann, R.W. Altered cholesterol biosynthesis causes precocious neurogenesis in the developing mouse forebrain. Neurobiol. Dis. 2016, 91, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Weiser, M.; Butt, C.; Mohajeri, M. Docosahexaenoic Acid and Cognition throughout the Lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, S.C. Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, M.; Takashima, N.; Matsumata, M.; Ikegami, S.; Kontani, M.; Hara, Y.; Kawashima, H.; Owada, Y.; Kiso, Y.; Yoshikawa, T.; et al. Arachidonic Acid Drives Postnatal Neurogenesis and Elicits a Beneficial Effect on Prepulse Inhibition, a Biological Trait of Psychiatric Illnesses. PLoS ONE 2009, 4, e5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakayori, N.; Maekawa, M.; Numayama-Tsuruta, K.; Katura, T.; Moriya, T.; Osumi, N. Distinctive effects of arachidonic acid and docosahexaenoic acid on neural stem/progenitor cells. Genes Cells 2011, 16, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Sakayori, N.; Kikkawa, T.; Tokuda, H.; Kiryu, E.; Yoshizaki, K.; Kawashima, H.; Yamada, T.; Arai, H.; Kang, J.X.; Katagiri, H.; et al. Maternal dietary imbalance between omega-6 and omega-3 polyunsaturated fatty acids impairs neocortical development via epoxy metabolites. Stem Cells 2016, 34, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.; Busquets, X.; Escribá, P.V. Brain Lipids in the Pathophysiology and Treatment of Alzheimer’s Disease. In Update on Dementia; InTech: Rijeka, Croatia, 2016. [Google Scholar]
- Ibarguren, M.; López, D.J.; Escribá, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [Green Version]
- Astarita, G.; Jung, K.-M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient Liver Biosynthesis of Docosahexaenoic Acid Correlates with Cognitive Impairment in Alzheimer’s Disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, D.; Simons, K. Lipid Rafts As a Membrane-Organizing Principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Staubach, S.; Hanisch, F.-G. Lipid rafts: Signaling and sorting platforms of cells and their roles in cancer. Expert Rev. Proteomics 2011, 8, 263–277. [Google Scholar] [CrossRef]
- Langelier, B.; Linard, A.; Bordat, C.; Lavialle, M.; Heberden, C. Long chain-polyunsaturated fatty acids modulate membrane phospholipid composition and protein localization in lipid rafts of neural stem cell cultures. J. Cell. Biochem. 2010, 110, 1356–1364. [Google Scholar] [CrossRef]
- Liu, R.-Z.; Mita, R.; Beaulieu, M.; Gao, Z.; Godbout, R. Fatty acid binding proteins in brain development and disease. Int. J. Dev. Biol. 2010, 54, 1229–1239. [Google Scholar] [CrossRef]
- Matsumata, M.; Inada, H.; Osumi, N. Fatty acid binding proteins and the nervous system: Their impact on mental conditions. Neurosci. Res. 2016, 102, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, T.E. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005, 19, 1028–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y. Role of Fabp7, a Downstream Gene of Pax6, in the Maintenance of Neuroepithelial Cells during Early Embryonic Development of the Rat Cortex. J. Neurosci. 2005, 25, 9752–9761. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhang, M.; Larsen, C.P.; Xu, F.; Hua, W.; Yamashima, T.; Mao, Y.; Zhou, L. DHA promotes the neuronal differentiation of rat neural stem cells transfected with GPR40 gene. Brain Res. 2010, 1330, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Shulman, G.I. Etiology of Insulin Resistance. Am. J. Med. 2006, 119, S10–S16. [Google Scholar] [CrossRef] [Green Version]
- Perona, J.S. Membrane lipid alterations in the metabolic syndrome and the role of dietary oils. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1690–1703. [Google Scholar] [CrossRef]
- Cohen, A.W.; Combs, T.P.; Scherer, P.E.; Lisanti, M.P. Role of caveolin and caveolae in insulin signaling and diabetes. Am. J. Physiol. Metab. 2003, 285, E1151–E1160. [Google Scholar] [CrossRef] [Green Version]
- Das, U.N. A defect in the activity of Δ6 and Δ5 desaturases may be a factor predisposing to the development of insulin resistance syndrome. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 343–350. [Google Scholar] [CrossRef]
- Kabayama, K. TNF-Induced insulin resistance in adipocytes as a membrane microdomain disorder: Involvement of ganglioside GM3. Glycobiology 2004, 15, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Fujita, A.; Cheng, J.; Hirakawa, M.; Furukawa, K.; Kusunoki, S.; Fujimoto, T. Gangliosides GM1 and GM3 in the Living Cell Membrane Form Clusters Susceptible to Cholesterol Depletion and Chilling. Mol. Biol. Cell 2007, 18, 2112–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, J.L.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickel, P.E. Lipid rafts and insulin signaling. Am. J. Physiol. Metab. 2002, 282, E1–E10. [Google Scholar] [CrossRef] [PubMed]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J.-I. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 13678–13683. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, E.; Schwenk, R.W.; Glatz, J.F.C.; Luiken, J.J.F.P.; van Eys, G.J.J.M. High fat diet induced diabetic cardiomyopathy. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 219–225. [Google Scholar] [CrossRef]
- Haag, M.; Dippenaar, N.G. Dietary fats, fatty acids and insulin resistance: Short review of a multifaceted connection. Med. Sci. Monit. 2005, 11, RA359–RA367. [Google Scholar]
- Weijers, R.N.M. Lipid Composition of Cell Membranes and Its Relevance in Type 2 Diabetes Mellitus. Curr. Diabetes Rev. 2012, 8, 390–400. [Google Scholar] [CrossRef]
- Shigematsu, S.; Watson, R.T.; Khan, A.H.; Pessin, J.E. The Adipocyte Plasma Membrane Caveolin Functional/Structural Organization Is Necessary for the Efficient Endocytosis of GLUT4. J. Biol. Chem. 2003, 278, 10683–10690. [Google Scholar] [CrossRef] [Green Version]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Garg, A. High-monounsaturated-fat diets for patients with diabetes mellitus: A meta-analysis. Am. J. Clin. Nutr. 1998, 67, 577S–582S. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.S.; Vögler, O.; Sánchez-Domínguez, J.M.; Montero, E.; Escribá, P.V.; Ruiz-Gutierrez, V. Consumption of virgin olive oil influences membrane lipid composition and regulates intracellular signaling in elderly adults with type 2 diabetes mellitus. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 256–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgerson, J.S.; Hauptman, J.; Boldrin, M.N.; Sjostrom, L. XENical in the Prevention of Diabetes in Obese Subjects (XENDOS) Study: A randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004, 27, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.A. Dietary n-3 PUFA and CVD: A review of the evidence. Proc. Nutr. Soc. 2014, 73, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-W.; Chien, Y.-S.; Chen, Y.-J.; Ajuwon, K.; Mersmann, H.; Ding, S.-T. Role of n-3 Polyunsaturated Fatty Acids in Ameliorating the Obesity-Induced Metabolic Syndrome in Animal Models and Humans. Int. J. Mol. Sci. 2016, 17, 1689. [Google Scholar] [CrossRef]
- Kunesová, M.; Braunerová, R.; Hlavatý, P.; Tvrzická, E.; Stanková, B.; Skrha, J.; Hilgertová, J.; Hill, M.; Kopecký, J.; Wagenknecht, M.; et al. The influence of n-3 polyunsaturated fatty acids and very low calorie diet during a short-term weight reducing regimen on weight loss and serum fatty acid composition in severely obese women. Physiol. Res. 2006, 55, 63–72. [Google Scholar]
- Guelzim, N.; Huneau, J.-F.; Mathé, V.; Tesseraud, S.; Mourot, J.; Simon, N.; Hermier, D. N-3 fatty acids improve body composition and insulin sensitivity during energy restriction in the rat. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 203–211. [Google Scholar] [CrossRef]
- Mendis, S.; Nordet, P.; Fernandez-Britto, J.E.; Sternby, N. Atherosclerosis in children and young adults: An overview of the World Health Organization and International Society and Federation of Cardiology study on Pathobiological Determinants of Atherosclerosis in Youth study (1985–1995). Prev. Control 2005, 1, 3–15. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Davis, N.E. Atherosclerosis—An inflammatory process. J. Insur. Med. 2005, 37, 72–75. [Google Scholar]
- Shah, S. Primary Prevention of Cardiovascular Disease. InnovAiT Educ. Inspir. Gen. Pract. 2012, 5, 195–203. [Google Scholar] [CrossRef]
- Insel, P.A.; Head, B.P.; Ostrom, R.S.; Patel, H.H.; Swaney, J.S.; Tang, C.-M.; Roth, D.M. Caveolae and Lipid Rafts: G Protein-Coupled Receptor Signaling Microdomains in Cardiac Myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Bakovic, M. Lipid rafts in health and disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Das, D. Lipid Raft in Cardiac Health and Disease. Curr. Cardiol. Rev. 2009, 5, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Hentschel, A.; Zahedi, R.P.; Ahrends, R. Protein lipid modifications-More than just a greasy ballast. Proteomics 2016, 16, 759–782. [Google Scholar] [CrossRef]
- Tang, Z.; Scherer, P.E.; Okamoto, T.; Song, K.; Chu, C.; Kohtz, D.S.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Molecular Cloning of Caveolin-3, a Novel Member of the Caveolin Gene Family Expressed Predominantly in Muscle. J. Biol. Chem. 1996, 271, 2255–2261. [Google Scholar] [CrossRef] [Green Version]
- Scherer, P.E.; Okamoto, T.; Chun, M.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc. Natl. Acad. Sci. USA 1996, 93, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.M.; Lisanti, M.P. The caveolin proteins. Genome Biol. 2004, 5, 214. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Oh, P.; Horner, T.; Rogers, R.A.; Schnitzer, J.E. Organized Endothelial Cell Surface Signal Transduction in Caveolae Distinct from Glycosylphosphatidylinositol-anchored Protein Microdomains. J. Biol. Chem. 1997, 272, 7211–7222. [Google Scholar] [CrossRef] [Green Version]
- Gratton, J.-P.; Bernatchez, P.; Sessa, W.C. Caveolae and Caveolins in the Cardiovascular System. Circ. Res. 2004, 94, 1408–1417. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guasch-Ferré, M.; Hu, F.B.; Martínez-González, M.A.; Fitó, M.; Bulló, M.; Estruch, R.; Ros, E.; Corella, D.; Recondo, J.; Gómez-Gracia, E.; et al. Olive oil intake and risk of cardiovascular disease and mortality in the PREDIMED Study. BMC Med. 2014, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-González, M.Á.; Toledo, E.; Arós, F.; Fiol, M.; Corella, D.; Salas-Salvadó, J.; Ros, E.; Covas, M.I.; Fernández-Crehuet, J.; Lapetra, J.; et al. Extravirgin olive oil consumption reduces risk of atrial fibrillation: The PREDIMED (Prevención con Dieta Mediterránea) trial. Circulation 2014, 130, 18–26. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef]
- Alemany, R.; Perona, J.S.; Sánchez-Dominguez, J.M.; Montero, E.; Cañizares, J.; Bressani, R.; Escribá, P.V.; Ruiz-Gutierrez, V. G protein-coupled receptor systems and their lipid environment in health disorders during aging. Biochim. Biophys. Acta 2007, 1768, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Corella, D.; Sorlí, J.V.; Estruch, R.; Coltell, O.; Ortega-Azorín, C.; Portolés, O.; Martínez-González, M.Á.; Bulló, M.; Fitó, M.; Arós, F.; et al. MicroRNA-410 regulated lipoprotein lipase variant rs13702 is associated with stroke incidence and modulated by diet in the randomized controlled PREDIMED trial. Am. J. Clin. Nutr. 2014, 100, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Ollerenshaw, J.D.; Heagerty, A.M.; Bing, R.F.; Swales, J.D. Abnormalities of erythrocyte membrane fatty acid composition in human essential hypertension. J. Hum. Hypertens. 1987, 1, 9–12. [Google Scholar]
- Villar, J.; Montilla, C.; Muñiz-Grijalvo, O.; Muriana, F.G.; Stiefel, P.; Ruiz-Gutiérrez, V.; Carneado, J. Erythrocyte Na(+)-Li+ countertransport in essential hypertension: Correlation with membrane lipids levels. J. Hypertens. 1996, 14, 969–973. [Google Scholar] [CrossRef]
- Russo, C.; Olivieri, O.; Girelli, D.; Guarini, P.; Pasqualini, R.; Azzini, M.; Corrocher, R. Increased membrane ratios of metabolite to precursor fatty acid in essential hypertension. Hypertension 1997, 29, 1058–1063. [Google Scholar] [CrossRef]
- Heyden, S. Polyunsaturated and Monounsaturated Fatty Acids in the Diet to Prevent Coronary Heart Disease via Cholesterol Reduction. Ann. Nutr. Metab. 1994, 38, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R.; Vögler, O.; Terés, S.; Egea, C.; Baamonde, C.; Barceló, F.; Delgado, C.; Jakobs, K.H.; Escribá, P.V. Antihypertensive action of 2-hydroxyoleic acid in SHRs via modulation of the protein kinase A pathway and Rho kinase. J. Lipid Res. 2006, 47, 1762–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, J.A.; Pearce, A.D.; Sinclair, A.J.; Vingrys, A.J.; Weisinger, R.S.; Weisinger, H.S. Increased blood pressure later in life may be associated with perinatal n-3 fatty acid deficiency. Lipids 2003, 38, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Weisinger, H.S.; Armitage, J.A.; Sinclair, A.J.; Vingrys, A.J.; Burns, P.L.; Weisinger, R.S. Perinatal omega-3 fatty acid deficiency affects blood pressure later in life. Nat. Med. 2001, 7, 258–259. [Google Scholar] [CrossRef]
- Forsyth, J.S. Long chain polyunsaturated fatty acid supplementation in infant formula and blood pressure in later childhood: Follow up of a randomised controlled trial. BMJ 2003, 326, 953. [Google Scholar] [CrossRef] [Green Version]
- Begg, D.P.; Sinclair, A.J.; Stahl, L.A.; Premaratna, S.D.; Hafandi, A.; Jois, M.; Weisinger, R.S. Hypertension induced by ω-3 polyunsaturated fatty acid deficiency is alleviated by α-linolenic acid regardless of dietary source. Hypertens. Res. 2010, 33, 808–813. [Google Scholar] [CrossRef]
- Arrigoni, E.; Del Re, M.; Fidilio, L.; Fogli, S.; Danesi, R.; Di Paolo, A. Pharmacogenetic Foundations of Therapeutic Efficacy and Adverse Events of Statins. Int. J. Mol. Sci. 2017, 18, 104. [Google Scholar] [CrossRef]
- Vaughan, C.J.; Gotto, A.M.; Basson, C.T. The evolving role of statins in the management of atherosclerosis. J. Am. Coll. Cardiol. 2000, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Potje, S.R.; Grando, M.D.; Chignalia, A.Z.; Antoniali, C.; Bendhack, L.M. Reduced caveolae density in arteries of SHR contributes to endothelial dysfunction and ROS production. Sci. Rep. 2019, 9, 6696. [Google Scholar] [CrossRef]
- Ugidos, I.F.; Pérez-Rodríguez, D.; Fernández-López, A. A role for lipids as agents to alleviate stroke damage: The neuroprotective effect of 2-hydroxy arachidonic acid. Neural Regen. Res. 2017, 12, 1273–1275. [Google Scholar]
- Ugidos, I.F.; Santos-Galdiano, M.; Pérez-Rodríguez, D.; Anuncibay-Soto, B.; Font-Belmonte, E.; López, D.J.; Ibarguren, M.; Busquets, X.; Fernández-López, A. Neuroprotective effect of 2-hydroxy arachidonic acid in a rat model of transient middle cerebral artery occlusion. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F. Domains in bacterial membranes and the action of antimicrobial agents. Mol. Biosyst. 2009, 5, 580. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.W.; Tang, D.Z.; Mandrell, R.; Kelly, M.; Spindel, E.R. Bombinin-like peptides with antimicrobial activity from skin secretions of the Asian toad, Bombina orientalis. J. Biol. Chem. 1991, 266, 23103–23111. [Google Scholar] [PubMed]
- Sani, M.-A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, S.; He, K.; Huang, H. Membrane thinning caused by magainin 2. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef]
- Laganowsky, A.; Reading, E.; Allison, T.M.; Ulmschneider, M.B.; Degiacomi, M.T.; Baldwin, A.J.; Robinson, C.V. Membrane proteins bind lipids selectively to modulate their structure and function. Nature 2014, 510, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Martens, C.; Shekhar, M.; Borysik, A.J.; Lau, A.M.; Reading, E.; Tajkhorshid, E.; Booth, P.J.; Politis, A. Direct protein-lipid interactions shape the conformational landscape of secondary transporters. Nat. Commun. 2018, 9, 4151. [Google Scholar] [CrossRef]
- Henderson, P.J.F. Sugar transport proteins. Curr. Opin. Struct. Biol. 1991, 1, 590–601. [Google Scholar] [CrossRef]
- Griffith, J.K.; Baker, M.E.; Rouch, D.A.; Page, M.G.P.; Skurray, R.A.; Paulsen, I.T.; Chater, K.F.; Baldwin, S.A.; Henderson, P.J.F. Membrane transport proteins: Implications of sequence comparisons. Curr. Opin. Cell Biol. 1992, 4, 684–695. [Google Scholar] [CrossRef]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colas, C.; Ung, P.M.-U.; Schlessinger, A. SLC transporters: Structure, function, and drug discovery. Medchemcomm 2016, 7, 1069–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; He, G.; Kakarla, P.; Shrestha, U.; Ranjana, K.C.; Ranaweera, I.; Mark Willmon, T.; Barr, S.R.; Hernandez, A.J.; Varela, M.F. Bacterial Multidrug Efflux Pumps of the Major Facilitator Superfamily as Targets for Modulation. Infect. Disord. Drug Targets 2016, 16, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Engelman, D.M. Membranes are more mosaic than fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef]
- Lemaire-Ewing, S.; Lagrost, L.; Néel, D. Lipid rafts: A signalling platform linking lipoprotein metabolism to atherogenesis. Atherosclerosis 2012, 221, 303–310. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, C.; Yin, L.; Wan, M.; Wang, X.; Li, L.; Liu, Y.; Wang, Z.; Fu, P.; Zhang, N.; et al. N ε -Fatty acylation of Rho GTPases by a MARTX toxin effector. Science 2017, 358, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Dumas, F.; Haanappel, E. Lipids in infectious diseases—The case of AIDS and tuberculosis. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1636–1647. [Google Scholar] [CrossRef]
- Teruel, M.N.; Meyer, T. Translocation and reversible localization of signaling proteins: A dynamic future for signal transduction. Cell 2000, 103, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Foteini, P.; Pippa, N.; Naziris, N.; Demetzos, C. Physicochemical study of the protein-liposome interactions: Influence of liposome composition and concentration on protein binding. J. Liposome Res. 2019, 29, 313–321. [Google Scholar] [CrossRef]
- Chaparro Sosa, A.F.; Kienle, D.F.; Falatach, R.M.; Flanagan, J.; Kaar, J.L.; Schwartz, D.K. Stabilization of Immobilized Enzymes via the Chaperone-Like Activity of Mixed Lipid Bilayers. ACS Appl. Mater. Interfaces 2018, 10, 19504–19513. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Bose, D.; Giri, R.P.; Mukhopadhyay, M.K.; Chakrabarti, A. Effects of GM1 on brain spectrin-aminophospholipid interactions. Biochim. Biophys. Acta Biomembr. 2019, 1861, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.; Bonacci, T.; Wang, X.; Truong, A.; Arceci, A.; Zhang, Y.; Mills, C.A.; Kernan, J.L.; Liu, P.; Emanuele, M.J. The E3 Ubiquitin Ligase SCF(Cyclin F) Transmits AKT Signaling to the Cell-Cycle Machinery. Cell Rep. 2017, 20, 3212–3222. [Google Scholar] [CrossRef] [Green Version]
- Calera, M.R.; Martinez, C.; Liu, H.; Jack, A.K.; Birnbaum, M.J.; Pilch, P.F. Insulin increases the association of Akt-2 with Glut4-containing vesicles. J. Biol. Chem. 1998, 273, 7201–7204. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.; Ramm, G.; Lopez, J.A.; James, D.E. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3-L1 adipocytes. Cell Metab. 2008, 7, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [Green Version]
- Majewski, N.; Nogueira, V.; Robey, R.B.; Hay, N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 2004, 24, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Sardiello, M. AKT modulates the autophagy-lysosome pathway via TFEB. Cell Cycle 2017, 16, 1237–1238. [Google Scholar] [CrossRef]
- Cho, W.; Stahelin, R.V. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Rafter, J.D.; Das, S.; Cho, W. The molecular basis of differential subcellular localization of C2 domains of protein kinase C-alpha and group IVa cytosolic phospholipase A2. J. Biol. Chem. 2003, 278, 12452–12460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prades, J.; Alemany, R.; Perona, J.S.; Funari, S.S.; Vögler, O.; Ruiz-Gutiérrez, V.; Escribá, P.V.; Barceló, F. Effects of 2-hydroxyoleic acid on the structural properties of biological and model plasma membranes. Mol. Membr. Biol. 2008, 25, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobnies, A.E.; Davies, S.M.A.; Kraayenhof, R.; Epand, R.F.; Epand, R.M.; Cornell, R.B. CTP: Phosphocholine cytidylyltransferase and protein kinase C recognize different physical features of membranes: Differential responses to an oxidized phosphatidylcholine. Biochim. Biophys. Acta 2002, 1564, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Enyedi, B.; Jelcic, M.; Niethammer, P. The Cell Nucleus Serves as a Mechanotransducer of Tissue Damage-Induced Inflammation. Cell 2016, 165, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Alwarawrah, M.; Wereszczynski, J. Investigation of the Effect of Bilayer Composition on PKCα-C2 Domain Docking Using Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alonso-Sande, M.; Higuera, M.; Torres, M.; Busquets, X.; et al. The triacylglycerol, hydroxytriolein, inhibits triple negative mammary breast cancer cell proliferation through a mechanism dependent on dihydroceramide and Akt. Oncotarget 2019, 10, 2486–2507. [Google Scholar] [CrossRef] [Green Version]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The Opposing Contribution of SMS1 and SMS2 to Glioma Progression and Their Value in the Therapeutic Response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Guardiola-Serrano, F.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Ibarguren, M.; López, D.J.; Terés, S.; Alvarez, R.; Alonso-Sande, M.; Busquets, X.; Escribá, P.V. The Novel Anticancer Drug Hydroxytriolein Inhibits Lung Cancer Cell Proliferation via a Protein Kinase Cα- and Extracellular Signal-Regulated Kinase 1/2-Dependent Mechanism. J. Pharmacol. Exp. Ther. 2015, 354, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchlis, V.D.; Bucher, D.; McCammon, J.A.; Dennis, E.A. Membranes serve as allosteric activators of phospholipase A2, enabling it to extract, bind, and hydrolyze phospholipid substrates. Proc. Natl. Acad. Sci. USA 2015, 112, E516–E525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, C.C. Cytosolic phospholipase A₂: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.H.; Gerber, S.H.; Murray, D.; Leslie, C.C. The calcium binding loops of the cytosolic phospholipase A2 C2 domain specify targeting to Golgi and ER in live cells. Mol. Biol. Cell 2004, 15, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.H.; Murray, D.; Leslie, C.C.; Falke, J.J. Specific translocation of protein kinase Calpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol. Biol. Cell 2006, 17, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Adjobo-Hermans, M.J.W.; Goedhart, J.; Gadella, T.W.J. Regulation of PLCbeta1a membrane anchoring by its substrate phosphatidylinositol (4,5)-bisphosphate. J. Cell Sci. 2008, 121, 3770–3777. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; Gatta, A.T.; Levine, T.P. Lipid transfer proteins: The lipid commute via shuttles, bridges and tubes. Nat. Rev. Mol. Cell Biol. 2019, 20, 85–101. [Google Scholar] [CrossRef]
- Kaplan, M.R.; Simoni, R.D. Transport of cholesterol from the endoplasmic reticulum to the plasma membrane. J. Cell Biol. 1985, 101, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Urbani, L.; Simoni, R.D. Cholesterol and vesicular stomatitis virus G protein take separate routes from the endoplasmic reticulum to the plasma membrane. J. Biol. Chem. 1990, 265, 1919–1923. [Google Scholar]
- Moser von Filseck, J.; Čopič, A.; Delfosse, V.; Vanni, S.; Jackson, C.L.; Bourguet, W.; Drin, G. INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 2015, 349, 432–436. [Google Scholar] [CrossRef]
- Vance, J.E.; Aasman, E.J.; Szarka, R. Brefeldin A does not inhibit the movement of phosphatidylethanolamine from its sites for synthesis to the cell surface. J. Biol. Chem. 1991, 266, 8241–8247. [Google Scholar] [PubMed]
- Heino, S.; Lusa, S.; Somerharju, P.; Ehnholm, C.; Olkkonen, V.M.; Ikonen, E. Dissecting the role of the golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc. Natl. Acad. Sci. USA 2000, 97, 8375–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiapparino, A.; Maeda, K.; Turei, D.; Saez-Rodriguez, J.; Gavin, A.-C. The orchestra of lipid-transfer proteins at the crossroads between metabolism and signaling. Prog. Lipid Res. 2016, 61, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F. Non-vesicular lipid trafficking at the endoplasmic reticulum-mitochondria interface. Biochem. Soc. Trans. 2018, 46, 437–452. [Google Scholar] [CrossRef]
- Jain, A.; Beutel, O.; Ebell, K.; Korneev, S.; Holthuis, J.C.M. Diverting CERT-mediated ceramide transport to mitochondria triggers Bax-dependent apoptosis. J. Cell Sci. 2017, 130, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Vigh, L.; Maresca, B.; Harwood, J.L. Does the membrane’s physical state control the expression of heat shock and other genes? Trends Biochem. Sci. 1998, 23, 369–374. [Google Scholar] [CrossRef]
- Török, Z.; Crul, T.; Maresca, B.; Schütz, G.J.; Viana, F.; Dindia, L.; Piotto, S.; Brameshuber, M.; Balogh, G.; Péter, M.; et al. Plasma membranes as heat stress sensors: From lipid-controlled molecular switches to therapeutic applications. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1594–1618. [Google Scholar] [CrossRef] [Green Version]
- Balogi, Z.; Multhoff, G.; Jensen, T.K.; Lloyd-Evans, E.; Yamashima, T.; Jäättelä, M.; Harwood, J.L.; Vígh, L. Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Prog. Lipid Res. 2019, 74, 18–30. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The Stress of Protein Misfolding: From Single Cells to Multicellular Organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704. [Google Scholar] [CrossRef] [Green Version]
- Multhoff, G.; Botzler, C.; Wiesnet, M.; Müller, E.; Meier, T.; Wilmanns, W.; Issels, R.D. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 1995, 61, 272–279. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Roth, A.G.; Petersen, N.H.T.; Mahalka, A.K.; Olsen, O.D.; Moilanen, I.; Zylicz, A.; Knudsen, J.; Sandhoff, K.; Arenz, C.; et al. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature 2010, 463, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Miyake, K.; Golenbock, D.T.; Triantafilou, K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002, 115, 2603–2611. [Google Scholar] [PubMed]
- Broquet, A.H.; Thomas, G.; Masliah, J.; Trugnan, G.; Bachelet, M. Expression of the Molecular Chaperone Hsp70 in Detergent-resistant Microdomains Correlates with Its Membrane Delivery and Release. J. Biol. Chem. 2003, 278, 21601–21606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehrmann, M.; Liebisch, G.; Schmitz, G.; Anderson, R.; Steinem, C.; De Maio, A.; Pockley, G.; Multhoff, G. Tumor-Specific Hsp70 Plasma Membrane Localization Is Enabled by the Glycosphingolipid Gb3. PLoS ONE 2008, 3, e1925. [Google Scholar] [CrossRef] [Green Version]
- Balogh, G.; Horváth, I.; Nagy, E.; Hoyk, Z.; Benkõ, S.; Bensaude, O.; Vígh, L. The hyperfluidization of mammalian cell membranes acts as a signal to initiate the heat shock protein response. FEBS J. 2005, 272, 6077–6086. [Google Scholar] [CrossRef]
- Balogh, G.; Maulucci, G.; Gombos, I.; Horváth, I.; Török, Z.; Péter, M.; Fodor, E.; Páli, T.; Benkő, S.; Parasassi, T.; et al. Heat Stress Causes Spatially-Distinct Membrane Re-Modelling in K562 Leukemia Cells. PLoS ONE 2011, 6, e21182. [Google Scholar] [CrossRef] [Green Version]
- Kunimoto, S.; Murofushi, W.; Kai, H.; Ishida, Y.; Uchiyama, A.; Kobayashi, T.; Kobayashi, S.; Murofushi, H.; Murakami-Murofushi, K. Steryl Glucoside is a Lipid Mediator in Stress-responsive Signal Transduction. Cell Struct. Funct. 2002, 27, 157. [Google Scholar] [CrossRef] [Green Version]
- Literáti-Nagy, Z.; Tory, K.; Literáti-Nagy, B.; Kolonics, A.; Török, Z.; Gombos, I.; Balogh, G.; Vígh, L.; Horváth, I.; Mandl, J.; et al. The HSP co-inducer BGP-15 can prevent the metabolic side effects of the atypical antipsychotics. Cell Stress Chaperones 2012, 17, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Dowhan, W. Phospholipid-assisted protein folding: Phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. EMBO J. 1998, 17, 5255–5264. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Umeda, M.; Dowhan, W. Phospholipid-assisted Refolding of an Integral Membrane Protein. J. Biol. Chem. 1999, 274, 12339–12345. [Google Scholar] [CrossRef] [Green Version]
- Debnath, D.; Bhattacharya, S.; Chakrabarti, A. Phospholipid assisted folding of a denatured heme protein: Effect of phosphatidylethanolamine. Biochem. Biophys. Res. Commun. 2003, 301, 979–984. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Z.; Cox, B.E.; Amarnath, V.; Epand, R.F.; Epand, R.M.; Davies, S.S. Phosphatidylethanolamines Modified by γ-Ketoaldehyde (γKA) Induce Endoplasmic Reticulum Stress and Endothelial Activation. J. Biol. Chem. 2011, 286, 18170–18180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, R.; Joseleau-Petit, D.; Chattopadhyay, M.K.; Richarme, G. Chaperone-like Properties of Lysophospholipids. Biochem. Biophys. Res. Commun. 2001, 289, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Tillu, V.A.; Collins, B.M. Caveolae. Curr. Biol. 2018, 28, R402–R405. [Google Scholar] [CrossRef] [Green Version]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipid Element | Protein Element | Pathogenic/Physiological Condition | Lipids Implicated in Pathogenicity | Therapeutic Approach Targeting the Lipid Fraction | Reference |
|---|---|---|---|---|---|
| Lipid rafts PUFAs | IL-2, FcR, PKC, NF-kB, AP-1 | Altered localization of receptors, mediators and transcription factors | PUFAs | Dietary supply of PUFAs alters T- and B-lymphocyte membranes | [185] |
| Lipid rafts PUFAs | PTKs (LCK), CD45, CD3, FcR | SLE | Increased amount of lipid rafts in activated T-cells | - | [185] |
| PE | Atg8/LC3 | Double membrane formation of the autophagosome | - | - | [129] |
| Palmitoyl moeity | TLRs | Innate immune response, regulation of immune receptor functions | - | - | [129,228] |
| Several lipid moieties | Several proteins | Plasmodium falciparum (malaria) | - | NMT validated as an attractive antimalarial drug target | [129] |
| Several lipid moieties | Several proteins | Trypanosoma brucei (human African trypanosomiasis) | - | NMT identified as a promising target for sleeping sickness (inhibitor DDD85646) | [129] |
| Fatty acylation | Rho-family GTPases (lysine residues) | Vibrio cholera | Toxin peptide catalyzing the fatty acylation of lysine residues of Rho-family GTPases | - | [229] |
| Chol | CR3 and others | Mycobacterium tuberculosis | Extractable lipids they are important virulence factors | Host Chol is required for receptor-mediated phagocytosis of M. tuberculosis by a macrophage. Blocking antibodies showed that Chol is required for mycobacterial entry via CR3. Statins showed promise in vitro and in vivo for the treatment of tuberculosis | [230] |
| Diverse lipid moieties | Several proteins | Herpes simplex virus | - | - | [129] |
| Lipid rafts | CD4 | HIV infection | PUFAs, increased amount of lipid rafts | Disruption of host cell lipid rafts with cyclodextrin prevents HIV infection. Inhibiting sphingolipid synthesis by the virus particle reduces its infective capacity. | [185] |
| Myristoylation | Gag protein | HIV infection | Targeting lipidated viral or host proteins may lead to new antiviral agents. | [129,230] | |
| Chol | Gp41 fusion protein | HIV infection | - | - | [129,230] |
| Phosphoinositides | - | HIV infection | Effect on positive membrane curvature | - | [230] |
| Lipid rafts, edges of Chol-rich domains | CD4-CCR5/CXCR4 | HIV infection | Effect on the budding out of the host cell | - | [230] |
| Diverse lipid components | Gag-Gag, GPCR | HIV infection | Effect on the budding out of the host cell | - | [230] |
| Diverse lipid components | Gag multimerization | HIV infection | Budding virus are enriched in several lipids compared to the plasma membrane composition of the infected cells from which they originate | - | [230] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, M.; Rosselló, C.A.; Fernández-García, P.; Lladó, V.; Kakhlon, O.; Escribá, P.V. The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy. Int. J. Mol. Sci. 2020, 21, 2322. https://doi.org/10.3390/ijms21072322
Torres M, Rosselló CA, Fernández-García P, Lladó V, Kakhlon O, Escribá PV. The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy. International Journal of Molecular Sciences. 2020; 21(7):2322. https://doi.org/10.3390/ijms21072322
Chicago/Turabian StyleTorres, Manuel, Catalina Ana Rosselló, Paula Fernández-García, Victoria Lladó, Or Kakhlon, and Pablo Vicente Escribá. 2020. "The Implications for Cells of the Lipid Switches Driven by Protein–Membrane Interactions and the Development of Membrane Lipid Therapy" International Journal of Molecular Sciences 21, no. 7: 2322. https://doi.org/10.3390/ijms21072322